
Abstract
The application of Ca isotopic analysis in biomedical studies has great potential to identify changes in Ca metabolism and bone metabolism. Reliable measurement of Ca isotope-amount ratios is challenging considering limited Ca amounts and significant procedural blank levels. In this study, Ca purification was performed using the DGA Resin, optimized for low procedural blanks and separation of Ca from matrix elements and isobaric interferences (Na, Mg, K, Ti, Fe, Ba), while maintaining quasi-quantitative recoveries which are sufficient since a 42Ca–48Ca double-spike (DS) was applied. Ca isotopic analysis was performed using multicollector thermal ionization mass spectrometry (MC TIMS). The obtained procedural Ca blank of ≤10 ng enables processing of limited Ca amounts down to 670 ng. Data reduction of the measured Ca isotope-amount ratios was performed using an in-house developed software solving the DS algorithm. Data quality was improved by extension of equilibration time of the sample-DS mixture and implementation of a normalization strategy for raw isotopic data. The reported δ(44Ca/40Ca)NIST SRM 915a of NIST SRM 915a processed as a sample was found to be 0.01 ‰ ± 0.08 ‰ (2 SD, n = 15). Ca isotope-amount ratios of the reference material NIST SRM 1400 (bone ash), NIST SRM 1486 (bone meal), GBW07601 (human hair), and IAPSO (seawater) were in good agreement within uncertainty with literature data. Novel data on additional reference materials for biological tissues (hair) is presented, which might indicate a potential fractionation of Ca incorporated into hair tissue when compared to the blood pool.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The application of Ca isotope-amount ratio analysis has proven to have a great potential to monitor human Ca metabolism and bone metabolism, and detect potential metabolism disorders. The metabolism of Ca in the human body is schematically illustrated in Fig. 1: (i) Ca is absorbed in the digestive tract from the diet, predominantly from dairy products, vegetables, and fruits [1,2,3,4], into the blood pool (including other extracellular fluids) [5]. (ii) From the blood pool, Ca is removed for bone formation, a process which induces a significant isotope fractionation favoring lighter Ca isotopes during bone mineralization, as compared to the blood pool. Ca loss from the bone, e.g., during bone remodeling, does not fractionate Ca isotopes to a significant extent [4]. Therefore, Ca with a lighter isotopic composition is reabsorbed back into the blood pool. For a healthy human adult, the processes of bone formation and bone resorption are in balance. (iii) In general, Ca can be excreted via skin, hair, sweat, and bile, but the main loss occurs via urinary and fecal excretion [4]. Previous animal [6,7,8,9,10] and human [1,2,3, 11,12,13,14,15] studies documented the fractionation of Ca isotope-amount ratios from bone (lighter) to blood and to urine (heavier).

Hypothetical diagram of the Ca homeostasis in the human body. Red dashed arrows indicate the Ca fluxes, for which an isotopic fractionation is documented. Possible or not suspected Ca isotopic fractionations are indicated with black dashed arrows and black solid arrows, respectively. This diagram is a modification of the diagrams from Morgan et al. [1], Lee et al. [16], and Tacail et al. [4]
Imbalances or disturbances in the human Ca metabolism and bone metabolism, e.g., increased bone resorption due to a disease like senile osteoporosis [2], multiple myeloma [17], kidney diseases, and diabetes [18] are detectable by shifts of the Ca isotopic values in the blood pool and the excreted urine, and potentially incorporated into hair, respectively. Bed rest studies indicated that the Ca isotopic values of urine [1, 3, 11] and blood [14] have the potential to be applied as diagnostic markers for net bone mineral balance. Each of these matrices poses challenges: (i) Processing of Ca by the kidney induces a significant Ca isotopic fractionation with urinary Ca becoming enriched in heavy Ca isotopes compared to blood. The Ca isotopic ratios vary depending on the amount of excreted urinary Ca resulting from additional Ca isotopic fraction during recycling in the kidney [1, 3, 8, 12]. (ii) Ca isotope-amount ratios of the blood pool show a high variability, possibly due to missing balancing by pooling [14]. Eisenhauer et al. found that Ca isotopic values of blood are the preferred biomarker for osteoporosis and that no further clinical parameters have to be considered to improve the diagnosis of this disease [2]. The application of Ca isotope-amount ratio data to aid in the detection of imbalances in the Ca metabolism and bone metabolism is more efficient than radiological methods, is less invasive, and does not expose the patient to hazards associated with radioactivity [14].
The endogenous Ca mass fraction in hair can represent intracellular Ca levels [19] and has been reported as an indicator for deficiencies in Ca metabolism [20,21,22] and bone metabolism [23]. A recent study showed that hair Ca mass fraction can be associated with Ca intake and bone mineral density [24]. Nonetheless, hair is considerably affected by exogenous Ca contamination as a result of hair treatments [20, 25]. Exogenous Ca is primary bound to the hair surface (i.e., cuticle) [26], and endogenous Ca can be detected under ambient extraction conditions [20]. In applying bone mineral density studies on bone diseases, future studies might investigate the potential of endogenous hair Ca as a repository of time-resolved Ca isotope-amount ratios as a complementary diagnostic tool.
The extent of natural abundance variations of stable Ca isotopes within an organism is small, with a range of about 2 ‰ to 4 ‰ in the δ(44Ca/40Ca) ratio of the different tissues [27]. Therefore, reliable measurements of Ca isotope-amount ratios with low uncertainty are required if isotope abundance data are to be applied to Ca metabolism and bone metabolism studies. Since in general only limited tissue material can be provided, low amounts of Ca need to be analyzed accordingly. The natural variations of Ca isotopes are measured by either multicollector thermal ionization mass spectrometry (MC TIMS) or multicollector inductively coupled plasma mass spectrometry (MC ICP-MS).
The major analytical challenges of Ca isotopic analysis are the instrumental isotopic fractionation (IIF), the simultaneous detection of the numerous stable isotopes (40, 42, 43, 44, 46, 48) with their large mass dispersion of stable Ca isotopes, the isobaric interferences, and the blank contribution — all influencing the uncertainty of the measurement procedure. In the case of Ca isotopes, two analytical approaches are commonly used for the correction of the IIF. When using MC TIMS, the double-spike method [28, 29] (applying either the pair 42Ca–48Ca or 43Ca–48Ca) is the method of choice (see Table 1). The large mass dispersion between the isotopes 40Ca and 48Ca (~ 20 % difference from mass 40 to 48) makes it impossible to measure all isotopes over this mass range simultaneously on commercially available MC TIMS instruments [30]. Hence, a peak-jump (i.e., dynamic) collection mode is required, which introduces additional variable parameters such as systematic offsets caused by changes in collector cup efficiency in the different configurations, ratio combinations, and integration times in the different magnetic jump lines [29]. The resulting compositions are commonly reported as δ(44Ca/40Ca) ratios. In the case of MC ICP-MS, the standard sample bracketing method [31] is the method of choice to correct for IIF using, in the best case, an isotope-certified reference material (CRM, e.g., NIST SRM 915a) as a reference standard (see Table 1). Then, only Ca isotopes between 42Ca and 48Ca are measured simultaneously due to the intense 40Ar+ ion beam and therefore in general δ(44Ca/42Ca) values are reported. Isobaric interferences (e.g., K, Ar, Ti) exist for all Ca isotopes, which have to be considered for reliable Ca isotopic analysis. Matrix-based non-spectral interferences (e.g., Na, Fe, P, Ba) have to be considered, as well [32]. Electronic Supplementary Material Table 1 lists the interferences that potentially occur for Ca isotopic analysis using MC ICP-MS and MC TIMS measurements. Previous studies have reported that Ca blank contributions of ≤ 1.5 % are negligible for Ca isotope-amount ratio analysis [6, 8, 32].
Existing measurement procedures for the determination of Ca isotope-amount ratios in biological samples are summarized in Table 1. In general, one can observe the following: (i) The Ca amount processed during sample purification — after acid digestion — has significantly decreased since the initial TIMS studies in 1997 from about 300 µg to about 1.5 μg today. Besides the improvement in performance of the (MC) TIMS instruments, the decreasing blank levels (300 ng to 30 ng) also play a role in realizing higher quality data. Measurements on the (MC) TIMS required a total Ca amount between 300 ng and 8000 ng for Ca isotopic analysis. (ii) In contrast, total Ca amounts between 25 µg and 100 μg have to be processed in Ca purification — after acid digestion — even in the latest MC ICP-MS studies. This is due to the fact that MC ICP-MS detects only the minor isotopes of Ca with natural abundances of ≤ 2 % [33], which requires high Ca amounts (1 µg g–1 – 10 μg g−1) for the Ca isotopic analysis. (iii) Overall, the repeatability expressed as 2 SD on the δ(44Ca/40Ca) ratio improved from approx. 0.5 ‰ to approx. 0.1 ‰ over time, with improving instrument performance of MC TIMS and MC ICP-MS. To date, the expected precision for δ(44Ca/40Ca) ratio is 0.1 ‰ [34]. The ability to measure Ca isotopes accurately is critical to leverage them as a clinical (or pre-clinical) tool to assess changes in Ca metabolism and bone metabolism.
In general, MC TIMS and MC ICP-MS are complementary techniques for isotopic analysis offering different ionization efficiencies and different overall transmission [35, 36]. The two techniques are comparable in terms of time consumption (incl. sample preparation) and precision [35, 37, 38]. The latter requires a proper IIF correction strategy for reliable isotope data. In the direct comparison, MC TIMS is less susceptible to isotopic fractionation (up to a factor of 10 smaller) [35] and does not require sensitivity matching within the IIF correction. With regard to Ca isotopes, MC TIMS analysis suffers from fewer (isobaric) interferences, which affect the measurement less, and MC TIMS is ahead in terms of limited biological tissue samples that need to be processed (see Table 1) [39].
Apart from studies designed to investigate the variability of Ca isotope-amount ratios in (human) body fluids and other tissues, routine applications involve numerous different analyses from one and the same biological sample. In general, minimal invasive techniques only provide limited sample amounts. Accordingly, in a routine application of Ca isotopic analysis, only small sample amounts will be available and thus rapidly reaching the limits of the analytical method (compare Tables 1 and 2). Hence, the aim of the present study was to develop a measurement procedure using DS MC TIMS for low-amount Ca isotopic analysis in limited biological tissues.
Experimental
Experimental work and Ca isotopic analysis using MC TIMS were carried out at the University of Calgary, Canada. All sample preparation procedures were accomplished in a class 1000 cleanroom in a clean bench to minimize blank levels and risk of contamination. Additional multi-elemental analysis using quadrupole ICP-MS was carried out at the Montanuniversität Leoben, Austria.
Materials and chemicals
Ultrapure hydrochloric acid (Aristar Ultra, VWR International, Mississauga, Ontario, Canada) and quartz distilled water (provided by Bob Loss at the John de Laeter Center for Mass Spectrometry at Curtin University in Western Australia) were used for preparation of the double- spike (DS). Ultrapure water (Aristar Ultra, VWR International) and double sub-boiled nitric acid were used for sample preparation and Ca purification. Analytical reagent-grade nitric acid (w = 65 %, Aristar Plus, VWR International) was purified by double sub-boiling using a sub-boiling distillation system (DST-1000 Savillex, Eden Prairie, MN, US). Perfluoroalkoxy (PFA) screw cap vials (Savillex) were pre-cleaned in a two-stage acid cleaning procedure using sub-boiled nitric acid (single and double, respectively). Pipette tips (Eppendorf, Mississauga, Canada; Corning Incorporated, Corning, NY, USA; Fisherbrand, Fisher Scientific, USA) were pre-cleaned in a two-stage washing procedure using diluted nitric acid (w = 10 % and w = 1 %, respectively), prepared from single sub-boiled nitric acid and high-quality water (PURELAB Plus, U.S.FILTER, ELGA LabWater, High Wycombe, UK). One-milliliter empty cartridges and frits (AC-100-R01, TrisKem International, Bruz, France) were pre-cleaned and stored in diluted nitric acid (w = 10 %). DGA Resin (part. No. DN-B25-S, TrisKem International, mean particle size of 50–100 μm) was soaked in diluted ethanol (w ≈ 30 %).
A multi-element matrix solution to test the Ca/matrix separation was prepared from a multi-element stock solution (ICP multi-element standard Merck VI, 10 mg L−1; Merck-Millipore, Darmstadt, Germany) and single-element standard of Ti, Na, and K (Merck-Millipore). Further, dilutions from the multi-element stock solution and the single-element standard Ti were used for external calibration in ICP-MS measurements. Indium (In, Merck-Millipore) with a mass fraction of 1 ng g−1 was added as an internal normalization standard.
Double filaments for MC TIMS analysis were prepared in-house using double-filament posts (Thermo Fisher Scientific, Bremen, Germany) and Re ribbon (99.99 % Re, H. Cross Co., Moonachie, NJ, USA). Diluted phosphoric acid (w = 5 %), used as activator, was prepared from ultrapure phosphoric acid (J.T. Baker, Phillipsburg, NJ, USA) and ultrapure water (VWR International).
NIST SRM 915a (calcium carbonate, NIST Gaithersburg, USA) was dissolved and further diluted quantitatively with diluted nitric acid (w = 3 %), prepared from double sub-boiled nitric acid and ultrapure water (VWR International), to a final mass fraction of about 4 μg g−1. IAPSO (seawater, OSIL, Hampshire, UK) was diluted quantitatively with nitric acid (w = 3 %) to a final mass fraction of about 10 μg g−1. For doping experiments, NIST SRM 987 (strontium carbonate, NIST) was dissolved in diluted nitric acid (w = 3 %).
Certified reference materials NIST SRM 915a (calcium carbonate, NIST), NIST SRM 1400 (bone ash, NIST), NIST SRM 1486 (bone meal, NIST), USGS42 (human hair, USGS, VA, USA), USGS43 (human hair, USGS), GBW07601 (human hair, NCRM, Langfang, China), GBW09101 (human hair, NCRM), and IAPSO (seawater, OSIL) were analyzed throughout this study.
Sample digestion
Between 10 mg and 100 mg of reference material (NIST SRM 1400, NIST SRM 1486, USGS42, USGS43, GBW07601, and GBW09101) were weighed into a PFA screw cap vial and mixed with 2 mL double sub-boiled nitric acid and 0.5 mL ultrapure hydrogen peroxide (w = 30 %, Aristar Plus, VWR International). The samples were digested for 2 h at 120 °C on a hot plate. After digestion, the solution was diluted quantitatively with ultrapure water (VWR International) to a final Ca mass fraction of 1 μg g−1.
The Ca mass fractions in the digests of the certified reference materials used in this study were quantified using the DS algorism (see the “Data processing” section). Table 3 summarizes the measured Ca mass fractions of the certified reference materials compared to the certified Ca mass fractions. In general, the acid digestion was complete with > 96 % Ca recovery for NIST SRM 1400, NIST SRM 1486, and GBW07601. Only the processing of GBW09101 showed visual signs of incomplete digestion (white residue). Nonetheless, the Ca recovery of GBW09101 was 88 % ± 4 % (U, k = 2). Further, Ca mass fractions of 1066 μg g−1 ± 53 μg g−1 (U, k = 2) and 1746 μg g−1 ± 87 μg g−1 (U, k = 2) were stated for USGS42 and USGS43, respectively.
Double-spike preparation
The 42Ca and 48Ca primary spikes were prepared from Oak Ridge National Laboratory (ORNL) calcium carbonate powder individually enriched in 42Ca (Series LO, Batch 139693, 7.5 mg) and 48Ca (Series RT, Batch 219801, 5 mg). The composition of the primary spikes are listed in Table 4. The calcium carbonate powder spikes were each transferred quantitatively into separate PFA containers and were dissolved quantitatively in 10 mL diluted nitric acid (w = 10 %), which was prepared from ultrapure nitric acid (w = 65 %, Aristar Ultra, VWR International) and quartz distilled water. All masses were measured using a milligram balance (AT201, Mettler-Toledo, Switzerland). Based on Rudge et al. [28], the optimum composition for the 42Ca–48Ca DS was determined to be 39.64 % of 42Ca single spike and 60.36 % of 48Ca single spike, resulting in a r(42Ca/48Ca) of 0.6567. Hence, the 42Ca–48Ca DS mixture was prepared from 1.067 mL of the 42Ca single spike (= 800 ng) and 2.400 mL of the 48Ca single spike (= 1200 ng) in a separate PFA container, resulting in an actual r(42Ca/48Ca) of about 0.6382. The composition of the 42Ca–48Ca DS is listed in Table 4. The 42Ca–48Ca DS was further diluted quantitatively with diluted nitric acid (w = 3 %) to a final mass fraction of about 1 μg g−1 in a separate PFA container, as the working solution.
The mass fraction of the DS was quantified using three mixtures of NIST SRM 915a, with known mass fraction, and DS (in a ratio of approx. 86:14). The Ca isotope abundance ratios were evaluated using the DS algorithm (see the “Data processing” section) applying NIST SRM 915a (with a known mass fraction) as the “spike” and DS as the “sample.” A more detailed explanation about the use of DS can be found elsewhere [28, 29, 54, 55].
Prior to Ca/matrix separation, sample aliquots of approx. 1000 ng Ca were spiked with DS in a ratio of approx. 86:14, as recommended by Rudge et al. [28]. The mixture of sample aliquot and DS was allowed to rest for minimum 24 h in a PFA container to achieve isotope equilibration between DS and sample prior Ca/matrix separation. Extended equilibration times were derived from previous studies on Zn isotopes [56, 57]. Future studies might consider heating to accelerate the isotope equilibration between DS and sample.
Separation protocol
Approximately 150 μL soaked DGA Resin (TrisKem International) was sandwiched between two frits in a 1-mL empty cartridge. The cartridge was connected with a widened Teflon gas line (4 mm O.D. Elemental Scientific, Omaha, USA) to a flared tubing (I.D. 1.02 mm, SCP Science, Quebec, Canada) and 10 cm of self-aspiration capillary (0.5 mm i.d., ES-2041, Elemental Scientific). Reagents were pumped through the cartridge at a flow rate between 150 µL min–1 and 500 μL min−1 using a peristaltic pump (MCP, ISOMATEC, Barrington, IL, USA).
Separation was performed according to the optimized procedure described in Table 5, samples were collected in PFA containers (see the “Optimized Ca/matrix separation” section).
To assess the influence of potential Sr++ interferences on Ca isotopic analysis using MC TIMS a doping test with various Ca/Sr ratios was carried out. Approximately 1000 ng of NIST SRM 915a was prepared with DS (approx. 86:14), doped with 0 ng, 10 ng, 100 ng, and 1000 ng of Sr, prepared from NIST SRM 987.
ICP-MS measurement
Multi-elemental analyses of the stepwise elution of Ca/matrix separation were performed using an ICP-MS (NexION 2000B, PerkinElmer, Ontario, Canada) coupled to an ESI SC-2 DX FAST autosampler, at the Montanuniversität Leoben. All stepwise elution of Ca/matrix separation (V = 0.5–1 mL) were diluted to a final volume of 5 mL of diluted nitric acid (w = 2 %).
ICP-MS measurements were accomplished performing a calibration in the range of 0.1 ng g–1 to 150 ng g−1 for Na, Mg, Al, Sr, and Ba; in the range of 1 ng g–1 to 1500 ng g−1 for Fe and Ti; and in the range 10 ng g–1 to 15,000 ng g−1 for Ca, respectively, prepared gravimetrically from Merck multi-element standard VI.
General instrumental settings for the multi-elemental measurements are described elsewhere [58].
MC TIMS measurement
Sample filaments were prepared by dissolving 1000 ng purified Ca in 2 μL of diluted phosphoric acid (w = 5 %), used as an activator, and then depositing the sample on a previously outgassed Re filament in a double-filament design. The Re filament was heated with 1 A min−1 current to dry the purified Ca sample. Finally, the electrical current was slowly preheated to maximum of 3 A until a weak red glow was visible, followed by shutting down of the current. A separate outgassed Re ionization filament was loaded in the sample carousel for a double-filament experiment. The filament assembly was loaded into the ion source of a Triton MC TIMS (Thermo Fisher Scientific) at the Stable Isotope Laboratory, University of Calgary, Canada. The instrument was equipped with one fixed axial and eight movable Faraday cup collectors (each with 1011 Ohm resistors) and an axial SEM detector. The instrument was operated in dynamic mode with two measurement cycles (1: 42–48, 2: 39–44; see Table 6) for Ca isotopic analysis. The signal of the 42Ca spike was placed in the center cup of the second line for the automated heating procedure: (i) Each ionization filament was heated to 1450 °C before (ii) the corresponding evaporation filament was heated to 1700 mA, which was maintained for 10 min; (iii) the evaporation filament was further heated to at 1900 mA, where an automated optimization of peak center and focus was performed for both lines; (iv) the evaporation filament kept heating until an signal intensity of 500 mV on 42Ca was archived; and then (v) 100 ratios were collected in 5 blocks of 20 ratios with an integration time of 4.2 s and an idle time of 3 s per line. A detailed description of the automated heating protocol is provided in the supplementary data (see Electronic Supplementary Material Table 2).
Data processing
Data reduction of Ca isotopes was performed using an in-house-programmed software (by Alex Tennant in 2016) in Python™. The software employs a root-finding algorithm to solve the DS equations as described in Mayer and Wieser [54] and Rudge et al. [28]. The raw isotope-amount ratios (r(48Ca/44Ca), r(42Ca/44Ca), r(40Ca/44Ca)) of the pure DS, pure NIST SRM 915a, and a mixture of both (approx. 86:14) have been entered as anchor values into the DS software, as well as the atomic masses of the isotopes 40Ca, 42Ca, 44Ca, and 48Ca according to IUPAC/CIAAW tables [33]. The raw ratios were measured by MC TIMS. Note that the raw ratios (r(48Ca/44Ca), r(42Ca/44Ca), r(40Ca/44Ca)) were normalized to the first-block average of r(42Ca/44Ca) to compensate for time-dependent IIF during the run on the MC TIMS. No blank correction was performed. All Ca isotopic data (δ(44Ca/40Ca), δ(44Ca/42Ca)) are reported relative to the NIST SRM 915a as δ-anchor.
Further, the amount of Ca in the sample was determined using the DS software, based on the known proportion of DS in the sample-DS mixture [54].
Results and discussion
Ca blank levels
Monitoring background levels of the omnipresent Ca is crucial in Ca isotopic analysis since significant amounts in the system bias the result of the Ca isotope-amount ratio measurement. Initial MC TIMS measurements indicated a procedural Ca blank in the range of 995 ng to 1739 ng, which are unacceptably high. Investigation of the working environment identified two major sources to the high procedural Ca blank levels: (i) Class 100 Cleanroom Nitrile Gloves (VWR International, Radnor, USA) and (ii) Kimwipes® (Kimberly-Clark Professional, Mississauga, Canada). Elimination of these two materials from all preparatory steps decreased the Ca procedural blank into the range of 20 ng to 59 ng. After further investigation of consumables and chemicals, high-quality water was replaced by ultrapure water and DigiTubes® were replaced by PFA vessels. The Ca/matrix separation was optimized for low DGA Resin and low acidity of the nitric acid (see the “Optimized Ca/matrix separation” section). The resulting procedural Ca blank of 6 ng ± 4 ng (SD = 1, n = 26) was measured. The background from filament material and activator was tested by measuring a Re double filament loaded only with 4 μL of diluted phosphoric acid (w = 5 %). The signal intensity on 40Ca was measured ≤ 3 mV. Hence, the contribution of the background from filament material and activator to the blank level was considered negligible.
The Ca blank level of ≤ 10 ng achieved within this study represents a reduction by a factor of three compared to recent studies using MC TIMS and is comparable to the lowest Ca blank levels reported in studies using MC ICP-MS(see Table 1). The procedural Ca blanks of this study enabled a reduction of Ca amounts to be processed to approx. 670 ng, while providing still ≤ 1.5 % blank contribution. This is a reduction in the processed Ca amounts by a factor of two compared to recent studies using MC TIMS. Throughout the present study, a Ca amount of approx. 1000 ng was processed, since enough sample material was available.
Optimized Ca/matrix separation
The DGA Resin provided the lowest Ca blanks (see Table 1); therefore, it had been selected for this study. Based on published data describing the retention behavior of DGA Resin for various elements [59, 60] and the separation protocol described by Retzmann et al. [61], the Ca/matrix separation using DGA Resin was optimized for minimal amounts of resin (150 μL) and low acid concentrations for sample loading and matrix wash (nitric acid c = 1.1 mol L−1 instead of c = 3 mol L−1 or higher; see Table 5). Figure 2 shows the stepwise elution of a multi-element matrix (containing approx. 1000 ng Ca; approx. 100 ng Na, K, Ti, and Fe; and approx. 10 ng of Mg, Al, Sr, and Ba). The results indicate that the (potentially) interfering elements Mg, Al, K, and Ti as well as matrix elements like Na, Fe, and Ba are removed from Ca within 3 mL of nitric acid (c = 1.1 mol L−1). It should be noted that a small portion of Ca (approx. 18 %) is also removed during the washing steps (3 mL) with nitric acid (c = 1.1 mol L−1). Based on previous investigations of the DGA Resin, the final Ca elution step has been shortened to 750 μL [61]. Overall, a quasi-quantitative elution of Ca (V = 0.75 mL) was achieved with a Ca recovery of 82 % ± 8 % (SD = 1, n = 15). This recovery is satisfactory considering the DS procedure that will correct for any mass-dependent fractionation that might occur on the resin as a result of less than 100 % recoveries.

Elution profile for multi-element matrix (containing approx. 1000 ng Ca; approx. 100 ng Na, Ti, K, and Fe; and approx. 10 ng of Mg, Al, Sr, and Ba). Relative uncertainty (Urel) of 15 % (k = 2)
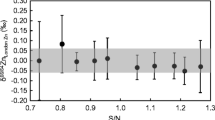
With the chosen loading conditions (nitric acid; c = 1.1 mol L−1) Sr is partly retained by the DGA Resin and co-elutes to a minor extent in the final Ca fraction (approx. 10 %). Consequently, potential interferences during the Ca isotopic analysis by 88Sr++, 86Sr++, and 84Sr++ were investigated. No significant influence of doubly charged Sr on the measured δ(44Ca/40Ca) ratios was observed (see Fig. 3). This is in agreement with what has been reported in the literature previously [62].

δ(44Ca/40Ca)NIST SRM 915a values measured for NIST SRM 915a (approx. 1000 ng) doped with alternating Sr amounts. Error bars correspond to U (k = 2)
Double-spike composition and improvement of data quality
The effect of changing the relative amount of Ca in the sample to Ca from the DS (recommended approx. 86:14 [28]) was investigated, since it might arise, e.g., for samples with unknown Ca content. Therefore, approx. 1000 ng of NIST SRM 915a was spiked with DS resulting in the sample-DS ratios of 96:4, 87:13, and 76:24. A significant shift in the δ(44Ca/40Ca)NIST SRM 915a value for sample-DS mixture ratio of 96:4 (− 0.25 ‰, see Fig. 4) was observed, which is in agreement with the modeled error surface for a 42Ca–48Ca DS [28]. For the sample-DS mixture ratios 87:13 and 76:24, no significant shifts were observed, which is in agreement with the modeled error surface for a 42Ca–48Ca DS [28]. For DS proportions between 10 % and 25 % in the sample-DS mixture, ≤ 5 % of the optimum error is expected [28]. Consequently, the experiments of this study were designed to remain within a close range to the recommended ratio in the literature [28].

δ(44Ca/40Ca)NIST SRM 915a values measured for NIST SRM 915a (approx. 1000 ng) spiked with varying DS amounts. Red outline indicates the ratio closest to the recommended sample-DS ratio in the literature [28]. Error bars correspond to U (k = 2)
Figure 5 shows the δ(44Ca/40Ca)NIST SRM 915a values of separated NIST SRM 915a — treated as sample — measured over a period of 1 year. Over this time, an improvement of the data quality and uncertainty was observed based on the following steps: (i) Initially, the mixture of NIST SRM 915a and DS (approx. 86:14), which was used as input value of the DS software, was prepared directly on the filament using NIST SRM 915a and DS stock solutions. The sample-DS mixture (approx. 86:14) to be separated was prepared from stock solutions, dried down, and re-dissolved under sonication in diluted nitric acid prior to separation. The observed δ(44Ca/40Ca)NIST SRM 915a values shifted towards lower values. (ii) In the first step of improvement, an ultrasonification step was added to the preparation procedure of the mixture of NIST SRM 915a and DS (approx. 86:14), which was used as input value of the DS software. This adjustment was performed to create comparable equilibration conditions of sample and DS for the mixtures used in the DS software and the sample to be separated. This resulted in a slight shift of δ(44Ca/40Ca)NIST SRM 915a values towards the δ-anchor value. (iii) In the second step of improvement, a normalization of the measured data was implemented. The raw data were normalized to the first-block average of r(42Ca/44Ca) to compensate time-dependent IIF within a run (see the “Data processing” section). This improved the repeatability of δ(44Ca/40Ca)NIST SRM 915a values and shifted them even closer to the δ-anchor value. (iv) In the third step of improvement, samples with α and β values (sample and mixture undergo instrumental mass fractionation with the fractionation factors α and β, respectively [28, 54], which are given by the DS software) outside the 2 SD range of average α and β values were identified as outlier and removed from the data set. (v) Finally, in the fourth step of improvement, the equilibration time of NIST SRM 915a-DS mixtures as input value of the DS software as well as sample-DS mixtures were extended to minimum 24 h, to ensure chemical equilibration of sample and DS. This further improved the repeatability of δ(44Ca/40Ca)NIST SRM 915a values, which resulted in an improved measurement uncertainty (see the “Uncertainty budget”). The average δ(44Ca/40Ca)NIST SRM 915a value reported from the fourth step of improvement is 0.01 ‰ ± 0.08 ‰ (2 SD, n = 15), which overlaps with the δ-anchor value. The reported uncertainty is even below the expected precision of 0.1 ‰ for δ(44Ca/40Ca) ratio [34] and the top level of what has been achieved so far (see Table 1).

Improvement steps on the data quality shown on the example of δ(44Ca/40Ca)NIST SRM 915a values (black diamond) of separated NIST SRM 915a, measured over a time period of 1 year. Error bars correspond to U (k = 2)
Uncertainty budget
The total combined uncertainty budget for the Ca isotopic analysis was calculated using a simplified Kragten approach [63] in order to elucidate the components that constitute the most significant impacts on the overall uncertainty in the analysis of the Ca isotopic ratios of biological materials. For this purpose, each input value of the DS software header and the analyzed sample (here mimicked by NIST SRM 915a) was altered individually by its standard deviation and then applied in the DS algorithm. Herein, the atomic masses, the r(48Ca/44Ca) and r(40Ca/44Ca) of the DS and NIST SRM 915a in the header file of the DS software as well as of the analyzed sample, the normalization strategy using r(42Ca/44Ca), and the magnet jump for r(40Ca/44Ca) were considered as contributors to the uncertainty. Note that correlated input values of the DS algorithm such as the r(48Ca/44Ca) and r(40Ca/44Ca) of the mixture and the samples were not considered in the uncertainty budget. A blank contribution of 1.5 % (= 15 ng) was considered. Herein, the Ca isotope-amount ratio of blood (δ(44Ca/40Ca)NIST SRM 915a ratio of Seronorm Whole Blood L-1: 0.89 ‰ [10]) was used as hypothetical Ca isotope-amount ratio of the blank contributing to the natural Ca isotope-amount ratio of the NIST SRM 915a — analyzed as sample — to mimic maximum blank contribution to biological samples. Further, the repeatability based on the multiple mixtures prepared from DS and NIST SRM 915a (δ(44Ca/40Ca)NIST SRM 915a ratio: SD = 0.04, δ(44Ca/42Ca)NIST SRM 915a ratio: SD = 0.02; n = 7) according to the improved procedure (see the “Double-spike composition and improvement of data quality” section) was considered as a contributor to the uncertainty budget.
The combined measurement uncertainty on the δ(44Ca/40Ca)NIST SRM 915a ratio for the NIST SRM 915a as sample was determined as ± 0.08 ‰ (U, k = 2). The main contributors to the uncertainty were the repeatability (90.7 %) and the blank contribution (9.2 %). The combined measurement uncertainty on the δ(44Ca/42Ca)NIST SRM 915a ratio for the NIST SRM 915a as sample was determined as ± 0.04 ‰ (U, k = 2). The main contributor to the uncertainty were the repeatability (91.5 %) and the blank contribution (8.4 %).
Following previous studies [6, 8, 32], a blank contribution of ≤ 15 ng (≤ 1.5 %) could be considered negligible to the uncertainty for a processed sample Ca amount of 1000 ng. Accordingly, further uncertainty budgets of the biological CRMs were estimated based on the repeatability.
Ca isotope-amount ratios of biological CRMs
Table 7 shows the δ(44Ca/40Ca)NIST SRM 915a and δ(44Ca/42Ca)NIST SRM 915a values of six biological CRMs (bone and hair tissue) and seawater, which were analyzed using the improved procedure. The relative variation of the δ(44Ca/40Ca)NIST SRM 915a values in the biological CRMs was 1.77 ‰, (3.00 ‰ including IAPSO (seawater)). The reported δ(44Ca/40Ca)NIST SRM 915a values for NIST SRM 1400 (bone ash) is − 1.12 ‰ ± 0.19 ‰ (U, k = 2), for NIST SRM 1486 (bone meal) is − 0.96 ‰ ± 0.13 ‰ (U, k = 2), and for IAPSO (seawater) is 1.88 ‰ ± 0.10 ‰ (U, k = 2). The latter two are in good agreement with previously published results [45, 64, 65] within uncertainty (see Table 7). The reported δ(44Ca/40Ca)NIST SRM 915a value for USGS42 (Tibetan human hair) is − 0.13 ‰ ± 0.21 ‰ (U, k = 2), for USGS43 (Indian human hair) it is 0.43 ‰ ± 0.21 ‰ (U, k = 2), for GBW07601 (Chinese human hair) is 0.65 ‰ ± 0.16 ‰ (U, k = 2), and for GBW09101 (human hair) it is 0.09 ‰ ± 0.15 ‰ (U, k = 2). The reported δ(44Ca/42Ca)NIST SRM 915a value for GBW07601 is in good agreement with previously published results [32, 42] within uncertainty (see Table 7). To the authors’ knowledge, the other three hair CRMs were measured for the first time for their Ca isotope-amount ratios. In general, the δ(44Ca/40Ca)NIST SRM 915a values of the CRM pairs USGS42 and USGS43 and GBW07601 and GBW09101 do not overlap within uncertainty, respectively. The values of USGS42 and GBW09101as well as of USGS43 and GBW07601 do overlap within uncertainty, respectively. The reported repeatability for the CRMs is about two times as large as the repeatability of the NIST SRM 915a (compare with the “Uncertainty budget” section). This is primarily due to the fact that the measured values before the fourth step of the improvement (compare with the “Double-spike composition and improvement of data quality” section) were also included. Nonetheless, inhomogeneity cannot be excluded since the hair CRM are pooled hair samples from several individuals.
Figure 6 shows the comparison of δ(44Ca/40Ca)NIST SRM 915a values of the hair CRMs with the δ(44Ca/40Ca)NIST SRM 915a values of CRMs for bone tissue and body fluids. Herein, one can see in an indication that the process of Ca incorporation into hair tissue from the blood pool potentially induces an isotope fractionation favoring lighter Ca isotopes. This indication must be treated with care, since no systematic study on a single individual has been performed yet. Figure 7 shows the plot of hair Ca mass fractions against the corresponding δ(44Ca/40Ca)NIST SRM 915a values. Herein, one can see a trend of lower δ(44Ca/40Ca)NIST SRM 915a values with decreasing Ca mass fraction. The hair CRMs with higher Ca mass fractions (> 1500 μg g−1) more likely represent the exogenous Ca signature of the environmental water (which are part of the dietary Ca source — comparable to the blood pool). The hair CRMs with lower Ca mass fractions (< 1100 μg g−1) have likely a larger proportion of endogenous Ca.

Variation of δ(44Ca/40Ca)NIST SRM 915a values of biological CRMs (N: NIST SRM, U: USGS, G: GBW) obtained in this study and from literature data (S: Seronorm L-1) body fluids [10]). Error bars correspond to U (k = 2)

The Ca mass fractions of human hair CRMs (N: NIST SRM, U: USGS, G: GBW) are plotted against their δ(44Ca/40Ca)NIST SRM 915a values. Reference values are provided as equivalent from published δ(44Ca/42Ca)NIST SRM 915a ratios [32, 42] under consideration of mass-dependent fractionation factor of 2.0483 [47, 48]. Error bars correspond to U (k = 2)
Future studies might systematically study the relation of the Ca isotope-amount ratios in hair and body fluids from the same individual and with respect to changes in Ca metabolism and bone metabolism.
Conclusion
With respect to biological samples (e.g., bone, body fluids, hair) available in limited quantities for routine analysis, existing measurement procedures for Ca isotopic analysis in biological material are reaching their limits, especially with regard to blank values and the blank value contribution. A careful consideration of the work environment, the consumables, and the applied Ca/matrix separation procedure enabled maintaining low Ca procedural blanks (≤ 10 ng) in the presented measurement procedure using DS MC TIMS for Ca isotopic analysis. The reduction in procedural Ca blank resulted in a reduction in the amount of Ca to be processed to approx. 670 ng, which allows routine Ca isotopic analysis of > 2.3 μg bone material, > 10 μL blood, > 6 μL serum, or > 1 μL urine. With the present measurement procedure, at a Ca mass fraction of about 450 μg g−1 in washed hair [22, 23, 63] and a weight of about 2 mg per 1 cm of hair, it is possible to resolve Ca isotope-amount ratios on a monthly scale from a single hair.
After optimization of the DS-sample preparation procedure (equilibration) and data reduction (normalization), the reported uncertainty of the presented DS MC TIMS measurement procedure for δ(44Ca/40Ca)NIST SRM 915a is ± 0.08 ‰ (U, k = 2). The repeatability is the major contributor (> 90 %) to the combined measurement uncertainty.
The reported δ(44Ca/40Ca)NIST SRM 915a values and δ(44Ca/42Ca)NIST SRM 915a values for NIST SRM 1400 (bone ash), NIST SRM 1486 (bone meal), GBW07601 (human hair), and IAPSO (seawater) are in good agreement with published data. Furthermore, δ(44Ca/40Ca)NIST SRM 915a values for three hair CRMs are reported for the first time: − 0.13 ‰ ± 0.21‰ (U, k = 2) for USGS42, 0.43 ‰ ± 0.21 ‰ (U, k = 2) for USGS43, and 0.09 ‰ ± 0.15 ‰ (U, k = 2) for GBW09101. Overall, this study presents a robust measurement procedure using DS MC TIMS for reliable low-level Ca isotopic analysis.
References
Morgan JLL, Skulan JL, Gordon GW, Romaniello SJ, Smith SM, Anbar AD. Rapidly assessing changes in bone mineral balance using natural stable calcium isotopes. Proc Natl Acad Sci. 2012;109(25):9989.
Eisenhauer A, Müller M, Heuser A, Kolevica A, Glüer CC, Both M, Laue C, Hehn U, Kloth S, Shroff R, Schrezenmeir J. Calcium isotope ratios in blood and urine: a new biomarker for the diagnosis of osteoporosis. Bone Rep. 2019;10:100200.
Heuser A, Frings-Meuthen P, Rittweger J, Galer SJG. Calcium isotopes in human urine as a diagnostic tool for bone loss: additional evidence for time delays in bone response to experimental bed rest. Front Physiol. 2019;10:12.
Tacail T, Le Houedec S, Skulan JL. New frontiers in calcium stable isotope geochemistry: perspectives in present and past vertebrate biology. Chem Geol. 2020;537:119471.
Chu N-C, Henderson GM, Belshaw NS, Hedges REM. Establishing the potential of ca isotopes as proxy for consumption of dairy products. Appl Geochem. 2006;21(10):1656–67.
Hirata T, Tanoshima M, Suga A, Tanaka Y, Nagata Y, Shinohara A, Chiba M. Isotopic analysis of calcium in blood plasma and bone from mouse samples by multiple collector-ICP-mass spectrometry. Anal Sci. 2008;24:1501–7.
Tacail T, Albalat E, Telouk P, Balter V. A simplified protocol for measurement of Ca isotopes in biological samples. J Anal At Spectrom. 2014;29:529–353.
Heuser A, Eisenhauer A, Scholz-Ahrens KE, Schrezenmeir J. Biological fractionation of stable Ca isotopes in Göttingen minipigs as a physiological model for Ca homeostasis in humans. Isot Environ Health Stud. 2016;52(6):633–48.
Tanaka Y-K, Mikuni-Takagaki Y, Hidaka K, Wada-Takahashi S, Kawamata R, Hirata T. Correction of mass spectrometric interferences for rapid and precise isotope ratio measurements of calcium from biological samples using ICP-mass spectrometry. Anal Sci. 2019;35(7):793–8.
Grigoryan R, Costas-Rodríguez M, Vandenbroucke RE, Vanhaecke F. High-precision isotopic analysis of Mg and Ca in biological samples using multi-collector ICP-mass spectrometry after their sequential chromatographic isolation – application to the characterization of the body distribution of Mg and Ca isotopes in mice. Anal Chim Acta. 2020;1130:137–45.
Skulan J, Bullen T, Anbar AD, Puzas JE, Shackelford L, LeBlanc A, Smith SM. Natural calcium isotopic composition of urine as a marker of bone mineral balance. Clin Chem. 2007;53(6):1155.
Heuser A, Eisenhauer A. A pilot study on the use of natural calcium isotope (44Ca/40Ca) fractionation in urine as a proxy for the human body calcium balance. Bone. 2010;46(4):889–96.
Morgan JLL, Gordon GW, Arrua RC, Skulan JL, Anbar AD, Bullen TD. High-precision measurement of variations in calcium isotope ratios in urine by multiple collector inductively coupled plasma mass spectrometry. Anal Chem. 2011;83(18):6956–62.
Channon MB, Gordon GW, Morgan JLL, Skulan JL, Smith SM, Anbar AD. Using natural, stable calcium isotopes of human blood to detect and monitor changes in bone mineral balance. Bone. 2015;77:69–74.
Rangarajan R, Mondal S, Thankachan P, Chakrabarti R, Kurpad AV. Assessing bone mineral changes in response to vitamin D supplementation using natural variability in stable isotopes of calcium in urine. Sci Rep. 2018;8(1):16751.
Lee S-H, Park S-J, Kim K-N, Cho D-Y, Kim Y-S, Kim B-T. Coronary calcification is reversely related with bone and hair calcium: the relationship among different calcium pools in body. J Bone Metab. 2016;23(4).
Gordon GW, Monge J, Channon MB, Wu Q, Skulan JL, Anbar AD, Fonseca R. Predicting multiple myeloma disease activity by analyzing natural calcium isotopic composition. Leukemia. 2014;28(10):2112–5.
Tanaka, Y-K, Yajima N, Higuchi Y, Yamato H, Hirata T. Calcium isotope signature: new proxy for net change in bone volume for chronic kidney disease and diabetic rats. Metallomics. 2017;9(12):1745–55.
Klevay LM, Bistrian BR, Fleming CR, Neumann CG. Hair analysis in clinical and experimental medicine. Am J Clin Nutr. 1987;46(2):233–6.
Bacsó J, Sarkadi L, Koltay E. On endogenous and exogenous calcium content of hair samples used in XRF and PIXE measurements. Int J Appl Radiat Isot. 1982;33(1):5–11.
MacPherson A, Bacsó J. Relationship of hair calcium concentration to incidence of coronary heart disease. Sci Total Environ. 2000;255(1):11–9.
Forte G, Alimonti A, Violante N, Di Gregorio M, Senofonte O, Petrucci F, Sancesario G, Bocca B. Calcium, copper, iron, magnesium, silicon and zinc content of hair in Parkinson’s disease. J Trace Elem Med Biol. 2005;19(2):195–201.
Miekeley N, de Carvalho Fortes LM, Porto da Silveira CL, Lima MB. Elemental anomalies in hair as indicators of endocrinologic pathologies and deficiencies in calcium and bone metabolism. J Trace Elem Med Biol 2001;15(1):46–55.
Park S-J, Lee S-H, Cho D-Y, Kim K-M, Lee D-J, Kim B-T. Hair calcium concentration is associated with calcium intake and bone mineral density. Int J Vitam Nutr Res. 2013;83(3):154–61.
Hinners TA, Terrill WJ, Kent JL, Colucci AV. Hair–metal binding. Environ Health Perspect. 1974;8:191–9.
Smart KE, Kilburn M, Schroeder M, Martin BGH, Hawes C, Marsh JM, Grovenor CRM. Copper and calcium uptake in colored hair. Int J Cosmet Sci. 2010;32(2):161–2.
Skulan J, DePaolo DJ. Calcium isotope fractionation between soft and mineralized tissues as a monitor of calcium use in vertebrates. Proc Natl Acad Sci. 1999;96(24):13709.
Rudge JF, Reynolds BC, Bourdon B. The double spike toolbox. Chem Geol. 2009;265(3–4):420–31.
Feng L, Zhou L, Yang L, Tong S, Hu Z, Gao S. Optimization of the double spike technique using peak jump collection by a Monte Carlo method: an example for the determination of Ca isotope ratios. J Anal At Spectrom. 2015;30(12):2403–11.
Fantle MS, Tipper ET. Calcium isotopes in the global biogeochemical Ca cycle: implications for development of a Ca isotope proxy. Earth-Sci Rev. 2014;129:148–77.
Wieser ME, Buhl D, Bouman C, Schwieters J. High precision calcium isotope ratio measurements using a magnetic sector multiple collector inductively coupled plasma mass spectrometer. J Anal At Spectrom. 2004;19(7):844.
Feng L, Zhou L, Yang L, Zhang W, Wang Q, Shuoyun T, Hu Z. A rapid and simple single-stage method for Ca separation from geological and biological samples for isotopic analysis by MC-ICP-MS. J Anal At Spectrom. 2018;33(3):413–21.
CIAAW. Calcium 2015 [Available from: https://www.ciaaw.org/calcium.htm].
Zhu HL, Zhang ZF, Wang GQ, Liu YF, Liu F, Li X, Sun WD. Calcium isotopic fractionation during ion-exchange column chemistry and thermal ionisation mass spectrometry (TIMS) determination. Geostand Geoanal Res. 2016;40(2):185–94.
Walczyk T. TIMS versus multicollector-ICP-MS: coexistence or struggle for survival? Anal Bioanal Chem. 2004;378(2):229–31.
Douthitt CB. The evolution and applications of multicollector ICPMS (MC-ICPMS). Anal Bioanal Chem. 2008;390(2):437–40.
Bürger S, Vogl J, Kloetzli U, Nunes L, Lavelle M. Chapter 14 Thermal ionisation mass spectrometry. Sector Field Mass Spectrometry for Elemental and Isotopic Analysis: The Royal Society of Chemistry; 2015. pp. 381–438.
Jakubowski N, Horsky M, Roos PH, Vanhaecke F, Prohaska T. Chapter 12 Inductively coupled plasma mass spectrometry. Sector Field Mass Spectrometry for Elemental and Isotopic Analysis: The Royal Society of Chemistry; 2015. pp. 208–318.
Chakrabarti R, Mondal S, Jacobson AD, Mills M, Romaniello SJ, Vollstaedt H. Review of techniques, challenges, and new developments for calcium isotope ratio measurements. Chem Geol. 2021;581:120398.
Reynard LM, Henderson GM, Hedges REM. Calcium isotope ratios in animal and human bone. Geochim Cosmochim Acta. 2010;74(13):3735–50.
Romaniello SJ, Field MP, Smith HB, Gordon GW, Kim MH, Anbar AD. Fully automated chromatographic purification of Sr and Ca for isotopic analysis. J Anal At Spectrom. 2015;30(9):1906–12.
Bao Z, Zong C, Chen K, Lv N, Yuan H. Chromatographic purification of Ca and Mg from biological and geological samples for isotope analysis by MC-ICP-MS. Int J Mass Spectrom. 2020;448:116268.
Skulan J, DePaolo DJ, Owens TL. Biological control of calcium isotopic abundances in the global calcium cycle. Geochim Cosmochim Acta. 1997;61(12):2505–10.
Clementz MT, Holden P, Koch PL. Are calcium isotopes a reliable monitor of trophic level in marine settings? Int J Osteoarchaeol. 2003;13(1–2):29–36.
Heuser A, Eisenhauer A. The calcium isotope composition (δ44/40Ca) of NIST SRM 915b and NIST SRM 1486. Geostand Geoanal Res. 2008;32(3):311–5.
Heuser A, Tütken T, Gussone N, Galer SJG. Calcium isotopes in fossil bones and teeth—diagenetic versus biogenic origin. Geochim Cosmochim Acta. 2011;75(12):3419–33.
Farkaš J, Déjeant A, Novák M, Jacobsen SB. Calcium isotope constraints on the uptake and sources of Ca2+ in a base-poor forest: a new concept of combining stable (δ44/42Ca) and radiogenic (εCa) signals. Geochim Cosmochim Acta. 2011;75(22):7031–46.
Huang S, Farkaš J, Yu G, Petaev MI, Jacobsen SB. Calcium isotopic ratios and rare earth element abundances in refractory inclusions from the Allende CV3 chondrite. Geochim Cosmochim Acta. 2012;77:252–65.
National Institute of Standards & Technology. Certificate of analysis - standard reference material 1400 bone ash. Gaithersburg: National Institute of Standards & Technology; 1992.
Driessens FCM, Verbeeck RK. Biominerals. Boca Raton: CRC Press; 1990.
Forte G, Bocca B, Senofonte O, Petrucci F, Brusa L, Stanzione P, Zannino S, Violante N, Alimonti A, Sancesario G. Trace and major elements in whole blood, serum, cerebrospinal fluid and urine of patients with Parkinson’s disease. J Neural Transmission. 2004;111(8):1031–40.
Harrington JM, Young DJ, Essader AS, Sumner SJ, Levine KE. Analysis of human serum and whole blood for mineral content by ICP-MS and ICP-OES: development of a mineralomics method. Biol Trace Elem Res. 2014;160(1):132–42.
Jeruszka-Bielak M, Brzozowska A. Relationship between nutritional habits and hair calcium levels in young women. Biol Trace Elem Res. 2011;144(1):63–76.
Mayer AJ, Wieser ME. The absolute isotopic composition and atomic weight of molybdenum in SRM 3134 using an isotopic double-spike. J Anal At Spectrom. 2014;29(1):85–94.
Yang L, Tong S, Zhou L, Hu Z, Mester Z, Meija J. A critical review on isotopic fractionation correction methods for accurate isotope amount ratio measurements by MC-ICP-MS. J Anal At Spectrom 2018.
Mohamed FAA. Identifying zinc inputs to Heard and McDonald Islands region using zinc concentrations and isotopic compositions. Calgary: University of Calgary; 2020.
Zimmermann T, Mohamed AF, Reese A, Wieser ME, Kleeberg U, Pröfrock D, Irrgeher J. Zinc isotopic variation of water and surface sediments from the German Elbe River. Sci Total Environ. 2020;707:135219.
Retzmann A, Zimmermann T, Pröfrock D, Prohaska T, Irrgeher J. A fully automated simultaneous single-stage separation of Sr, Pb, and Nd using DGA Resin for the isotopic analysis of marine sediments. Anal Bioanal Chem. 2017;409(23):5463–80.
Sasaki Y, Zhu Z-X, Sugo Y, Kimura T. Extraction of various metal ions from nitric acid to n-dodecanen by diglycolamide (DGA) compounds. J Nucl Sci Technol. 2007;44(3):405–9.
Pourmand A, Dauphas N. Distribution coefficients of 60 elements on TODGA resin: application to Ca, Lu, Hf, U and Th isotope geochemistry. Talanta. 2010;81:741–53.
Retzmann A, Walls D, Miller K, Wieser M, Irrgeher J, Prohask T. Assessing the potential of online ICP–MS analysis to optimize Ca/matrix separation using DGA Resin for subsequent isotopic analysis. Monatsh Chem. 2021;152:401–10.
Feng L-p, Zhou L, Yang L, DePaolo DJ, Tong S-Y, Liu Y-S, Owens TL, Gao S. Calcium isotopic compositions of sixteen USGS reference materials. Geostand Geoanal Res. 2017;41(1):93–106.
Kragten J. Tutorial review. Calculating standard deviations and confidence intervals with a universally applicable spreadsheet technique. Analyst. 1994;119(10):2161–5.
Amini M, Eisenhauer A, Böhm F, Holmden C, Kreissig K, Hauff F, Jochum KP. Calcium isotopes (δ44/40Ca) in MPI-DING reference glasses, USGS rock powders and various rocks: evidence for ca isotope fractionation in terrestrial silicates. Geostand Geoanal Res. 2009;33(2):231–47.
Huang S, Farkaš J, Jacobsen SB. Stable calcium isotopic compositions of Hawaiian shield lavas: evidence for recycling of ancient marine carbonates into the mantle. Geochim Cosmochim Acta. 2011;75(17):4987–97.
Steuber T, Buhl D. Calcium-isotope fractionation in selected modern and ancient marine carbonates. Geochim Cosmochim Acta. 2006;70(22):5507–21.
Hippler D, Witbaard R, van Aken HM, Buhl D, Immenhauser A. Exploring the calcium isotope signature of Arctica islandica as an environmental proxy using laboratory- and field-cultured specimens. Palaeogeogr Palaeoclimatol Palaeoecol. 2013;373:75–87.
Acknowledgements
The authors would like to thank Courtney Kruschel for her support in the lab and Alex Tennant for his support with the DS software.
Funding
This work was funded by the Chemical Monthly Fellowship (2018) of the Austrian Academy of Sciences (ÖAW), by the Discovery Research Grant awarded by the Natural Sciences and Engineering Research Council of Canada (NSERC), and by the Faculty of Science Grand Challenges Fund of the University of Calgary.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
No human participants or animals are involved in the present study. All analyzed biological tissue samples are commercially available certified reference materials.
Conflict of interest
The authors declare no competing interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published in the topical collection celebrating ABCs 20th Anniversary.
Supplementary information
ESM 1
(DOCX 26 kb)
Rights and permissions
About this article

Cite this article
Retzmann, A., Walls, D., Miller, K.A. et al. A double-spike MC TIMS measurement procedure for low-amount Ca isotopic analysis of limited biological tissue samples. Anal Bioanal Chem 414, 675–689 (2022). https://doi.org/10.1007/s00216-021-03650-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-021-03650-8