
Abstract
Accurate measurement of human epidermal growth factor receptor 2 (HER2) copy number variation (CNV) is very important for guiding the tumor target therapy in breast cancer. Digital PCR (dPCR) is a sensitive and an absolute quantitative method, which can be used to detect HER2 CNV. Three HER2 exon-specific digital PCR assays along with three new reference genes assays (homo sapiens ribonuclease P RNA component H1 (RPPH1), glucose-6-phosphate isomerase (GPI), and chromosome 1 open reading frame 43 (C1ORF43), on different chromosomes) were established and validated by using standard reference material, 8 different cell lines and 110 clinical Formalin-fixed and paraffin-embedded (FFPE) samples. DPCR can achieve precise quantification of HER2 CNV by calculating the ratio of HER2/reference gene. The positive and negative coincidence rates were 98% (53/54) and 95% (53/56), respectively, compared with fluorescence in situ hybridization (FISH) diagnostic result 110 of FFPE samples. The common reference gene CEP17 used for FISH diagnostic was not suitable as single reference gene for HER2 CNV measurements by dPCR. The best practice of HER2 CNV determination by dPCR is to conduct the three duplex assays of H1 (HER2 exon 4) with the proposed three new reference genes, with a positive cut-off value of H1/RPPH1 ≥ 2.0 or H1/averaged reference gene ≥ 2.0. The proposed dPCR method in our study can accurately provide absolute copy number of HER2 and reference gene on an alternative chromosome, thus avoiding false negative caused by polysomy of chromosome 17. The improved molecular typing and diagnosis of breast cancer will better guide clinical medication.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common malignant tumor in women [1], which is a serious threat to patients’ life and health. Breast cancer has become the most frequent cancer in the world according to the recent report on the global cancer burden for 2020 released by World Health Organization’s International Agency for Research on Cancer (IARC) (https://www.iarc.who.int/wp-content/uploads/2020/12/pr292_E.pdf). HER2-positive breast cancer accounts for about 20% ~ 30% of invasive breast cancer, which is characterized by high invasiveness, high risk of recurrence, rapid progression, and poor prognosis [2]. Detection of HER2 gene expression in tumor cells of breast cancer patients is beneficial to the choice of treatment and prognosis. For example, successful anti-tumor treatment with trastuzumab (Herceptin) is strongly correlated with HER2 overexpression in the tumor.
Currently, fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) are the major methods for detection of HER2 overexpression at gene and protein level in clinical practice, respectively. The American Society of Clinical Oncology defines HER2-positive tumors as those with an average copy number of HER2 genes per nucleus > 6 (single probe test) or HER2/CEP17 ratio > 2.2 (double probe test) detected by FISH [3, 4], but discordance may occur in cancers with increased CEP17 copy number. Those with HER2/CEP17 ratio < 1.8 or HER2 copy number < 4 per nucleus were defined to be negative. Therefore, those with 1.8 < HER2/CEP17 ratio < 2.2 followed in an equivocal region is another issue need to improve.
CEP17 is a common reference probe approved by the US Food and Drug Administration (FDA) for the detection of HER2 amplification in the FISH. However, an increase of CEP17 signal detected in breast cancer patients would affect the analytical result based on the HER2/CEP17 ratio. It is estimated that this occurs in 2% to 9% of breast cancers [5, 6]. In a comprehensive study, Wolff et al. [3] stated that approximately 20% of current HER2 testing may be inaccurate, which underscores the importance of developing more accurate and sensitive methods to determine HER2 status.
A number of reports highlight the superior performance of digital PCR (dPCR) for copy number variation (CNV) analysis [7, 8]. DPCR, as an absolute copy number measurement method, is ideally fit for detection of HER2 amplification [9,10,11,12,13,14]. It has been reported that dPCR can detect a 1.25-fold difference in copy number in cell line DNA [8], which will increase the sensitivity and reduce the equivocal region significantly. However, further validation in clinical samples is needed. The aim of this paper is to (1) develop highly accurate and sensitive dPCR assays to detect copy number concentration of HER2, (2) explore a potential new reference genes to determine HER2 CNV, (3) test suitability of the common reference gene CEP17, and (4) examine the proposed new method by testing clinical samples from breast cancer patients.
Materials and methods
Cell lines culture and genomic DNA extraction
HER2-positive cell line (HCC1954, CRL-2338™) was purchased from ATCC (USA), and HER2-negative cell (PLCL7) was provided by Fudan University. The 1640 culture medium was used for two cell lines, with 10% Fetal bovine serum (FBS, Invitrogen, #10099141) and 1% Penicillin–Streptomycin Solution (Invitrogen, #15140122). Genomic DNA was extracted by using the QIAGEN DNA purification kit (QIAGEN, #13362) according to the manufacturer’s instructions. The sizes of the major fragments of the extracted genomic DNAs were higher than 10 kb.
Clinical samples
One hundred and eleven FFPE clinical breast tumor samples were provided by PLA Rocket Force Characteristic Medical Center. All the samples have been assessed previously by IHC and FISH at PLA Rocket Force Characteristic Medical Center. The DNA was extracted by using the GeneRead DNA FFPE kit (QIAGEN, #180134).
All patients were informed that the samples were not only used for pathological diagnosis by FISH and immunohistochemistry, but also for non-profit scientific research. The dPCR analysis was performed on existing samples collected during standard diagnostic tests, posing no extra burden to patients.
IHC and FISH
IHC and FISH testing of the HER2 breast cancer samples were completed by PLA Rocket Force Characteristic Medical Center. The method is described in detail in Electronic Supplementary Material. The results of FISH and IHC of the 111 clinical specimens were shown in Electronic Supplementary Material Table S1.
DPCR assay optimization
The primers and probes for three exon-specific assays (H1: exon 4, H2: exon 16, H3: exon 24) for HER2 gene and for reference genes assays were designed with Primer Express 3.0.1 software (Electronic Supplementary Material Table S2). To determine CNV of HER2, three potential reference genes, Homo sapiens ribonuclease P RNA component H1 (RPPH1), glucose-6-phosphate isomerase (GPI), and chromosome 1 open reading frame 43 (C1ORF43), were chosen according to previous reports [15]. CEP17, the most frequent reference gene in HER2 clinical diagnostics, was also tested [10]. For the six proposed new assays (H1, H2, H3, RPPH1, GPI, and C1ORF43), each PCR was firstly conducted for the optimization based on the quantification of copy number concentration and separation of positive and negative cluster. The optimization of annealing temperature and concentration of primers and probes was shown in Electronic Supplementary Material Figs. S1 and S2, respectively. In brief, 20 μL reaction mixtures were prepared, containing the template DNA (2 ng ~ 42 ng), 2 × ddPCR supermix for probes without UNG (BioRad, CA, USA), primers, and probes. Droplets were generated on a QX200 droplet generator (BioRad, CA, USA). The optimized primer and probe concentration was in Electronic Supplementary Material Table S2. The PCR was performed at 95 ℃ for 10 min; 94 ℃ for 30 s, 60 ℃ for 1 min with 40 cycles; and 98 ℃ for 10 min by using a thermocycler (Viti). Then, the thermal cycled plate was analyzed on QX200 droplet reader (BioRad, CA, USA). Data analysis was performed with the QuantaSoft software (version 1.7.4., Bio-Rad). For detailed dPCR workflow, please refer our previous report [16].
Dynamic range and limit of blank, detection, and quantification of HER2 CNV
To determine the dynamic range of the above dPCR assays for HER2 CNV, a series of dilutions was prepared by mixing the HER2-positive cell line genomic DNA with a constant concentration of wild-type PLCL7 genomic DNA. Each dilution was analyzed in three replicates with three duplex assays (H1 with RPPH1, H2 with RPPH1 and H3 with RPPH1, respectively). The ratios of copy number concentration of HER2 to RPPH1 (H1 to RPPH1, H2 to RPPH1 and H3 to RPPH1, respectively) was calculated.
The limit of blank (LoB), limit of detection (LoD) and limit of quantification (LoQ) for HER2 CNV were calculated according to Eqs. (1) and (4) reported in the guideline of EP17-A [17].
Method validation with certified reference material
HER2-positive standard reference materials (SRM2373, including five different cell line genomic DNA) purchased from National Institute of Standards and Technology (NIST) were used to validate the dPCR assays. Both the primer and probe for HER2 and reference gene DCK listed on the NIST’s certificate was synthesized and tested firstly by using the SRM2373. Then, the proposed six assays, including three HER2 exon specific assays and four reference gene assays, in a duplex way (H1/RPPH1, H1/GPI, H1/C1ORF43, H2/CEP17, H3/CEP17) with three replicates, were validated using the same reference material. And duplex assay of HER2 and reference gene CEP17 in previous report [10] was compared with our proposed assays. The reaction mixture, with a total volume of 20 µL, comprised 2 µL of template DNA. The PCR and analysis procedures were described above.
Suitability testing of the new reference genes
At present, there is no uniform application of well-behaved reference gene in detection of HER2-positive breast cancer. Even CEP17, the most commonly used reference gene, has been reported an amplification occurred [5, 6]. Therefore, three new potential reference genes were chosen according to previous study [15], and specific PCR primer and probe were designed for the three reference genes. In order to verify whether they are applicable in most cells, eight cell lines were used for the analysis. The established methods were used to quantify the copy number concentrations of the three reference genes in the approximate equivalent genomic DNA. The test was performed in a duplex format (H1/RPPH1, H2/GPI, H3/C1ORF43) with three replicates.
Method validation of clinical samples
To verify the applicability of the proposed dPCR method, 111 clinical FFPE samples, including 54 positive and 57 negative samples according to FISH diagnostic results, were analyzed. One FISH negative sample (P32) was excluded as none of the assays were amplified and the sample was not improved after re-extraction. To simplify the analysis, two duplex assays (one is H1/RPPH1, another one is H2/CEP17) were used to determine the HER2 status for 110 samples (except P32). Each clinical sample was analyzed with three replicates. The ratios of H1/RPPH1, H2/RPPH1, H1/CEP17, and H2/CEP17 were calculated. For some HER2-negative samples, C1ORF43 or GPI were further tested if RPPH1 and CEP17 were inconsistent.
Results and discussion
Validation of dPCR assay for HER2 copy number variation
The position of three target exons in HER2 and amplification results of dPCR is shown in Fig. 1a. The exon-specific assay generated two clusters (positive and negative) for each exon (H1, H2, H3), and only one cluster was generated for each corresponding no-template control (NTC), suggesting there was no contamination during the analysis. The same is true for three reference genes (Fig. 1b–d).

The location of three exons of HER2 and the optimized results of dPCR assay for HER2 exon-specific (a) and reference genes RPPH1 (b), GPI (c), and C1ORF43 (d), respectively. The event number on x axis is the number of accepted droplets. Ch1 amplitude on the Y axis is the fluorescence intensity of channel 1 for FAM, and Ch2 amplitude is the fluorescence intensity of channel 2 for VIC/HEX
To facilitate the detection, three non-competitive duplex assays (a combination of H1 with RPPH1, H2 with GPI, and H3 with C1ORF43, respectively, one measuring HER2 copy number concentration with FAM fluorescence and another one is measuring reference gene with VIC fluorescence) were used for detection of HER2 CNV (Electronic Supplementary Material Fig. S3). The copy number concentration of HER2 in HER2-positive cell lines was about 36 times higher than that in HER2-negative cell lines (Electronic Supplementary Material Table S3). However, the results of three reference genes were not significantly different for both HER2-positive and HER2-negative cell line. This suggests the three exon-specific assays can be used to detect the HER2 amplification. The copy number concentrations of the three HER2 exons were consistent with each other for HER2-positive and HER2-negative cell line, respectively (Electronic Supplementary Material Table S3), indicating no significant difference occurred in the three target exons in each cell line. The ratios of HER2/RPPH1 calculated for both HER2-negative and HER2-positive cell lines were 0.94 and 36, respectively. These two cell lines were used for evaluating the following detection limit of HER2 CNV by using H1/RPPH1, H2/RPPH1, and H3/RPPH1.
Limit of blank, detection, and quantification
To determine the LoB of HER2 CNV, the copy number ratio of HER2 to RPPH1 was measured in sixty-two duplex assays of H1/RPPH1, H2/RPPH1, and H3/RPPH1, respectively, obtained from 4 HER2-negative cell line genomic DNA. LoB was estimated according to the Eq. (1) in EP17-A [17] due to the distribution of 62 blank measurements for each duplex experiment was symmetrical (Electronic Supplementary Material Fig. S4). The estimated LoB of 0.98, 0.98, and 0.99 were obtained for H1/RPPH1, H2/RPPH1, and H3/RPPH1, respectively. To determine the LoD, 12 replications were performed on each sample in 5 different runs with a low ratio ranging from 1.0 to 1.4 of HER2/RPPH1 on three different days to ensure the total assay variation was reflected. After Shapiro–Wilk test, the 60 measurement results of samples with low concentration were distributed as a Gaussian distribution, so LoD was calculated according to Eq. (2) [17]. Finally, the LoD of the three assays were 1.21, 1.23 and 1.22, respectively (Table 1).
Sixty measurements from 5 level samples of HER2/RPPH1 were used to determine LoQ. The SDs of the 60 measurements of three assays were 0.14, 0.15, and 0.13. The estimate (bias + 2SDs = 1.474, 1.499, 1.482) is less than the defined goal for total error (1.50), so the LoQ = LoD.
Dynamic range of HER2 copy number concentration
The linear range was investigated by measuring the ratio of H1/RPPH1, H2/GPI, and H3/C1ORF43. We tested the linearity ratio of each exon-specific assay using a dilution series of HER2-positive template ranging from 1.0 to 36 (Fig. 2). Good linearity (0.84 < slope < 1.01; R2 > 0.99) was obtained for all three ratio of HER2/reference gene across the entire dilution series.

Detection of H1/RPPH1, H2/GPI, and H3/C1ORF43 ratio in the presence of serial dilutions of HER2-positive template ranging from 1.0 to 36. The mean and standard deviation (error bar) are shown for each dilution (n = 3). H1, H2, and H3 representing the assay for HER2 exon 4, 16, and 24, respectively
Validation by reference materials
We first used the assays of HER2 and reference gene DCK listed on the NIST certificate (https://www-s.nist.gov/srmors/certificates/2373.pdf) to determine the copy number concentration and ratio of NIST reference materials. The results showed that they were consistent with the information values on the NIST certificate (Electronic Supplementary Material Fig. S5a and 5b), which showed that our dPCR process worked properly.
Then, the proposed three HER2 exon-specific assays and reference genes (RPPH1, GPI, and C1ORF43) were validated by using the NIST reference materials (SRM A-E). It was found that the copy number concentration of each HER2-specific assay of NIM was consistent with the NIST’s information values (Electronic Supplementary Material Fig. S6a) in all five SRMs. The copy number concentration of RPPH1 in A, B, C, and D were consistent with NIST (Electronic Supplementary Material Fig. S6b). However, we did find a significant increase of RPPH1 in SRM E. The copy number concentration of GPI and C1ORF43 was different in the NIST SRMs which were from five different cell lines, indicating none of the three tested reference genes were stable in all the five cell lines. However, when the three reference genes were averaged, significant difference was only observed in E (Fig. 3b). The averaged copy number concentration of three reference genes was significantly higher than the NIST’s value in SRM E, indicating an amplification of RPPH1, GPI, and C1ORF43 in SRM E. The copy number ratio of averaged three exons to averaged three reference genes agreed well with the reference value of NIST in SRM A-D (Fig. 3c).

Five NIST standard reference materials (A–E) were used for validation of our proposed dPCR method and previously reported method. a HER2 copy number concentration quantified by the proposed assays in this study (labeled as HER2-NIM, an averaged value of the three exon specific assays ± 95% confidence interval (CI)), previously reported assay (labeled as HER2-Method of previous paper, mean ± 95% CI) and NIST’s assay (labeled as HER2-NIST, mean ± 95% CI). b Reference gene copy number concentration quantified by the proposed assays (labeled as Reference gene-NIM, the averaged value of RPPH1, GPI, and C1ORF43 ± 95% CI), previously reported assay (labeled as Reference gene-Method of previous paper, the mean value of CEP17 ± 95% CI) and NIST’s assay (labeled as Reference gene-NIST, mean ± 95% CI). c Ratio of HER2/Reference gene characterized by this study (labeled as Ratio-NIM, mean ± 95% CI), previously reported assay (labeled as Ratio-Method of previous paper, mean ± 95% CI) and on NIST’s certificate (labeled as Ratio-NIST, reference value ± 95% CI)
Additionally, our proposed assay was compared to the duplex assay (HER2/CEP17) in a reported previous paper [10] and labeled as Method of previous paper in Fig. 3. The copy number concentrations of HER2 in SRM B, C, D, and E were consistent determined by three methods. However, it was significantly lower than that determined by methods of national institute of metrology (NIM) and NIST in SRM A. Interestingly, the CEP17 copy number concentration was consistent with that determined by methods of NIM and NIST in SRM D, but significant higher in other four SRMs. This leads to a significant lower in the ratio of HER2/CEP17, which indicates CEP17 is also not stable in SRM A, B, C, and E.
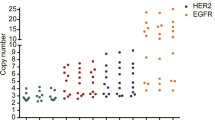
Suitability of reference genes in different cell lines
In order to verify the applicability of the three potential reference genes as well as CEP17, the copy number concentrations of HER2 and four reference genes (RPPH1, GPI, C1ORF43, and CEP17) as well as their ratios were analyzed in 8 randomly selected cell lines (Fig. 4). However, it is meaningless to compare the absolute copy number concentration of each target gene in different cell lines since the input of each cell line DNA amount may be different. Therefore, we compared the copy number concentration of HER2 and four reference genes in the same cell line. The results showed that the copy number concentrations of HER2 and reference genes were consistent in 8 cell lines (Fig. 4a). However, the ratio of HER2/CEP17 increased in PLCL5 and SW620, and ratio of HER2/RPPH1 decreased in HCT116 (Fig. 4b).

The mean of three HER2 assays and reference genes copy number concentration (a) and ratio (b) of HER2/reference genes in 8 different cell lines. Copy number concentration of HER2 was calculated by averaging the three HER2 exon-specific assays H1, H2, and H3. There was no significant difference among the results for the three assays targeting the three exons of HER2 in each cell line so the averaged concentration of the three HER2 assays was used in the analysis
Validation by analysis of clinical samples
The copy number concentrations of H1, H2, CEP17, and RPPH1 for all samples were listed in Electronic Supplementary Material Table S4–S5, respectively. The ratios of H1/RPPH1, H2/RPPH1, H1/CEP17, and H2/CEP17 were shown in Fig. 5. A significant difference was observed between HER2/RPPH1 and HER2/CEP17 in the FISH positive samples (Fig. 5a, p > 0.0001). However, most ratios were ≥ 2.0, thereby H1/RPPH1 ≥ 2.0 was used as the cut-off value to distinguish positive. Consequently, only one sample (P23# with a ratio of 1.7) was selected out as a negative (a FISH false positive, Electronic Supplementary Material Table S4). P23 was further confirmed to be a negative when referring the ratio of H2/RPPH1 (equal to 1.2). After reviewing the copy number concentration of H1 and H2, we found that H1 was higher than H2 in most samples, indicating that H1 (HER2 exon 4) is more sensitive in degraded sample due to the amplification length of H1 (53 bp) is shorter than that of H2 (63 bp). The cut-off value of H1/RPPH1 ≥ 2.0 is confirmed to be suitable for the HER2-positive sample.

The distribution of copy number ratios of HER2/RPPH1 and HER2/CEP17 in 54 positive samples (a) and 56 negative samples (b). (ns: no significant; ***: significant, 0.0001 < P < 0.05; solid line in the violin plot is the median and the dashed line is the quartile). H1 and H2 represent the results from the assay of HER2 exon 4 and 16, respectively. CEP17 and RPPH1 represent the results from the reference gene assay
We also tested if CEP17 can be used for detection of HER2 CNV in the clinical samples. When using the H1/CEP17 ≥ 2.0 as the cut-off value, 2 samples with a ratio < 2.0 were selected out. When referring the H2/CEP17 ≥ 2.0 as the cut-off value, 9 samples (labeled with * in Electronic Supplementary Material Table S4) were selected out. To further reviewing the copy number concentration, except P23, the other 8 samples can be attributed to false negative due to their stronger amplification of both HER2 and CEP17. This can be explained by polysomy of chromosome 17. The amplification of CEP17 in HER2-positive samples leads to a decrease of the ratio of HER2/CEP17, which can be further confirmed by the significant difference between HER2/CEP17 and HER2/RPPH1 (Fig. 5a). Therefore, CEP17 is not suitable as the reference gene for HER2 CNV. It is interesting to note that the copy number concentration of CEP17 in sample P23 was quite close to that of RPPH1, which further confirms that it is a false positive.
For the 56 FISH negative samples, no significant difference was observed between the ratio of HER2/RPPH1 and HER2/CEP17 (Fig. 5b). Most ratios of HER2/RPPH1 were < 2.0, thereby the cut-off value were firstly defined to be H1/RPPH1 or H2/RPPH1 < 2.0. The result showed 11 samples were selected out due to their ratio ≥ 2.0 (highlighted with blue, yellow, and green in Electronic Supplementary Material Table S5). To further check if these were positive, copy number concentration of RPPH1 was referred, and the other two reference genes (GPI and C1ORF43) were tested for consideration.
For the possible positive samples, the averaged copy number ratio of H1/RPPH1, H2/RPPH1, H1/GPI, H1/C1ORF43 < 2.0 was used as the cut off value. According to such a cut-off value, 6 samples (highlighted with blue in Electronic Supplementary Material Table S5) can be confirmed as HER2 negative as a significant decrease of RPPH1 was observed, which caused by either unsuccessful amplification or deletion. One sample (P52, highlighted with yellow) with a ratio equal to 2.05 was confirmed as positive, as a relative higher copy number concentration of HER2 and normal copy number concentration of all three reference genes was observed. However, 4 samples highlighted with green were needed for further check as no additional copy number information of GPI and C1ORF43 were obtained due to sample limitation. Therefore, the copy number concentration of HER2 and more than two reference genes as well as their ratio should be considered in determining the status of HER2, rather than relying on only one reference gene.
When using the ratio of H1/CEP17 < 2.0 as the cut-off value, 13 samples with ratio of H1/CEP17 ≥ 2.0 were selected out (all labeled with * in Electronic Supplementary Material Table S5). By reviewing the copy number concentration of the other three reference genes, 11 samples were confirmed as false positive due to deletion of CEP17. It is interesting to note the rest two samples (P46 and P50) can be confirmed as positive as copy number concentration of RPPH1 and CEP17 was consistent. Thereby this further confirms that CEP17 should not be used as a single reference gene.
Best practice of HER2 CNV measurements by dPCR
It is well known that FISH relies on the experience of a pathologist and is a challenge to tumor heterogeneity. In the latest Chinese Guideline for HER2 Detection in Breast Cancer [18], the detection results of double probe FISH can be divided into five categories: HER2/CEP17 ≥ 2.0, (1) if average HER2 copy number/cell ≥ 4.0, positive; (2) if average HER2 copy number/cell < 4.0, negative; HER2/CEP17 < 2.0, (3) if average HER2 copy number/cell ≥ 6.0, positive; (4) if 4.0 ≤ average HER2 copy number/cell < 6.0, need refer IHC; (5) if average HER2 copy number/cell < 4.0, negative. It is especially difficult to make a decision when the result falls into category 4.
The proposed dPCR method provides the HER2 and reference gene copy number concentration as well as the ratio, which makes the results very objective. Moreover, dPCR using extracted genomic DNA samples can avoid heterogeneity of tissue sample. The suitability of exon-specific assays for detection of HER2 was confirmed by the NIST SRM and clinical samples, especially H1 is more sensitive in clinical samples. Therefore, H1 should be considered first and H2 can be used as supplement. For the choice of reference genes, it was observed in some clinical samples that both a deletion of CEP17 or RPPH1 caused the ratio of HER2/CEP17 or HER2/RPPH1 to be higher than 2.0 and an amplification of CEP17 leading to a decrease of HER2/CEP17. This indicates that either CEP17 or RPPH1 should not be used as a single reference gene for determining the HER2 CNV. RPPH1 combined with two other reference genes are the best choice. Thereby the best practice of HER2 CNV by dPCR is to conduct the three duplex assays of combing H1 and three reference genes.
The established dPCR method was used to test 110 samples of invasive breast cancer, of which 56 samples had a ratio of H1/RPPH1 ≥ 2.0, indicating HER2 positive; The mean value of HER2/ reference gene in 54 samples was < 2.0, indicating HER2 negative. By traditional methods (IHC combined with FISH), the positive and negative coincidence rates were 98% (53/54) and 95% (53/56), respectively. During the analysis, we found it difficult to define a good reference gene for determination of HER2 status, although RPPH1 was stable in most samples. Therefore, it is recommended to measure RPPH1, GPI and C1ORF43.
Due to the small number of samples tested in this study, the applicability of the HER2/Reference ≥ 2.0 cut-off value for HER2-positive samples needs to be further evaluated in future work. Additionally, this method has great potential application in liquid biopsy for early diagnostic of breast cancer [19,20,21]. Thus, our future work will also focus on the suitability of the proposed method in liquid biopsy.
Conclusion
In this study, a highly sensitive HER2 CNV method by dPCR was established and evaluated by NIST SRM as well as clinical FFPE samples. Compared with IHC and FISH technologies, dPCR is not interfered by personnel operation, detection reagents, and pathologist’s subjective interpretation. It can achieve precise quantification of the HER2 CNV by calculating the ratio of HER2/reference and provide powerful help for the molecular typing and diagnosis of breast cancer, so as to better guide clinical medication. In addition, the proposed dPCR method in our study can accurately provide the absolute copy number of HER2 and reference genes on an alternative chromosome, thus avoiding the false negative caused by polysomy of chromosome 17.
References
Janelsins MC, Kohli S, Mohile SG, Usuki K, Ahles TA, Morrow GR. An update on cancer- and chemotherapy-related cognitive dysfunction: current status. Semin Oncol. 2011;38(3):431–8. https://doi.org/10.1053/j.seminoncol.2011.03.014.
Wei Z, Li B, Zefei J. Research status of HER-2 gene mutation in breast cancer. Chin J Breast Dis. 2016;10:362–5.
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, van de Vijver M, Wheeler TM, Hayes DF. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25(1):118–45. https://doi.org/10.1200/jco.2006.09.2775.
Vranic S, Teruya B, Repertinger S, Ulmer P, Hagenkord J, Gatalica Z. Assessment of HER2 gene status in breast carcinomas with polysomy of chromosome 17. Cancer. 2011;117(1):48–53. https://doi.org/10.1002/cncr.25580.
Watters AD, Going JJ, Cooke TG, Bartlett JM. Chromosome 17 aneusomy is associated with poor prognostic factors in invasive breast carcinoma. Breast Cancer Res Treat. 2003;77(2):109–14. https://doi.org/10.1023/a:1021399923825.
Bartlett JM, Going JJ, Mallon EA, Watters AD, Reeves JR, Stanton P, Richmond J, Donald B, Ferrier R, Cooke TG. Evaluating HER2 amplification and overexpression in breast cancer. J Pathol. 2001;195(4):422–8. https://doi.org/10.1002/path.971.
Huggett JF, Foy CA, Benes V, Emslie K, Garson JA, Haynes R, Hellemans J, Kubista M, Mueller RD, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT, Bustin SA. The digital MIQE guidelines: minimum information for publication of quantitative digital PCR experiments. Clin Chem. 2013;59(6):892–902. https://doi.org/10.1373/clinchem.2013.206375.
Weaver S, Dube S, Mir A, Qin J, Sun G, Ramakrishnan R, Jones RC, Livak KJ. Taking qPCR to a higher level: Analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods (San Diego, Calif). 2010;50(4):271–6. https://doi.org/10.1016/j.ymeth.2010.01.003.
Whale AS, Huggett JF, Cowen S, Speirs V, Shaw J, Ellison S, Foy CA, Scott DJ. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. 2012;40(11): e82. https://doi.org/10.1093/nar/gks203.
Zhu Y, Lu D, Lira ME, Xu Q, Du Y, Xiong J, Mao M, Chung HC, Zheng G. Droplet digital polymerase chain reaction detection of HER2 amplification in formalin fixed paraffin embedded breast and gastric carcinoma samples. Exp Mol Pathol. 2016;100(2):287–93. https://doi.org/10.1016/j.yexmp.2015.11.027.
Otsuji K, Sasaki T, Tanaka A, Kunita A, Ikemura M, Matsusaka K, Tada K, Fukayama M, Seto Y. Use of droplet digital PCR for quantitative and automatic analysis of the HER2 status in breast cancer patients. Breast Cancer Res Treat. 2017;162(1):11–8. https://doi.org/10.1007/s10549-016-4092-5.
Garcia-Murillas I, Turner NC. Assessing HER2 amplification in plasma cfDNA. Methods Mol Biol (Clifton, NJ). 2018;1768:161–72. https://doi.org/10.1007/978-1-4939-7778-9_10.
Shah PS, Murarka S, Joshi A, Mehta B, Parmar V, Shah N, Patel K, Sands J. Single-day HER2neu amplification assessment using chip-based digital PCR in formalin-fixed paraffin-embedded breast carcinoma tissue. Breast Cancer (Dove Med Press). 2018;10:121–9. https://doi.org/10.2147/bctt.S161264.
Nie M, Zheng M, Li C, Shen F, Liu M, Luo H, Song X, Lan Y, Pan JZ, Du W. Assembled step emulsification device for multiplex droplet digital polymerase chain reaction. Anal Chem. 2019;91(3):1779–84. https://doi.org/10.1021/acs.analchem.8b04313.
Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29(10):569–74. https://doi.org/10.1016/j.tig.2013.05.010.
Dong L, Meng Y, Wang J, Liu Y. Evaluation of droplet digital PCR for characterizing plasmid reference material used for quantifying ammonia oxidizers and denitrifiers. Anal Bioanal Chem. 2014;406(6):1701–12. https://doi.org/10.1007/s00216-013-7546-1.
Daniel WT, Kristian L, M.D, Marina K, David AA, Patricia EG, Robert LJ, Martin HK, Rudolf ML, Thomas JP, Scassellati GA, Heinz S, Jane T. Protocols for determination of limits of detection and limits of quantitation; Approved Guideline. EP17-A 24
[Guideline for HER2 detection in breast cancer, the 2019 version]. Zhonghua Bing Li Xue Za Zhi = Chin J Pathol. 2019; 48 (3):169–175. https://doi.org/10.3760/cma.j.issn.0529-5807.2019.03.001.
Ahn S, Kim HJ, Kim M, Chung YR, Kang E, Kim EK, Kim SH, Kim YJ, Kim JH, Kim IA, Park SY. Negative conversion of progesterone receptor status after primary systemic therapy is associated with poor clinical outcome in patients with breast cancer. Cancer Res Treat. 2018;50(4):1418–32. https://doi.org/10.4143/crt.2017.552.
De La Cruz LM, Harhay MO, Zhang P, Ugras S. Impact of neoadjuvant chemotherapy on breast cancer subtype: does subtype change and if so, how? : IHC profile and neoadjuvant chemotherapy. Ann Surg Oncol. 2018;25(12):3535–40. https://doi.org/10.1245/s10434-018-6608-1.
Xian Z, Quinones AK, Tozbikian G, Zynger DL. Breast cancer biomarkers before and after neoadjuvant chemotherapy: does repeat testing impact therapeutic management? Hum Pathol. 2017;62:215–21. https://doi.org/10.1016/j.humpath.2016.12.019.
Funding
This work was supported by the National Science & Technology Pillar Program (2017YFF0204605) and a basic research funding was sponsored by the National Institute of Metrology, People’s Republic of China (AKYZD2202).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
The study was approved by the ethics committee of the PLA rocket force characteristic medical center and was performed in accordance with ethical standards.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, X., Xing, D., Liu, Z. et al. Establishment and evaluation of digital PCR methods for HER2 copy number variation in breast cancer. Anal Bioanal Chem 415, 725–733 (2023). https://doi.org/10.1007/s00216-022-04466-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-022-04466-w