
Abstract
We present an analytical method developed and validated to study the potential uptake of 13 selected drugs (ten pharmaceuticals, one illicit drug, and two transformation products) into lettuce plants from contaminated water and soil. Some of the selected drugs (i.e., cocaine, methadone, cis-diltiazem, valsartan, and valsartan acid), which are commonly present in treated wastewater, were investigated for the first time in plant tissues. The method is based on ultrasonic solvent extraction with acetonitrile–methanol (1:1, v/v) and subsequent automated extract cleanup and analysis by means of online solid-phase extraction–liquid chromatography–tandem mass spectrometry. Optimum extraction conditions were selected after evaluation of analyte recoveries with four different extraction techniques (ultrasonic solvent extraction, solid–liquid extraction, pressurized liquid extraction, and a “quick, easy, cheap, effective, rugged, and safe” based method) and six different solvent mixtures. Furthermore, two different solid-phase extraction cleanup sorbents were evaluated. The method developed has high sensitivity (with limits of detection between 0.1 and 12.6 ng per gram dry weight and limits of quantification between 0.5 and 42.0 ng per gram dry weight), satisfactory accuracy (with analyte relative recoveries above 80% for all analytes but acridone and oxcarbazepine), and good repeatability (with relative standard deviations below 9% for all analytes). As part of the validation procedure, the analytical method was applied to the analysis of lettuce plants irrigated with water fortified with the selected compounds for the entire growing period. The results obtained evidenced the transfer of all the investigated drugs into lettuce leaves.

ᅟ
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Water scarcity and the increasing water demand for agriculture have forced water authorities to seek alternative water resources. Consequently, countries in arid and semiarid regions where water is a limited commodity (e.g., countries bordering the Mediterranean Sea) are increasingly using treated municipal wastewater to cover water demand [1, 2]. Although the safety and the suitability of reclaimed wastewater for agricultural irrigation have already been demonstrated [3, 4], it can also be a source of anthropogenic chemicals, such as pharmaceutically active compounds and personal care products, in the environment [5] because they are not completely removed during wastewater treatment processes. Thus, the consumption of fresh crops irrigated with reclaimed water could represent a direct route of human exposure to wastewater-derived contaminants [6].
In recent years, numerous analytical methods have been developed to determine the occurrence of pharmaceutically active compounds, drugs of abuse, pesticides, and hormones in environmental aqueous matrices (drinking water, groundwater, surface water, and wastewater) [7,8,9,10,11]. Most of these methods are based on solid-phase extraction (SPE) of the analytes and liquid chromatography (LC)–mass spectrometry (MS) detection. SPE is a quite versatile technique useful to extract and preconcentrate a wide spectrum of organic compounds from environmental samples that can be connected in series to the LC–MS system. This allows the development of fully automated methods that have been proved to be more sensitive and reproducible than off-line analytical approaches, because the whole sample is transferred into the LC–MS system and sample handling is reduced [12,13,14].
In contrast, few analytical methods exist in the peer-reviewed literature for the determination of such organic contaminants in environmental solid matrices, such as soils and plants, being even scarcer for the later [15,16,17,18,19]. Different sample preparation approaches have been used to extract pharmaceutically active compounds from plant tissues. Boxal et al. [20] extracted ten drugs of common veterinary use from lettuce and carrots with different solvent mixtures, by solid–liquid extraction (SLE) or Soxhlet extraction. Pharmaceutically active compounds commonly found in treated wastewater, including carbamazepine, diclofenac, and ibuprofen, and various pesticides were successfully extracted by means of pressurized liquid extraction (PLE) from lettuce using acetone–hexane and ethyl acetate (EtAc)–hexane [21]. Ultrasonic solvent extraction (USE), for instance, with acetonitrile (ACN)–water, or methyl tert-butyl ether (MTBE) followed by ACN, was used to extract various wastewater-derived contaminants (pharmaceutically active compounds, personal care products, steroid hormones) from different plant tissues (radish and ryegrass [22], lettuce and spinach [23], and lettuce and tomato [24]). An SLE method using methanol (MeOH) and MeOH–water has been recently developed by Riemenschneider et al. [25] to extract several organic contaminants from lettuce, cabbage, and tomato. The extraction of pharmaceutically active compounds with “quick, easy, cheap, effective, rugged, and safe” (QuEChERS)-based approaches was also explored on several occasions [25, 26], and it was reported to provide good analyte recoveries in celery and lettuce matrices [26]. SPE is usually the technique of choice to clean up and purify the plant extracts obtained [20, 22, 23, 25].
Measurement of polar organic compound concentrations in plant tissues is of great interest to investigate their fate in edible crops. However, it is a big challenge because of the low concentrations at which these compounds may be present [usually in the nanograms per gram dry weight (dw) range] and the complexity of these matrices, as they contain pigments, fats, cellulose, and waxes in different proportions, which may interfere with analyte extraction and instrumental analysis [23, 27]. Co-extracted chlorophyll and waxy or fatty materials from plant tissues may interfere with the accuracy and precision of the results if they are not removed during sample treatment [28]. The analytical methods reported to date in the peer-reviewed literature to analyze pharmaceutically active compounds in plant tissues require considerable time and effort for the sample preparation process or specialized extraction equipment [15].
In this context, the main objective of the present study was to develop and validate an analytical method based on LC–tandem MS (MS/MS) detection for the simultaneous determination of 13 drugs of widespread use and commonly present in treated wastewater in lettuce leaves. [10, 11]. The performance of four techniques (i.e., SLE, PLE, USE, and QuEChERS-based extraction) to extract the selected drugs from this matrix was compared. Lettuce was selected as the matrix of study for several reasons: (1) it is the most cultivated leaf vegetable worldwide, (2) it is a quick-growing crop, (3) it is easy to grow in greenhouse conditions, (4) it is usually consumed without being cooked or heated, which preserves the retained contaminants, (5) it has an extensive complex root structure that may facilitate the uptake of organic contaminants from soil [29], and (6) its vegetative part consists merely of green leaves, which simplifies sample preparation. The drugs selected included ten pharmaceuticals belonging to six different therapeutic classes—that is, nonsteroidal anti-inflammatory drugs (diclofenac and ibuprofen), antibiotics (trimethoprim), anticonvulsants (carbamazepine, oxcarbazepine, and lamotrigine), antihypertensive drugs (cis-diltiazem and valsartan), benzodiazepines (midazolam), and opioid agonists (methadone)—one illicit drug (cocaine), and two transformation products (acridone and valsartan acid). After validation, the optimized analytical approach was applied to the analysis of the selected drugs in lettuce plants grown in soil pots and irrigated with a mixture of these drugs for the entire growing period. To our knowledge, cocaine, methadone, cis-diltiazem, valsartan, and valsartan acid have not been previously investigated in plant tissues, and this work represents the first report on the potential accumulation of these compounds in lettuce.
Materials and methods
Chemicals reagents
High-purity standards used for method development and analytical purposes were purchased from Sigma-Aldrich (St Louis, MO, USA), and Cerilliant (Round Rock, TX, USA). Cocaine hydrochloride salt and methadone hydrochloride salt used in the mixtures used for irrigation were provided as a concession for research purposes (2009C00124) by the Division of Narcotic Drugs and Psychotropic Substances of the Spanish Agency of Pharmaceuticals and Medical Products. Medical formulations were used for irrigation in the case of midazolam (EFG injectable solution, 5 mg of active principle per milliliter, Normon, Madrid, Spain), valsartan (40 mg, Kern Pharma, Barcelona, Spain), valsartan acid (purity 95% or greater, synthesized and purified following the procedure described by Nödler et al. [30]), and oxcarbazepine (Trileptal® 300 mg, Novartis, Basel, Switzerland). The chemical structures of the target drugs as well as their purity, suppliers, and selected physicochemical properties are provided in Table S1.
Isotopically labeled compounds used as surrogate standards (diclofenac-d 4, ibuprofen-d 3, trimethoprim-d 3, carbamazepine-d 10, valsartan-d 3, valsartan acid-d 4, lamotrigine-13 C 3, midazolam-13 C 6, cocaine-d 3, methadone-d 3) were purchased from Cerilliant, Alsachim (Illkirch-Graffenstaden, France), or Toronto Research Chemicals (Toronto, ON, Canada).
Individual stock standard solutions with a concentration of 1000 μg mL-1 were prepared in MeOH and stored at -20 °C. A fresh irrigation solution was prepared before each use by dilution of a water-based stock standard mixture of the target analytes (2 μg mL-1) with tap water to a final concentration of 200 ng mL-1. Working mixtures containing the target analytes and/or the isotopically labeled compounds were prepared in MeOH at appropriate concentrations to be used in the validation studies and for calibration purposes. The Bond Elut QuEChERS method extraction kit and the Bond Elut QuEChERS dispersive SPE kit for general fruits and vegetables were purchased from Agilent Technologies (Santa Clara, CA, USA). High-performance LC (HPLC)-grade solvents were purchased from Merck (Darmstadt, Germany): ACN (purity 99.9% or greater), MeOH (purity 99.9% or greater), EtAc (purity 99.5% or greater), hexane (purity 99.0% or greater), acetone (purity 99.7% or greater), and water. Chromasolv MTBE (purity 99.9% or greater), formic acid (purity 96% or greater, ACS reagent), and sodium thiosulfate pentahydrate salt (purity 99.5% or greater) were supplied by Sigma-Aldrich. Polytetrafluoroethylene (PTFE) filters (13 mm, 0.2 μm) were purchased from Millipore (Carrigtwohill, Cork, Ireland). Oasis HLB online SPE cartridges (10 mm × 1 mm, 30-μm particle size, 2.5 mg) were purchased from Waters (Milford, MA, USA), and HySphere Resin GP online SPE cartridges (10 mm × 2 mm, 5–15-μm particle size, 13 mg) were purchased from Spark Holland (Emmen, Netherlands).
Sample preparation
Lettuce (Lactuca sativa L. ‘Maravilla de Verano-Canasta’) was selected for the study as it can be cultivated in both outdoor and greenhouse setups at any time of the year and it is very resistant to fungal diseases. A mature lettuce head was purchased from a local organic supermarket to perform method optimization and validation studies. Grown plants irrigated with a mixture of the selected drugs (see “Experimental design for the uptake study” for further details) were investigated to evaluate drug uptake. Before extraction, lettuce leaves were gently hand washed with tap water to remove soil residues and subsequently rinsed with deionized water. Then, the leaves were gently blot dried with a paper towel, and stored at –20 °C for at least 48 h. Frozen leaves were then freeze-dried for 3 days or longer if needed (until no ice was left) to remove residual water using a LyoAlfa 6 system (Telstar Technologies, Terrassa, Spain). Freeze-dried samples were ground to powder with a laboratory grinder with a stainless steel grinding chamber (Snijders, Tilburg, Netherlands) and stored in 20-mL glass vials in the dark at –20 °C until extraction.
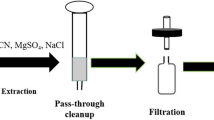
Sample extraction and cleanup
Four extraction techniques were assessed and compared in this study: SLE, PLE, USE, and QuEChERS-based extraction. For each technique, six different extraction solvent mixtures were tested: ACN–MeOH (1:1, v/v), MTBE–MeOH (1:1, v/v), hexane–acetone (1:1, v/v), MTBE–ACN (1:1, v/v), EtAc–MeOH (1:1, v/v), and 0.5% formic acid in ACN–MeOH (1:1, v/v). Detailed extraction protocols are described in the electronic supplementary material. The effects of a cleanup step on analyte recovery were also evaluated. For this, lettuce leaf extracts obtained with the different extraction techniques and extraction solvent mixtures were filtered through 0.2-μm PTFE filters, liquid–liquid extracted with hexane (hexane washed), or preconcentrated by SPE on either Oasis HLB or HySphere Resin GP cartridges followed by LC–MS/MS analysis (see the electronic supplementary material for further details).
In the optimized approach, 0.5 g of a freeze-dried lettuce leaf sample was placed in 50-mL polypropylene centrifuge tubes and spiked with a known amount of the isotopically labeled compounds (100 ng g-1 dw, equivalent to 0.5 ng mL-1 in the final extract). Spiked samples were left in the fume hood for 1 h to allow the MeOH of the spiking solution to evaporate, and then placed in a fridge overnight. Analytes were extracted from lettuce leaves by means of two consecutive USE cycles using 16 mL of a mixture ACN:MeOH (1:1, v/v) and a Fisherbrand FB15064 ultrasonic water bath (Fisher Scientific, Pittsburgh, PA, USA). For the first extraction cycle, 2 mL of water was also added to the lettuce sample. Each 20-min USE cycle was performed after vortex mixing of the extraction solvent and the sample for 2 min. After each USE cycle, the sample was centrifuged at 4000 rpm for 30 min, and the supernatant was transferred to a 40-mL glass vial. Extraction solvents were combined and evaporated until a volume of 2 mL under a gentle stream of nitrogen at 24 °C with use of a TurboVap® LV (Biotage, Uppsala, Sweden). Then, the 2-mL extract was diluted with HPLC water to 100 mL, and filtered through a 0.2-μm PTFE filter. A 10-mL aliquot of the filtrate was transferred to a vial for extract cleanup by online SPE.
For cleanup, 5 mL of the extract was preconcentrated on Oasis HLB cartridges by means of an automated online SPE sample processor (Symbiosis Pico; Spark Holland). Online SPE cartridges were conditioned with 2 mL of ACN and 2 mL of MeOH, and equilibrated with 2 mL of water, at a flow rate of 5 mL min-1. Extract loading was performed at a slower flow rate (1 mL min-1) to enhance analyte retention onto the sorbent. After extract loading, the cartridge was washed with 1 mL of water to complete the transfer of the sample and remove interferences such as inorganic salts, and trapped analytes were eluted from the cartridge to the LC column with the LC mobile phase. This cleanup step contributed to improving method sensitivity and reducing matrix interferences.
LC–MS/MS analysis
LC–MS/MS analyses were performed in a system consisting of an HPLC Symbiosis Pico system, operated in extraction LC mode, connected in series to a 4000 QTRAP hybrid triple quadrupole–linear ion trap mass spectrometer equipped with a TurboIonSpray source (Sciex, Redwood City, CA, USA), operated in the positive electrospray ionization (ESI) mode. The analytical instrumentation was controlled by Analyst 1.4.2 (Sciex), into which the Symbiosis software (Spark Holland) was integrated.
HPLC separation of the analytes was performed with a 125 mm × 2 mm Purospher STAR RP-18 end-capped column (5-μm particle size) (Merck, Darmstadt, Germany) and a mobile phase consisting of ACN and water, both acidified with 0.1% formic acid, at a flow rate of 0.3 mL min-1. The initial conditions (5% ACN) were maintained for 2 min before the organic solvent content was increased from 5% to 50% during the next 19 min and then to 100% in the following 2 min. Pure organic conditions were held for 5 min before returning to the initial conditions, which were maintained for 10 min to allow column reequilibration before the next injection. This gradient provided a chromatographic analysis time of 38 min.
MS/MS analyses were performed in the selected reaction monitoring (SRM) mode. Thus, the transitions from the precursor ion to two of its main fragment ions were registered for each target compound. Only one SRM transition was selected for each surrogate standard. Selected SRM transitions for each analyte and isotopically labeled compound and the optimal instrumental conditions set for their analysis are summarized in Table 1.
Method validation
The performance of the optimized analytical approach was evaluated in terms of linearity, trueness (analyte recovery), precision (repeatability), sensitivity, and matrix effects.
Quantification was performed by the internal standard method. For this, a fixed and known amount of surrogate standard (100 ng g-1 dw) was added to the samples, calibration standards, and blanks. The calibration curve was then constructed by linear weighted least-squares regression (1/x as the weighting factor) by plotting the ratio of the analyte signal to the surrogate standard signal as a function of the analyte concentration. Calibration standards were prepared by spiking compound-free lettuce extracts with the appropriate amount of standards, and expanded within the calibration range 0.0025–2.5 ng mL-1 (equivalent to 0.5–500 ng g-1 dw in lettuce leaves).
Extraction recoveries were evaluated from replicate analysis (n = 6) of compound-free lettuce leaf samples fortified with the 13 target compounds before the extraction process at two concentrations (50 and 400 ng g-1 dw). Absolute recoveries were determined by comparing the peak areas obtained in the spiked lettuce leaf samples with those obtained in standard solutions at equivalent concentrations (0.25 and 2 ng mL-1, respectively). Analyte relative recoveries were evaluated by comparing the analyte absolute recovery with that obtained for the surrogate standard used for quantification. Method precision, expressed as method repeatability, was calculated as the relative standard deviation (RSD) of the relative recoveries obtained in the recovery study.
Method detection limits and method quantification limits were estimated as the concentration of analyte that provided a signal-to-noise ratio of 3 and 10, respectively. For those analytes found at high concentration in the samples or that were not found in the lettuce samples, method detection limits and method quantification limits were estimated from the signal-to-noise ratio observed in the samples fortified at the lowest level (50 ng g-1 dw) in the recovery study.
To evaluate the matrix effects (the potential ion suppression or ion enhancement produced by the matrix components), compound-free lettuce samples (n = 3) were extracted with the optimized protocol, and the extract was spiked with the target compounds at 1 ng mL-1 (equivalent to 200 ng g-1 dw) just before online SPE–LC–MS/MS analysis. The peak areas obtained in these samples were compared with those obtained in online SPE–LC–MS/MS analysis of HPLC water spiked at an equivalent concentration (1 ng mL-1).
Quality controls were prepared by spiking analyte-free lettuce extracts at a concentration of 50 ng g-1 dw, and they were analyzed every six samples. Blanks consisting of the lettuce used in the validation study fortified only with the surrogate standard mixture were also analyzed to confirm the absence of the target analytes in this matrix, and blanks consisting of HPLC-grade water were also injected after every quality control sample to confirm the absence of carryover or memory effects.
Experimental design for the uptake study
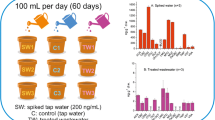
In May 2016, nine plants (three controls and six replicates) of lettuce (Lactuca sativa L. ‘Maravilla de Verano-Canasta’) at approximately the four-leaf stage were purchased from a local plant nursery and transplanted individually into 0.20-m-deep poly(ethylene terephthalate) pots filled with about 2000 ± 20 g of organic substrate for horticulture. Lettuce plants were left to grow on the terrace of a three-story building under natural sunlight (Institute of Environmental Assessment and Water Research, Barcelona, Spain). After 5 days of acclimation, the plants were irrigated daily with 100 mL of tap water containing the selected drugs at a concentration of 200 ng mL-1 by pouring the water directly onto the soil. Control plants were irrigated with 100 mL of tap water. Sodium thiosulfate [1 mL of a 1.8% (m/v) solution] was used to remove residual chlorine (approximately 0.6 mg L-1) present in the tap water used for irrigation. Application of fertilizers and pesticides and irrigation were performed as needed to ensure optimal crop growth. No rainfall occurred during the experimental period. Mature lettuces were harvested after 60 days by cutting them just below the cotyledonary node, and were processed for analysis in triplicate as described earlier in “Sample preparation” and “Sample extraction and cleanup” sub-sections.
Results and discussion
Optimization of sample extraction and cleanup
The most efficient extraction approach (described in the electronic supplementary material) was selected after evaluation of analyte absolute recoveries in filtered extracts. For this, analyte peak areas obtained after LC–MS/MS analysis of filtered extracts and standard solutions prepared with a mixture of H2O–MeOH at equivalent concentrations (100 ng mL-1) were compared. Besides the extraction efficiency, other factors such as extraction time and cost were also considered in the final decision.
Analyte absolute recoveries indicated the ACN–MeOH extraction mixture to be one of the best options to extract the target compounds from lettuce leaves, as can be observed in Fig. S4. Good recoveries were achieved with this solvent mixture in USE, SLE, and QuEChERS-based extraction. Overall, the poorest extraction yields were obtained when a mixture of hexane–acetone was used.
Although the analytes were successfully extracted by SLE with different extraction mixtures—that is, ACN–MeOH (1:1, v/v) and MTBE–MeOH (1:1, v/v)—this technique was not selected because the recoveries obtained for most analytes presented high variability (RSD >20%) and it is a very time-consuming technique (extraction time of 16 h).
QuEChERS-based extraction has been the technique of choice of several authors [26, 31, 32] to extract pharmaceuticals and personal care products from plant tissues. However, it was not used further in our study because we obtained recovery values of less than 50% for more than half of the target compounds (see Fig. S4).
PLE was not selected because the acidic compounds diclofenac, ibuprofen, valsartan, and valsartan acid were not recovered at all by means of the PLE approaches tested. This could be attributed to the low extraction temperature used (up to 80 °C) to avoid thermal degradation of the analytes, or the use of the incorrect extraction solvent, because these are factors that can severely affect the extraction efficiency [33]. Other authors used water as an extraction solvent to recover pharmaceuticals from plant tissues [26]. For instance, Chuang et al. [26] reported recoveries greater than 70% for PLE of carbamazepine and trimethoprim with different ACN–MeOH–H2O mixtures. However, according to our results, these compounds were also efficiently PLE- extracted with a different polar solvent mixture, i.e., MeOH–acetone.
Therefore, taking all the aforementioned findings into consideration, we finally selected USE for further optimization. Since preliminary tests were performed using individual solvents sequentially, the USE- extraction recoveries provided by solvent mixtures were also evaluated. For this, spiked lettuce samples were extracted with two USE cycles using either ACN–MeOH (1:1, v/v), or hexane–acetone (1:1, v/v), or the aforementioned mixtures in sequence. The results, shown in Fig. S5, confirmed the ACN–MeOH mixture as the best extraction solvent mixture. Unfortunately, this extraction solvent mixture proved very difficult to evaporate, as it left a viscous residue that was difficult to evaporate and resuspend (experimentally observed and reported by Bragança et al. [31] as well). For this reason, 2 mL of water was added to the extraction solvent mixture. To solve the problem of suspended particles, all samples were filtered through 0.2-μm PTFE filters.
Although a good signal was obtained for most target analytes in LC–ESI-MS/MS analysis of filtered USE extracts, a cleanup approach was considered to avoid precipitation of matrix components in the ionization source, and thus frequent cleanup of the source, and ultimately, to increase method robustness. Following the recommendations provided in peer-reviewed analytical studies [28, 34], the suitability of a cleanup step based on hexane washes was evaluated. Analyte recoveries in hexane-washed extracts are provided in Fig. S6. The results obtained indicated that this procedure was not effective for extract cleanup because of the losses of many analytes during the process. Furthermore, this step resulted in very high RSD values because waxy or fatty components were not completely removed [28] and/or the need of intensive manual sample handling.
An SPE-based cleanup of the extract was also evaluated, as this technique may increase method sensitivity because of extract preconcentration and removal of matrix interferences that are not retained in the SPE sorbent, and can be completely automatized, which reduces sample handling and consequently improves method precision. The extraction recoveries of the analytes on two generic SPE sorbents (i.e., Oasis HLB and HySphere Resin GP) was evaluated through the analysis of HPLC water fortified with the target analytes at a concentration of 0.1 ng mL-1. The results obtained are shown in Fig. 1. As it can be observed in this figure, both sorbents provided similar average extraction recoveries. However, Oasis HLB was chosen for the excellent RSD values (n = 3) provided, less than 10%, except for diclofenac (15%).

Absolute recoveries of target analytes in the solid-phase extraction sorbents evaluated. ACR acridone, CBZ carbamazepine, CDM cis-diltiazem, COC cocaine, DCF diclofenac, IBU ibuprofen, LMG lamotrigine, MDZ midazolam, MTD methadone, OxCBZ oxcarbazepine, TMP trimethoprim, VSA valsartan acid, VST valsartan
LC–ESI-MS/MS analysis
Optimum MS conditions under positive and negative ionization were obtained for each target analyte after infusion of individual standard solutions (0.5–1 μg mL-1) into the mass spectrometer. The carboxylic acid bearing compounds diclofenac, ibuprofen, valsartan, and valsartan acid were found to be amenable to both positive and negative ESI, with the latter providing higher signal intensity. However, they were all analyzed in the positive polarity mode so that all target compounds could be determined in a single analytical run, reducing analysis time and cost, and contributing with the latter to the development of a “green” analytical approach.
Furthermore, the analysis of ibuprofen in positive ESI mode allowed us to register two SRM transitions, which is an important criterion required by EU legislation for reliable identification of organic residues in food [35]. The ibuprofen molecule fragments poorly in negative ESI mode, providing only one SRM transition for its analysis (Gros et al., 2006).
Different LC mobile phases with ACN, MeOH, or a mixture of both (1:1) as organic constituents and with and without pH modifiers in the aqueous phase (0.1% formic acid for pH 2.7, 10 mM ammonium formate for pH 4, and 10 mM ammonium acetate for pH 6) were tested to maximize the signal of the target analytes. The use of acidified MeOH provided the highest MS signal for all target compounds but ibuprofen, and good chromatographic peak shape in all cases. However, MeOH had to be replaced by ACN to produce narrow and symmetric peaks when we switched to online SPE–LC–MS/MS mode. This was because ACN has a higher elution strength than MeOH, which is required to properly elute the target compounds from the SPE cartridge.
A representative total ion chromatogram and representative extracted ion chromatograms of the target analytes obtained after online SPE–LC–ESI(+)-MS/MS analysis of a lettuce sample fortified at a concentration of 50 ng g-1 dw are shown in Fig. 2.


Total ion chromatogram (a) and extracted ion chromatograms (b) obtained for the target analytes after online solid-phase extraction –liquid chromatography–positive electrospray ionization tandem mass spectrometry analysis of a lettuce leaf sample fortified at a concentration of 50 ng per gram dry weight
Method validation
Method performance was evaluated in terms of linearity, analyte recovery, repeatability, sensitivity, and matrix effects as described in “Method validation” in “Materials and methods.” Validation results are provided in Table 2.
The MS signal response was linear from the analyte limit of quantification to 500 ng g-1 dw for all analytes, but for acridone, cis-diltiazem, diclofenac, lamotrigine, and valsartan acid, for which the linearity range extended only up to 400 ng g-1 dw or even 300 ng g-1 dw in the case of valsartan acid. Coefficients of determination (r 2) higher than 0.998 were obtained for all calibration curves. All curves were constructed by linear weighted least-squares regression with 1/x as the weighting factor, and including at least six calibration points, for which the predicted concentration did not differ by more than 20% (exceptionally 25%) from the theoretical concentration.
Recoveries of the overall method were calculated by analysis of lettuce leaf samples (n = 6) fortified at 50 and 400 ng g-1 dw. As shown in Table 2, the absolute and relative analyte recoveries obtained at both concentrations were, in general, in good agreement. The lowest overall method absolute recoveries were observed for ibuprofen and valsartan acid (around 30%). These low recoveries can be explained by pronounced ionization suppression effects observed in the case of ibuprofen (81%) (see Table 2) and the low recoveries provided by the SPE cartridge for valsartan acid (30%) (see Fig. 1). However, extraction losses and matrix effects can be well compensated by the use of isotopically labeled surrogates as indicated by relative recovery values, which were between 81% and 119% in all cases, but for acridone, oxcarbazepine, and cis-diltiazem. The deviations observed in the relative recoveries of the aforementioned compounds can be attributed to the fact that isotopically labeled analogues were not available for these compounds, and thus isotopically labeled analogues of structurally related compounds (e.g., carbamazepine-d 10 for oxcarbazepine) or closely eluted compounds with similar absolute recoveries (e.g., carbamazepine-d 10 for acridone and midazolam-d 3 for cis-diltiazem) were used. Method repeatability was satisfactory for all target compounds, with RSD values below 9% for all analytes. The online SPE cleanup process contributed to improving the precision of the method because it reduced sample handling. This parameter was negatively affected when the extracts were hexane-washed to remove undesired matrix components (see Fig. S6).
All target analytes but cis-diltiazem, methadone, and valsartan acid were subject to ionization suppression effects, as shown in Table 2. The MS signal was reduced between 12% (trimethoprim) and 81% (ibuprofen). In addition to ibuprofen, ionization of acridone, lamotrigine, and oxcarbazepine was seriously reduced (by more than 50%) by lettuce matrix components. Besides correcting for analyte losses, isotopically labeled compounds added as surrogate standards also contributed to compensate for matrix-derived ionization effects.
With regard to method sensitivity, the method developed allowed detecting and quantifying most of the target analytes in lettuce leaves at concentrations as low as 0.1 ng g-1 dw, and 0.5 ng g-1 dw in the case of diclofenac. The highest method detection limit and method quantification limit were obtained for lamotrigine, as 12.6 ng g-1 dw and 42.0 ng g-1 dw, respectively.
Uptake of selected drugs by lettuce plants
The optimized and validated analytical method was applied to determine the occurrence of selected drugs in the leaves of the lettuce plants used in the uptake study. The leaves of each individual plant were analyzed in triplicate. Leaves of control plants (i.e., lettuces that were not irrigated with the target drugs) were found to be free of drugs; however, all experimental plants accumulated the investigated drugs to some extent. The concentrations measured are summarized in Fig. 3. All analytes were detected in all investigated samples, but ibuprofen and valsartan acid that were detected in only a few plants at concentrations below the method quantification limit. The compounds that accumulated the most in lettuce leaves were carbamazepine followed by methadone, cis-diltiazem, and cocaine, presenting average concentrations above 100 ng g-1 dw in all the samples analyzed (1497, 475, 215, and 170 ng g-1 dw, respectively). Since carbamazepine concentrations exceeded the method linearity range for this compound, samples were diluted and reanalyzed. Carbamazepine was reported by other authors to potentially move inside the plant, and to accumulate preferably in plant leaves rather than in roots [36]. The lowest concentrations were observed for acridone, diclofenac, ibuprofen, valsartan acid, and valsartan, with average values below 30 ng g-1 dw. This could be attributed to a low potential for uptake, translocation, or accumulation of these compounds by lettuce plants, due to plant physiology itself or compound degradation in the soil. In the case of diclofenac, these low concentrations are in agreement with previous studies that reported slightly higher amounts of diclofenac in plant roots than in leaves [34, 36, 37]. The results from this study clearly showed that lettuce is capable of taking pharmaceuticals up from soil and water, and accumulating them in edible parts.

Average concentration (nanogram per gram dry weight) of the target analytes in lettuce leaves after triplicate analysis of the six lettuces grown in the uptake study (n = 18). d.w. dry weight
Conclusions
After comparison of different techniques to extract drugs from lettuce leaves, a novel analytical method based on USE followed by a SPE-based extract purification approach connected in series to the LC–ESI-MS/MS instrument was optimized and validated for the reliable quantification of 13 drugs. The drugs selected have been reported to be commonly present in wastewater treatment plant effluent water, and consequently they are likely to be taken up and accumulated by crops irrigated with reclaimed water. Satisfactory relative recoveries (72–103%) were obtained for all compounds but those for which isotopically labeled analogues were not available (acridone, cis-diltiazem, and oxcarbazepine), with RSD values below 9% in all cases. The validated method allows the detection and quantification of the selected compounds in lettuce leaves in the low nanogram per gram dry weight range. This method was applied to evaluate drug uptake by lettuces. The results revealed that all investigated drugs could enter lettuce after irrigation of the plant with water fortified (contaminated) with the drugs selected. This work represents the first step to provide novel knowledge of the behavior of these organic compounds in the water–soil–plant system, as some of them were investigated for the first time in plant tissues (i.e., cocaine, methadone, midazolam, cis-diltiazem, valsartan, and valsartan acid). Future work will be directed to further investigate the fate of the selected compounds in the plant root system and in the soil, and the effect that a contaminated soil may have on new plant individuals, so that the main mechanisms driving the uptake and accumulation of these drugs in the plant can be identified, and the potential risk of consuming edible plants contaminated with these products to human health can be correctly assessed.
References
Benotti MJ, Snyder SA. Pharmaceuticals and endocrine disrupting compounds: Implications for ground water replenishment with recycled water. Ground Water. 2009;47(4):499–502.
Fatta-Kassinos D, Kalavrouziotis IK, Koukoulakis PH, Vasquez MI. The risks associated with wastewater reuse and xenobiotics in the agroecological environment. Sci Total Environ. 2011;409(19):3555–63.
York DW, Holden R, Sheikh B, Parsons L. Safety and suitability of recycled water for irrigation of edible crops. In: Proceedings of the 23rd Annual Water Reuse Symposium, Water Reuse Association, Dallas, Texas. 2008.
Lonigro A, Rubino P, Lacasella V, Montemurro N. Faecal pollution on vegetables and soil drip irrigated with treated municipal wastewaters. Agric Water Manag. 2016;174:66–73.
Daughton CG, Ternes TA. Pharmaceuticals and personal care products in the environment: agents of subtle change? Environ Health Perspect. 1999;107 Suppl 6:907–38.
Zheng W, Wiles KN, Dodge L. Uptake and accumulation of pharmaceuticals and hormones in vegetables after irrigation with reuse water. Champaign: Illinois Sustainable Technology Center; 2016. Available at http://hdl.handle.net/2142/90131. Accessed Jan 2017.
Li WC. Occurrence, sources, and fate of pharmaceuticals in aquatic environment and soil. Environ Pollut. 2014;187:193–201.
Luo Y, Guo W, Ngo HH, Nghiem LD, Hai FI, Zhang J, et al. A review on the occurrence of micropollutants in the aquatic environment and their fate and removal during wastewater treatment. Sci Total Environ. 2014;473-474:619-41.
Narvaez VJF, Jimenez CC. Pharmaceutical products in the environment: Sources, effects and risks. Vitae. 2012;19(1):93–108.
Pal A, Gin KYH, Lin AYC, Reinhard M. Impacts of emerging organic contaminants on freshwater resources: Review of recent occurrences, sources, fate and effects. Sci Total Environ. 2010;408(24):6062–9.
Pal R, Megharaj M, Kirkbride KP, Naidu R. Illicit drugs and the environment - a review. Sci Total Environ. 2013;463-464:1079-92.
Camilleri J, Baudot R, Wiest L, Vulliet E, Cren-Olivé C, Daniele G. Multiresidue fully automated online SPE-HPLC-MS/MS method for the quantification of endocrine-disrupting and pharmaceutical compounds at trace level in surface water. Int J Environ Anal Chem. 2015;95(1):67–81.
Trenholm RA, Vanderford BJ, Snyder SA. On-line solid phase extraction LC-MS/MS analysis of pharmaceutical indicators in water: a green alternative to conventional methods. Talanta. 2009;79(5):1425–32.
Valsecchi S, Polesello S, Mazzoni M, Rusconi M, Petrovic M. On-line sample extraction and purification for the LC-MS determination of emerging contaminants in environmental samples. Trends Environ Anal Chem. 2015;8:27–37.
Wu X, Dodgen LK, Conkle JL, Gan J. Plant uptake of pharmaceutical and personal care products from recycled water and biosolids: a review. Sci Total Environ. 2015;536:655–66.
Snow DD, Cassada DA, Papastavros E, Bartelt-Hunt SL, Li X, Zhang Y, et al. Detection, occurrence and fate of emerging contaminants in agricultural environments. Water Environ Res. 2013;85(10):869–88.
Babić S, Mutavdžić PD. Analysis of PhACs in solid environmental samples (soil, sediment, and sludge). In: Mira Petrovic DB, Sandra P, editors. Comprehensive analytical chemistry. Amsterdam: Elsevier; 2013. p. 129–67.
Ho YB, Zakaria MP, Latif PA, Saari N. Determination of veterinary pharmaceuticals residue in soil and biological materials: a review of current analytical methods. In: Aris AZ, Tengku Ismail TH, Harun R, Abdullah AM, Ishak MY, editors. From sources to solution: proceedings of the International Conference on Environmental Forensics 2013. Singapore: Springer; 2014. p. 181–6.
Białk-Bielińska A, Kumirska J, Borecka M, Caban M, Paszkiewicz M, Pazdro K, et al. Selected analytical challenges in the determination of pharmaceuticals in drinking/marine waters and soil/sediment samples. J Pharm Biomed Anal. 2016;121:271–96.
Boxall ABA, Johnson P, Smith EJ, Sinclair CJ, Stutt E, Levy LS. Uptake of veterinary medicines from soils into plants. J Agric Food Chem. 2006;54(6):2288–97.
Calderón-Preciado D, Jiménez-Cartagena C, Peñuela G, Bayona JM. Development of an analytical procedure for the determination of emerging and priority organic pollutants in leafy vegetables by pressurized solvent extraction followed by GC-MS determination. Anal Bioanal Chem. 2009;394(5):1319–27.
Carter LJ, Harris E, Williams M, Ryan JJ, Kookana RS, Boxall ABA. Fate and uptake of pharmaceuticals in soil-plant systems. J Agric Food Chem. 2014;62(4):816–25.
Wu X, Conkle JL, Gan J. Multi-residue determination of pharmaceutical and personal care products in vegetables. J Chromatogr A. 2012;1254:78–86.
Zheng W, Wiles KN, Holm N, Deppe NA, Shipley CR. Uptake, translocation, and accumulation of pharmaceutical and hormone contaminants in vegetables. ACS symposium series. Washington: American Chemical Society; 2014. p. 167-81.
Riemenschneider C, Seiwert B, Goldstein M, Al-Raggad M, Salameh E, Chefetz B, et al. An LC-MS/MS method for the determination of 28 polar environmental contaminants and metabolites in vegetables irrigated with treated municipal wastewater. Anal Methods. 2017;9(8):1273–81.
Chuang YH, Zhang Y, Zhang W, Boyd SA, Li H. Comparison of accelerated solvent extraction and quick, easy, cheap, effective, rugged and safe method for extraction and determination of pharmaceuticals in vegetables. J Chromatogr A. 2015;1404:1–9.
Ferrer I, García-Reyes JF, Fernandez-Alba A. Identification and quantitation of pesticides in vegetables by liquid chromatography time-of-flight mass spectrometry. Trends Anal Chem. 2005;24(7):671–82.
Jones-Lepp TL, Sanchez CA, Moy T, Kazemi R. Method development and application to determine potential plant uptake of antibiotics and other drugs in irrigated crop production systems. J Agric Food Chem. 2010;58(22):11568–73.
Mc Farlane JC, Trapp S (eds). Plant contamination: modeling and simulation of organic chemical processes. 1994. CRC Press, Boca Raton.
Nödler K, Hillebrand O, Idzik K, Strathmann M, Schiperski F, Zirlewagen J, et al. Occurrence and fate of the angiotensin II receptor antagonist transformation product valsartan acid in the water cycle - A comparative study with selected β-blockers and the persistent anthropogenic wastewater indicators carbamazepine and acesulfame. Water Res. 2013;47(17):6650–9.
Bragança I, Plácido A, Paíga P, Domingues VF, Delerue-Matos C. QuEChERS: a new sample preparation approach for the determination of ibuprofen and its metabolites in soils. Sci Total Environ. 2012;433:281–9.
Ferro G, Polo-López MI, Martínez-Piernas AB, Fernández-Ibáñez P, Agüera A, Rizzo L. Cross-contamination of residual emerging contaminants and antibiotic resistant bacteria in lettuce crops and soil irrigated with wastewater treated by sunlight/H2O2. Environ Sci Technol. 2015;49(18):11096–104.
Carabias-Martínez R, Rodríguez-Gonzalo E, Revilla-Ruiz P, Hernández-Méndez J. Pressurized liquid extraction in the analysis of food and biological samples. J Chromatogr A. 2005;1089(1-2):1–17.
Tanoue R, Sato Y, Motoyama M, Nakagawa S, Shinohara R, Nomiyama K. Plant uptake of pharmaceutical chemicals detected in recycled organic manure and reclaimed wastewater. J Agric Food Chem. 2012;60(41):10203–11.
European Commission. Commission Decision 2002/657/EC of 12 August 2002 implementing the Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results, Off J Eur Communities 2012;L221/8.
Wu X, Conkle JL, Ernst F, Gan J. Treated wastewater irrigation: Uptake of pharmaceutical and personal care products by common vegetables under field conditions. Environ Sci Technol. 2014;48(19):11286–93.
Herklotz PA, Gurung P, Vanden Heuvel B, Kinney CA. Uptake of human pharmaceuticals by plants grown under hydroponic conditions. Chemosphere. 2010;78(11):1416–21.
Acknowledgements
N.M. gratefully acknowledges the University of Bari for financial support provided through a predoctoral grant (29th Doctorate Program in Biodiversity, Agriculture and Environment). This work was financially supported by the Government of Catalonia (Consolidated Research Groups B2014 SGR 418—Water and Soil Quality Unit and B2014 SGR 291—Catalan Institute for Water Research), and by the EU’s Seventh Framework Programme for research, technological development and demonstration (grant agreement no. 603437—SOLUTIONS), and it reflects only the authors’ views. The EU is not liable for any use that may be made of the information contained herein. Merck is acknowledged for the gift of liquid chromatography columns.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Published in the topical collection Advances in LC-MS/MS Analysis with guest editors Damià Barceló and Mira Petrovic.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 595 kb)
Rights and permissions
About this article

Cite this article
Montemurro, N., Postigo, C., Lonigro, A. et al. Development and validation of an analytical method based on liquid chromatography–tandem mass spectrometry detection for the simultaneous determination of 13 relevant wastewater-derived contaminants in lettuce. Anal Bioanal Chem 409, 5375–5387 (2017). https://doi.org/10.1007/s00216-017-0363-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0363-1