
Abstract
In this article, a dataset from a collaborative non-target screening trial organised by the NORMAN Association is used to review the state-of-the-art and discuss future perspectives of non-target screening using high-resolution mass spectrometry in water analysis. A total of 18 institutes from 12 European countries analysed an extract of the same water sample collected from the River Danube with either one or both of liquid and gas chromatography coupled with mass spectrometry detection. This article focuses mainly on the use of high resolution screening techniques with target, suspect, and non-target workflows to identify substances in environmental samples. Specific examples are given to emphasise major challenges including isobaric and co-eluting substances, dependence on target and suspect lists, formula assignment, the use of retention information, and the confidence of identification. Approaches and methods applicable to unit resolution data are also discussed. Although most substances were identified using high resolution data with target and suspect-screening approaches, some participants proposed tentative non-target identifications. This comprehensive dataset revealed that non-target analytical techniques are already substantially harmonised between the participants, but the data processing remains time-consuming. Although the objective of a “fully-automated identification workflow” remains elusive in the short term, important steps in this direction have been taken, exemplified by the growing popularity of suspect screening approaches. Major recommendations to improve non-target screening include better integration and connection of desired features into software packages, the exchange of target and suspect lists, and the contribution of more spectra from standard substances into (openly accessible) databases.

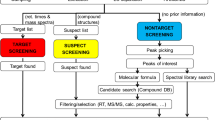
Matrix of identification approach versus identification confidence
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The evolution of accurate mass (AM) high resolution mass spectrometry (HRMS) coupled to gas or liquid chromatography (GC or LC, respectively) has initiated a new trend in analytical data processing in recent years. Targeted analytical methods are now often complemented with non-target or untargeted data acquisition and screening methods, where tandem mass spectrometry (MS–MS) is used to obtain fragmentation information to support identification. Earlier articles on the use of AM HRMS (hereafter shortened to HRMS, where the accurate mass is implied) in the environmental context (e.g. Refs. [1–4]) mention the three main approaches towards substance identification in non-target HRMS analysis, which are summarised here with some additional nuances:
-
1.
Target screening involves a reference standard measured in-house under the same analytical conditions such that retention time (RT), HRMS, and, where possible, (HR)MS–MS information is available for identification and confirmation. Quantitative target results should be distinguished from semi-quantitative results by using the term “quantitative target analysis/screening”.
-
2.
Suspect screening is performed when prior information (from a variety of sources, discussed in greater detail below) indicates that a given structure may be present in the sample. Thus, although no reference standard is available, the exact mass and isotope pattern calculated from the molecular formula plus or minus the expected adduct(s) of the suspect substance can be used to screen for this substance in the sample.
-
3.
Non-target screening involves all remaining components detected in a sample where no prior information is available. Because no structural information is available in advance, a full non-target identification starting from the exact mass, isotope, adduct, and fragmentation information needs to be performed.
The data analysis for target and suspect substances in non-target acquisition data can be performed in two main ways (or a combination of both). Traditionally in target analysis the presence or absence of each substance is determined individually using the extracted ion chromatogram (XIC). However, the evolution of non-target methods means that often a screening for target and/or suspect compounds is performed after peak detection with a suitable algorithm, such that the exact masses of the appropriate adduct of the target or suspect are searched within a given mass and (for targets) RT error. Whereas the first approach treats targets and suspects preferentially (i.e., they can be detected in cases where the peak is of insufficient quality for automated peak detection), in the latter case the target and suspect compounds are effectively a subset of all the “non-target components”. Here, the term “component” is used to refer to the group of exact masses (i.e. adduct and isotopologues) associated with one compound. Irrespective of the extraction technique, evidence from the measurement data is needed to confirm the identification, including the isotope pattern, presence of additional adducts, RT, fragmentation information, and other experimental evidence (e.g. presence of related substances, time trends). Several papers discuss this in greater detail (e.g. Refs. [1, 3–5]). Although the concept of identification points (IPs) given in EU Guideline 2002/657/EC [6] can be used to represent the evidence available for an identification where a reference standard (and thus a RT) is available, these are in urgent need of revision because they were released before HRMS(–MS) became as prominent as it is today. The concept of “identification levels” in HRMS analysis has been discussed recently in the environmental community and beyond (e.g. Refs. [7–9]), building on earlier attempts by the Metabolomics Society [10] to deal with the varying levels of confidence in identifications resulting from these three strategies for identifying substances using non-target acquisition data.
The concepts of identification strategy and confidence are merged in Fig. 1, showing that target, suspect, and non-target compounds start by definition at Levels 1 (reference standard available), 3 (tentative candidate(s)), and 5 (no information), respectively. If sufficient MS (exact mass, isotope, adduct), MS–MS (i.e., fragmentation), and experimental information (e.g. retention behaviour, presence of related substances) is available, suspect and non-target components can gain in confidence through to Level 2 (library match and/or diagnostic fragments) and even Level 1 after purchase of the corresponding standard for identifications (green arrows in Fig. 1). Compounds with a confirmed identity then become target compounds in future investigations. However, should the evidence of the sample and of the reference standard (target) or tentative candidate (suspect) not match, then the component associated with the target or suspect should become a “non-target of interest” (Level 5)—see red arrows. If the HRMS and RT information matches a target compound but HRMS–MS is not available because of lack of intensity, this is still regarded as a target identification, but should be reported with fewer identification points (e.g. 2 IP, as opposed to 4.5 IP where HRMS–MS from an isolated precursor is available [6]).

Matrix of identification approach versus identification confidence
It is important to note that this elaborate combination of strategy, confidence, and evidence is necessary with soft ionisation (SI) HRMS–MS analysis at this stage for one main reason: the lack of comprehensive spectral libraries for soft ionisation techniques, in contrast with the comparatively comprehensive GC–electron ionisation (EI) MS libraries available [11, 12] with over 200,000 substances. The reasons for this are varied, including the lack of reproducibility between SI instruments and different settings and the relative newness of the technique, and are discussed in greater detail elsewhere (e.g. Refs. [13–15]). In consequence of this difference in library resources, the identification of “unknown” environmental substances of interest measured with GC–EI MS can often be performed with a spectral library, whereas substances measured with alternative ionisation techniques usually require the parallel approaches of target, suspect, and non-target screening. This is not just limited to LC-based techniques; the dependence of strategy on ionisation technique is revealed for the environmental context in recent work comparing quadrupole time-of-flight (QTOF) GC–MS using EI and atmospheric pressure chemical ionisation (APCI) [16] and GC and ultra-high performance LC (UPLC) coupled to QTOFMS for a universal screening approach [17].
Several strategies for the selection of candidates for suspect and non-target screening of environmental substances using HRMS–MS information have been developed and put into practice recently. Suspect screening has been performed using predicted transformation products (TPs) [18–20], registered pesticides and their TPs in Switzerland [21], surfactants in wastewater [3] and fracking fluids [22], site-specific chemicals [5], and high-consumption pesticides and pharmaceuticals in sediments [23]. Other lists, including the potentially persistent and bioaccumulative organics in commerce [24], pharmaceuticals [25], and impurities, by-products, and transformation products [26] from Howard and Muir, can also be used in suspect screening. For non-target identification, candidate discrimination criteria used include retention time information, also represented as partitioning coefficients [19, 23] or the chromatographic hydrophobicity index (CHI) [5, 27], in silico fragmentation prediction with bond dissociation methods, for example MetFrag [28], or rule-based approaches including Mass Frontier [29], and also the number of references available for a substance in such databases as ChemSpider or SciFinder as a measure of relative importance in the environment [30, 31]. It is clear from the detailed data processing schemes in recent non-target papers (e.g. Figs. 1 and 3 in [5], Fig. 1 in [21]) that non-target screening of environmental samples is becoming increasingly complex.
In response to this trend of increasing complexity and the need articulated by members to compare and harmonise non-target screening methods in Europe, the NORMAN Association (www.norman-network.net) instigated a collaborative non-target screening trial in 2013 on a sample extract from the River Danube. Each participating institute was requested to analyse the test sample using established MS techniques in their laboratory and declare (1) how many substances were present in the sample and (2) how many of these could be provisionally identified using target, suspect, and non-target screening approaches. Analytical standards for the calculation of retention index information were also provided. After the trial, a workshop was held with all participants to develop agreements on harmonised terminology, workflows, and reporting formats. This dataset forms a unique opportunity to review the state-of-the-art of non-target acquisition and identification techniques using high resolution mass spectrometry with a comparable dataset from several environmental institutes in Europe and to comment on current and future trends.
Materials and methods
Sampling and trial participation
The sample used in the collaborative trial was collected from location JDS57, downstream of Ruse/Giurgiu (RO/BG; rkm 488; coordinates N43.890150, E26.017067) on September 18, 2013 as part of the Third Joint Danube Survey, organised by the International Commission for the Protection of the Danube River (ICPDR) [33, 34]. The sample preparation included a large-volume solid-phase extraction (LVSPE) of 1000 L water [35]. Briefly, the sampler cartridge was filled with 160 g Macherey Nagel Chromabond® HR-X (neutral resin) and 100 g each of Chromabond® HR-XAW (anionic) and HR-XCW (cationic exchange resin). The resins were extracted with 500 mL each of ethyl acetate and methanol (HR-X), and 500 mL methanol with 2 % 7 mol L−1 ammonia in methanol (HR-XAW) or 500 mL methanol with 1 % formic acid (HR-XCW). The extracts were then combined, neutralised, filtered (Whatman GF/F), and reduced to a final volume of 1 L using rotary evaporation. Aliquots of 1.5 mL, equivalent to 1.5 L river water, were transferred into vials and evaporated to dryness under nitrogen. These were sent to each participant with a laboratory blank, which was created via circular pumping of 5 L LC–MS-grade water through the LVSPE to mimic leaching from 1000 L water passing through the LVSPE. Sample stability (at least three time points over 1.5 months) and homogeneity testing (three replicates) was performed using 20 and 50 substances for GC–MS and LC–MS, respectively, to confirm the suitability of the sample for the trial. The substances are listed with the results in the Electronic Supplementary Material (ESM), Tables S1 and S2.
The samples were dispatched on December 9, 2013 with standard mixtures for use in calculating retention-index information: C10 to C25 alkane standards for GC–MS techniques and 10 substances for LC–MS techniques. The LC–MS standard mixture (the ChemSpider [32] identity number and the logarithm of the octanol–water partitioning coefficient (log P) calculated with ChemAxon [36] are given in brackets) consisted of metformin (3949; −1.36), chloridazon (14790; 1.11), carbetamide (133997; 1.65), monuron (8470; 1.93), metobromuron (17276; 2.24), chlorbromuron (24141; 2.85), metconazole (77764; 3.59), diazinon (2909; 4.19), quinoxyfen (2635909; 4.98), and fenofibrate (3222; 5.28). All participants were requested to measure these standards with the sample and report the results by March 15, 2014. As such, this dataset formed a test set for retention information comparison, data storage, and re-processing of raw mass spectral data for retrospective analysis. Of the 26 institutes from 15 countries who received samples, 19 submitted results; seven institutes for GC–MS and 17 institutes for LC–MS (five institutes submitted for both). The participants ranged from institutes performing non-target methods for the first time through to experienced research groups. The overall time committed by the participants varied from two days to six weeks. Each institute that submitted results is represented by a number.
LC–HRMS
An overview of the LC–HRMS(–MS) methods used is given in Tables 1 and 2.
One of the 17 LC participants used a serial coupling of zwitterionic hydrophilic interaction (HILIC) and reversed-phase (RP) chromatography (LCLC; HILIC-C18) [37], and the remainder used C18 reversed-phase columns (one C8) with either high-performance liquid chromatography (HPLC) or long ultra-HPLC (UHPLC) runs. The solvent was usually water–methanol or water–acetonitrile, with no or varied modifiers (formic acid or ammonium acetate, formate or hydroxide). Between 2 and 100 μL of the sample was injected. Electrospray ionisation (ESI) only was used, with different collision-induced dissociation (CID) or higher-energy CID (HCD) energies, and participants who measured in both positive and negative mode did this in separate runs. All-ion approaches (fragmentation without precursor selection) were used by some participants. The scan ranges started between m/z 50 and 100, ending between m/z 1000 and 2000, and the resolution ranged from ~18,000 (at m/z 300) to 140,000 (at m/z 200). Five instruments from the Orbitrap family were used, and 12 were from the TOF family (different vendors). The data processing for the target compounds was generally performed using vendor software, while the peaks that were not assigned as targets were identified through suspect and non-target screening, performed with a much wider variety of software and methods. While many participants also quantified target substances, this is not the focus of this article.
Many different information sources were used by participants (shown in Table 3) including open and vendor libraries with mass spectra available, extensive compound databases, selective compound databases containing water-body relevant substances (STOFF-IDENT and DAIOS), as well as suspect lists from literature. These were generally supported or rejected via a combination of accurate mass (including isotope and/or adduct information), RT, MS–MS, library match, and predicted properties. Approaches to consider retention time information included the log P method using standards provided to each participant (Grosse et al., in prep.), often in combination with STOFF-IDENT [41], the CHI approach [27], and a QSPR approach (Aalizadeh et al., in prep) using support vector machines (SVM) and k-nearest neighbours (kNN). Software used for fragmentation prediction included MetFrag [28], MetFusion [55], Mass Frontier [29], MassFragment in UNIFI and MassLynx (Waters [56]), SmartFormula 3D (Bruker Daltronics [57]), and Molecular Structure correlator) [58, 59].
GC–MS
A summary of the GC–MS methods is given in the ESM, Tables S3 and S4. One participant submitted high resolution data, and three of seven GC participants used GC×GC. Two used chemical ionisation (CI) methods in addition to EI: APCI and positive and negative CI. Four participants chose a large volume injection to obtain sufficient signal intensity (see results) and the scan range started in the range m/z 35–50, ending in the range m/z 350–800. A range of different solvents were used. Target compound identification was usually performed using vendor software. Non-target identification was performed using the NIST database (Table 3) either with or without AMDIS [60], combined in most cases with the Kovats retention index.
Data reporting and comparison
Participants were requested to submit their results in a data collection template (DCT), a multi-tab spreadsheet, to ensure sufficient information was available for evaluation. Pre-treatment steps to increase comparability included reclassification of methods and reported identifications to be consistent with the definition of target, suspect, and non-target provided in the introduction. Several substance identifiers were used to perform the comparison, including the SMILES code [61], Chemical Abstract Services (CAS) number, names, molecular formulas, and the InChI Key [62]. OpenBabel [63] was used to perform conversions, and ChemSpider [32] and PubChem [40] were used to fill data gaps. Entries for the same target repeated multiple times per participant were merged to create one entry only, with multiple identifications for the same substance and peak as a target, suspect, and/or non-target selected according to the hierarchy target>suspect>non-target. However, multiple identities for the same peak (e.g. co-eluting targets) were not merged.
Results and discussion
Stability testing
Different batches of the sample used in the trial were subjected to stability and homogeneity testing to determine whether the sample was suitable for such a trial and provide a baseline for comparison; tables of results are provided in the ESM, Table S1 (GC) and S2 (LC). For GC–MS, 20 substances were selected to cover the full run time and polarity range. Homogeneity testing was performed on Lots 15, 33, and 50 (three replicates of each) with the coefficient of variation usually below 1 % and with a maximum of 7 %. For the stability (tested on Days 1, 10, and 30), the highest relative concentration was 1.033 on Day 30. For LC–MS, 50 substances were selected and while the homogeneity (three aliquots) had coefficients of variation in the range 1–16 %, very similar values were obtained for reproducibility testing (three injections). Stability was tested on Days 1, 7, 15, and 48 and revealed no degradation of any compound selected. Generally an increasing trend to higher factors (Day X/Day 1) was observed. The rather high variation of the stability factors (in the range 0.5–2) was caused predominantly by measurement sensitivity and was regarded as sufficient for the trial because the main focus here is not a comparison of quantitative results.
LC–HRMS overview
In total, 17 institutes submitted LC–HRMS results and 15 of these reported target substances (for which the identity was confirmed with a reference standard). The breakdown of targets, suspects, and non-targets is given in Table 4, including additional categories (whether the target was quantified or not, isomer mix, identified non-target, and formula only) to reflect the identification confidence and how the substances were identified. Participants 6 and 13, with large numbers of assigned formulas, were able to do this automatically through their software. The distribution reflects the target and suspect lists and the databases available at the different institutes, and reveals the huge amount of information that can be gained by increasing the exchange of knowledge between institutes. The time invested by the participants (two days to six weeks of the total three months given for the trial) is also reflected in these results.
In total 625 target results were reported, corresponding to 347 unique compounds, and the majority (451 results) were quantified. A total of 631 suspect and tentatively identified non-target substances were reported, 553 of which had a defined structure associated with the peak (the remaining substances were reported as a substance class or unspecified isomer). Altogether 649 unique compounds were identified as target, suspect or non-target. The most frequently reported included (targets + suspects) carbamazepine (13 + 2), atrazine (9 + 4), sulfamethoxazole (11 + 1), DEET (6 + 6), metformin (10 + 2), terbutylazine (6 + 6), caffeine (7 + 5), atrazine-desethyl (8 + 3), and tramadol (7 + 3). Metoprolol, terbutylazine-desethyl, phenazone (also known as antipyrine), 4 and 5-methylbenzotriazole, atrazine-2-hydroxy, venlafaxine, isoproturon, telmisartan, perfluorooctane sulfonate (PFOS), and metolachlor were all reported nine times. One institute additionally reported an isobaric substance (propisochlor) as a suspect instead of metolachlor. A further seven and four substances were reported eight and seven times, respectively. These results indicate that the “well known compounds” are obviously more common target compounds and found more often, but the fact that many parent and TPs occur in this list indicates that many groups consider the formation of TPs already in their target screening. Terbutylazine was the most frequently reported of the isobaric compounds; see Fig. 2 and text below for more discussion on co-eluting isobars. Of the 24 substances specified as suspects for the trial (detected at this location in previous surveys [64] and given in the ESM, Table S5), all but three were detected at least once by the LC participants; one of these missing three was reported by a GC participant (bisphenol A), and the remaining two were not reported by any participant (naproxen and N,N-dimethyl-1-decanamide).

Isobaric parents and transformation products in the collaborative trial sample, the number of times reported, and the number of references in ChemSpider [32]
The participants were requested to report intensities in the sample and the blank using the DCT, and different approaches were used to consider the results of the blank sample. All but one participant reported intensities in the sample, and all but four reported intensities in the blank. Dibutylphthalate and/or isomers were reported eleven times by seven participants; three participants reported this in both the sample and blank, two in the sample only, one in the blank only, and one did not report either intensity. Of the other frequently reported substances, metformin and terbutylazine were reported in the blank once (several orders of magnitude lower than in the sample), carbamazepine, desethylatrazine, and atrazine twice (two or more orders of magnitude lower), DEET three times (one to two orders of magnitude lower), and caffeine four times (a factor of 2–600 lower). Most of the reported alkyl phosphate species (e.g. tributyl phosphate and triphenyl phosphate, see ESM Table S5) were found in both sample and blank, as were the polyethylene glycol suspects. The participants who applied a blank subtraction used intensity cut-offs at different ratios to determine whether the substance was regarded as present in the blank or not, and this affected the non-target peak lists provided. As an example, one participant subtracted all non-target peaks where the same peak was detected in the blank and the intensity was within two orders of magnitude of the intensity measured in the blank. Because only one each of blank and sample was provided, it was challenging to determine whether the intensities were sample-specific, especially given that sample enrichment was used. As such, both false positive and false negative results are possible.
Target and suspect screening was performed using both peak lists and XICs. Generally, the use of XICs gave more hits because this also captured target and/or suspect substances where the peak was not suitable for the peak picking algorithm used. However, the peak picker and the parameters used have a large effect on the quality of peak picking results and this would be interesting to investigate more closely in the future. In this trial, the strategy and results depended strongly on the software used by the participant, because most used vendor software to perform target and suspect screening. Many participants mentioned using manual or visual confirmation of the target and/or suspect results.
Analytical methods in LC–HRMS non-target screening
The results summarised in Table 4 reveal that non-target acquisition and screening has developed at an incredibly rapid pace in recent years, with many institutes now regularly screening several hundred compounds using multi-residue methods. The participants agreed that generic methods, based on C18 columns, generic gradients, and either HPLC or long-run UHPLC, were best for non-target approaches, to allow sufficient time for fragment acquisition. Many institutes also analysed the sample several times to gain additional information, running e.g. positive and negative separately rather than performing polarity switching, acquiring MS and all-ion MS–MS data to gain all fragments at once, running with and without internal standards, or acquiring data-dependent MS–MS of target compounds first, followed by re-measurements to acquire MS–MS of suspects and non-targets of interest. This reveals that the number of compounds being investigated is almost on the edge of instrument capabilities, and there is a great interest in the development of appropriate data analysis strategies for all-ion MS–MS data.
One participant used HILIC combined with RP-LC to improve the separation of the highly polar substances, and the peak distribution shown in Fig. S1a in the ESM reveals the potential for greater separation of the polar substances that otherwise may elute in the dead volume of RP columns, shown in Fig. S1b in the ESM. The identified substances reported by this participant from the HILIC column (metformin at 16.85 min, melamine at 7.48 min) were also detected by other participants with the C18 column (12 and four times in total, respectively) in the range 0.3–2.9 min. This reveals that HILIC methods may help address the limitations of non-target screening of very polar compounds with RP columns alone, e.g. interferences observed with non-targets in the low RT range [3] and in regions of high matrix interference [23], but this has to be investigated further.
Although there was a great variety in the scan range used by the participants, all but one identified substance fell in the range m/z 100–900. The target compound piperazine ([M+H]+ m/z 87.0921) was reported by one participant. The lowest m/z suspects were five single-hit substances in the range m/z 100–114, whereas the first multiply-reported substance (benzotriazole) was detected at m/z [M+H]+ 120.0556. The highest m/z target reported was iohexol (m/z 821.8879), and one suspect was reported above this, namely tilmicosin (m/z 869.5690). The conclusion to harmonise the non-target methods was to suggest a minimum scan range of m/z 100–1000, but to measure with the largest scan range possible without negatively affecting the overall analytical performance, and the results reveal that this would have captured the most relevant substances in this sample.
Several other interesting aspects could be investigated with this comprehensive dataset. Although many options are now available for ionisation, many participants chose to report substances exclusively in positive ESI mode (excluding negative mode), and no LC participant used alternative ionisation methods, for example APCI or APPI. A desire for better functionality to compare and merge peak lists measured using complementary ionisation techniques was expressed to increase the ease of use of multiple ionisation techniques for non-target screening, because this is not yet time-efficient for routine analysis. Similar functionality is also needed to merge results from complementary chromatographic approaches (e.g. HILIC-RP). The delicate balance between sample volume injected and chromatographic performance was also visible. Although the participant with the highest sample injection volume (200 μL) also reported the most target compounds, neither melamine nor metformin were included, indicating that the high amount of solvent may have hindered the detection of these highly polar substances for chromatographic reasons. From the available data, it is clear that comprehensive non-target screening approaches can be applied with great success using either Orbitrap or TOF-based instruments. Instrument-specific aspects, including which compounds can only be measured successfully with the higher resolution of the Orbitraps, which ionise better or worse in specific instruments, which methods perform best for low intensity substances, which solvents and modifiers are better, and the effect of detection limits on detection etc., would require a custom-made spiked environmental sample to create known case studies. The parallel submission of a spiked sample was encouraged by all participants for future trials, to investigate some of these phenomena more closely. However, the nature of the dataset available enables a detailed analysis of several specific topics in non-target screening, which are illustrated with several examples in the following text.
Example: isobaric substances
Isobaric substances (where isobaric is used to refer to substances with the same exact mass) form some of the most challenging cases in high-throughput screening. With high-accuracy data, isobaric substances often have the same molecular formula (but not always, because some can have the same exact mass within the instrumental accuracy [65]), and in environmental screening these can often be very similar substances structurally. The case of terbutylazine, sebutylazine, simazine, propazine, and their TPs, shown in Fig. 2, is a good example. Fragmentation information can be used to distinguish propazine (2 × –C3H7) from terbutylazine and sebutylazine (both –C2H5, –C4H9) in the top row of Fig. 2, or simazine (2 × –C2H5) from the desethyl TPs (both –C4H9) in the second row. As a result of the structural similarity, these substances also often co-elute, especially in generic chromatographic methods including those used in non-target methods. Two participants reported both simazine and desethylterbutylazine for the same m/z and RT, but with diagnostic fragments indicating the presence of both. In this sample, neither fragmentation nor retention information could separate terbutylazine and TPs from sebutylazine and TPs with the generic chromatography used, although clearly more institutes reported the terbutylazine-related substances. This is logical in terms of substance use: terbutylazine is widely used, whereas neither sebutylazine nor propazine are currently registered in the EU. However, propazine is present as a by-product in other triazines and could be observed when other triazine compounds are present if concentrations were sufficient. It is also interesting to reflect on the prioritisation that the number of references would give here: most institutes reported terbutylazine (10) and its TP terbutylazine-desethyl (9) instead of the isobaric simazine (4); the former were also provided as suspects (ESM Table S5); terbutylazine-desethyl was also reported by one GC participant. Without knowing terbutylazine to be present, sorting candidates purely by the number of references would clearly favour simazine (518 references versus 92 in ChemSpider, see Fig. 2). Thus care must also be taken with this strategy, and additional knowledge (especially the presence of related substances, fragmentation information, and retention time) should also be considered. This reinforces the point made by many participants that a better integration of identification strategies is necessary for non-target screening approaches and that the incorporation of use data, rather than the number of references alone, is desirable in the future.
Example: co-eluting substances
The case of 4 and 5-methylbenzotriazole summarises many challenges with the data evaluation. All participants reported these target substances together, agreeing that they required special methods to be separated (e.g. Ref. [66]) and could not be separated chromatographically with a generic (multi-residue) screening method. Because the data could not be compared by name (entries included 4/5-Methyl-Benzotriazole, 4&5-Methyl-Benzotriazole, 4- and 5-methylbenzotriazole through to Tolyltriazole), or CAS numbers, SMILES or InChI Keys (given for either of the isomers, e.g. CAS 136-85-6 and 29385-43-1), in the end the comparison was performed by sorting by molecular formula C7H7N3 because no other substances were reported with this formula. The limitation in representing structural uncertainties in InChI Keys is currently the subject of a new working group within IUPAC to improve data comparison of such cases. Other cases included the example of simazine, terbutylazine, and related compounds shown in Fig. 2 and the (often) co-eluting substances tramadol and O-desmethylvenlafaxine. Tramadol was reported much more frequently; two participants reported both substances for the same peak, and at least one other participant ruled out the presence of O-desmethylvenlafaxine manually as the diagnostic fragments m/z 133.0649 and 107.0492 were absent in the measured spectrum. Another interesting case is dibutylphthalate, for which seven participants reported at least one of three isomers (di-n-butyl phthalate, di-isobutylphthalate, p-dibutylterephthalate) for 1–3 peaks per participant, using different strategies (reporting one isomer multiple times versus trying to “guess” which isomer was which, through to reporting with a generic name). Again, this reveals a great need for reporting structural uncertainties in a way compatible with high-throughput screening. This also reveals the need for a retention time index in LC–MS–MS; the question of which phthalate isomer was which was solved elegantly by the GC–MS participants with a combination of chemical ionisation and retention index information (see below).
Dependence on target and suspect lists
The results of the target and suspect screening in the collaborative trial revealed a clear dependence on the target and suspect lists used, which were in turn also highly dependent on the main focus of each laboratory. This is revealed by the large range of target compounds reported: 354 of 622 targets were reported once only, and the maximum reported by one institute was 167, which is a very large number of compounds considering the low concentrations observed in the sample. Several participants reported multiple suspect identities for one peak, such that the number of suspect substances and non-target peaks in Table 4 may be an overestimation in some cases, especially where suspect matching was performed on the exact mass and isotope pattern only. Many participants took the suspect screening the necessary steps further, performing selection or rejection of the matching suspects using retention time and fragmentation information (measured versus library or versus predicted fragments where no library spectrum was available) before reporting the (tentative) identification. The danger of screening based on mass only was revealed by a few cases where the suspect list contained the exact mass of the compound represented as a salt, although this can certainly not be detected as such in the instrument. For example, tris[4-(diethylamino) phenyl]methylium acetate, C31H42N3.C2H3O2, CCN(CC)c1ccc(cc1)[C+](c2ccc(cc2)N(CC)CC)c3ccc(cc3)N (CC)CC.CC(=O)[O–] was reported at m/z 516.3565 by one participant, whereas the monoisotopic mass of the substance without the acetate salt is 456.3379 Da. Another example is a substance with the formula C7H8N4O2, which was reported once by three participants, but with a different identity each time (theophylline, 1,7-dimethylaxanthine, or theobromine). Whereas the foremost was reported as a target confirmed by reference standard, the other two compounds were detected additionally by one GC participant with distinct EI MS spectra and good NIST library match values: similarity 700, probability 87 % for 1,7-dimethylxanthine, and similarity 823, probability 80 % for theobromine, although only the latter was reported officially because a cut-off of 800 was used. This again reveals the need for spectra and other additional information to support suspect screening. The concept of performing smarter suspect screening (“screen smart”, carefully selecting the compounds to be screened to suit the investigation) was discussed as often being more efficient than “screen big” with large, unselective suspect lists. Although the latter has the advantage of large coverage, this is at the expense of a high rate of false positives and thus often a larger burden of proof. Participants also agreed that it would be useful in the future to exchange target and suspect compound lists between institutes using an open platform, for example STOFF-IDENT [41]; the NORMAN Association is investigating this as a planned activity for 2015.
Molecular formula assignment, adduct detection, and homologous series
Moving on from target and suspect screening, an important aspect of non-target identification is the detection of the adduct state and assignment of molecular formulas to components of interest, and this is illustrated using homologous series identified by participants in the trial sample. Whereas the GC participants could use library searching to identify these common substances, the LC participants performed suspect screening using (i) lists of surfactants published previously [3], (ii) tentative identification through recognition of diagnostic fragments (e.g. 89.0593 and 133.0857 corresponding to C4H8O2 and C6H12O3, respectively), through to (iii) retrospective screening for surfactants after the discovery of diagnostic fragments of surfactants during non-target identification efforts. The polyethylene glycol (PEG) homologues provide an interesting example of adduct detection and molecular formula assignment. These were detected as the [M + H]+, [M + NH4]+, and/or [M + Na]+ species by several participants; see ESM Fig. S2 for the distribution of the [M + H]+ and [M + NH4]+ species measured by one participant and Figs. S3 and S4 in the ESM for the adducts and isotopes detected for some of these species. Where at least two adduct species are present, the neutral mass of the compound can be determined unequivocally; if only one species is present the adduct state must be assumed when calculating the molecular formula. Table 5 shows the calculation of the molecular formulas for three PEG species detected as different adducts by one participant.
This example highlights the importance of considering adducts and applying a comprehensive peak grouping before non-target identification, which is a feature now offered by many open source and vendor software packages to different degrees of comprehensiveness. Once the correct neutral mass was identified, molecular formulas were calculated with the elements C, H, N, O, and S. From five to seven formulas with a high score (>90) were proposed; the top four are shown in Table 5. The score considers the mass, isotope match, and isotope spacing; however, the last three columns of Table 5 show that these four formulas could be reduced to only one or two possible formulas if fragmentation information was also considered in the selection of the molecular formula. Although several approaches now include fragment information in formula selection, including SIRIUS [67], MOLGEN-MS/MS [68], and mzMine [69], these were not used in the trial, whereas of the vendor software used by different participants, only SmartFormula3D from Bruker associated fragmentation information with the precursor ions for formula calculation.
Retention time information in LC–HRMS
An important objective of the trial was to assess the use of retention time information in the LC screening approaches. Most participants measured the substances provided (given in Methods) and used the results to calculate a retention time index (RTI) for the LC data. Seven participants reported these RTI for some or all of their reported values in the DCTs. The results of 26 target compounds that were detected at least four times were used to investigate the RTI further. The calculated RTI was found to have below 3 % root mean square deviation (RSD) for eleven of these targets, in the range 3–6 % RSD for another eleven targets, and above 6 % RSD for the remaining four of the 26 target compounds. The RTIs of target substances reported by two participants were used to calculate log D values that were in turn used to search the STOFF-IDENT database [41], where empirical matching criteria are included. Above 80 % (50 of 60, 81 of 96) of the target compounds that were in STOFF-IDENT were also within the calculated log D range using the RTI standards. Although the majority of substances matched, the non-match of 10 and 15 manually validated target substances (per participant, respectively) reveals that a consensus approach should be considered when using retention time prediction approaches [70].
A different approach was used by another participant (14) to reject suspects on the basis of retention time (and other) information. Their initial suspect screening yielded 1060 and 538 suspect hits in positive and negative mode, respectively, which was reduced to 81 and 75 suspect matches, respectively, using additional mass accuracy, isotope, and peak score criteria. Of these, a further 27 and 40 suspects were rejected using retention time models developed using a SVM kNN QSPR approach (Aalizadeh et al., in prep), and a further four and five were rejected because they were not ESI-amenable. Finally 78 suspects were reported, as given in Table 4 (48 positive, 30 negative).
Although all participants agreed that the retention information is useful and essential for identification efforts, many participants expressed a need to have this information better integrated into their current software. Additionally, the non-targets found by some participants were clearly compounds such as sulfonated surfactants and polyfluorinated compounds, which often perform poorly in retention time prediction and other QSPR calculations [27, 71]. A further example of how useful retention information can be to confirm the identification of suspect compounds is given below for GC–MS.
Non-target identification example
Several tentatively-identified non-target compounds were proposed by participants, despite the limited time (Table 4), and these compounds could be found retrospectively in the raw files from other participants. One participant reported a double peak at m/z [M−H]− 199.0431 at 19.48 min (small intensity) and 19.98 min (large intensity) of a 40 min run, tentatively identified as mesitylene sulfonic acid (2,4,6-trimethylbenzene sulfonic acid, ChemSpider ID 69438) or isomer, molecular formula C9H12SO3, with the fragments m/z 184.0196 and 135.0815. Another participant found a signal at m/z 199.0428 with a double peak at 5.91 and 6.15 min of a 25 min run (small and large intensity, respectively) with peaks at m/z 79.9566 (SO3 −), 80.9644 (SO4H−), 119.0500 (C9H11 −), 135.0811 (C9H12O−, or –SO2), 183.0115 (–CH4), and 184.0194 (–CH3) with HCD 90 (see Fig. 3). A total of 135 candidates with this formula are in ChemSpider, but using MetFrag to rank on the basis of the in silico fragmentation of the candidates, trimethyl benzene sulfonic acid isomers and a few ethyl isomers are all ranked equally, with a score of one and four explained peaks. Although the larger peak is also present in the blank and seems to come from the sorbents used in the large volume sampling, this is two orders of magnitude lower than the intensities observed in the samples for both participants. However, the standard for mesitylene sulfonic acid was available at the second institute and although the retention time matched well (5.96 min), the fragments did not match; only two fragments (m/z 79.9573 and 135.0824) were seen in the standard, at varying collision energies including HCD 90. Thus the tentative identification cannot be confirmed. However, the peak seen by the second participant at m/z 183.0115 is diagnostic for linear alkylbenzene sulfonic acid (LAS) species [3] and the structure below may be able to explain the spectra. Although not yet confirmed, this example reveals how measurements of the same sample by two different laboratories can be used to help in the identification.

Spectrum of the non-target found at 6.15 min by one participant, a tentatively identified candidate (4-isopropyl-benzenesulfonic acid, insert), and the resulting fragments. The original tentative identification of mesitylene sulfonic acid can be rejected on the basis of mismatching fragments: see ESM Figs. S5 and S6
GC–MS
For the GC–MS results, four institutes reported target compounds (confirmed with reference standards), with 64 results reported for 55 unique compounds. The target substances detected multiple times were all polycyclic aromatic hydrocarbons (PAHs): phenanthrene (four), anthracene and naphthalene (three each), and fluorene, fluoranthene, and pyrene (twice each). The remaining identifications were performed using library information, usually the NIST MS library [11] versions 02 (175,214 spectra of 147,350 compounds), 08 (220,460 EI MS spectra of 192,108 compounds), and 11 (243,893 EI MS spectra of 212,961 compounds), although one participant also used the Wiley library, 7th Edition [12]. Match factors in the range 700–800 (of a maximum of 999) were used as the lower limit for library matches (some also in combination with the probability estimates) and most participants also confirmed these matches manually. An additional 295 compounds were reported from library matches (identified but not confirmed with reference standards), such that in all, 255 unique compounds were reported with a single identity as a target substance or library match. A further 47 peaks were reported as library matches but with the isomer unidentified, and 35 reported substances remained unknown (many participants did not report unknown substances). Merging the target and library match compound lists revealed that phenanthrene was detected by six of seven participants, and dibutylphthalate, diethylphthalate, tris(1-chloro-2-propanol)phosphate (TCPP), and caffeine (five times each) and 4-oxoisophorone, benzothiazole, cyclohexylisocyanate and triphenylphosphine oxide, and 1,1'-(1,4'phenylene)bis-ethanone (four each) were also detected frequently. Several substances overlapped with the LC–MS targets and suspects, notably the several phthalate and alkyl phosphate species, caffeine, and carbamazepine; in total 46 substances were reported as identified in both LC and GC, including the suspects and library matches, of the 858 unique compounds reported in total (~5.4 %).
The sample preparation was not ideal for GC analysis and the participants also used different solvents. Therefore, the relatively few substances detected and limited comparability between GC participants is not completely unreasonable. A separate sample for GC–MS analysis is recommended for future trials. Although some participants also quantified their target substances, this data is not presented here because the dataset was too small for a meaningful evaluation.
High resolution GC–MS
One participant used high resolution GC–APCI QTOFMS during the trial, combining the results with EI MS in an interesting approach. As mentioned above, the workflow to identify substances with the GC–APCI QTOFMS approach effectively followed the target, suspect, non-target approaches used for LC–HRMS screening, rather than the standard target screening plus NIST database search used generally in EI MS. Further participants used GC×GC, while another participant used the combination of chemical ionisation and EI MS to identify the molecular mass corresponding with the EI MS spectrum, shown in Fig. 4. The base peak m/z 149 (indicative of phthalates) was observed three times but was only associated with the m/z [M + H]+ 279 twice, supporting the proposed NIST matches of dibutylphthalate isomers. However, consideration of the experimental KRI with the literature values in NIST prompted an alteration of the tentatively-proposed identities in Fig. 4 (shown by the dashed boxes and blue arrows), because diisobutylphthalate (shown left) had a lower retention index than dibutylphthalate (shown right). Final confirmation using a reference standard, however, is still necessary. The HRMS–MS spectrum obtained via APCI QTOFMS–MS is shown in Fig. 4b.

(a) Use of complementary ionisation techniques (EI, PCI), library match, and retention information (experimental KRI versus measured values in NIST) by one participant to identify two dibutylphthalate isomers and reverse the order of proposed database matches on the basis of retention information. (b) The corresponding APCI QTOFMS–MS spectrum of the suspect dibutylphthalate reported by another participant, which could be either isomer (both structures: see insert)
As for the LC participants, the GC participants mentioned that the data processing is a limiting factor for using GC×GC and/or soft ionisation in combination with EI MS and the need for exact mass spectra, especially MS–MS spectra, was expressed. This reinforces the observation that the workflow used for identification is determined by the ionisation technique used (EI versus softer ionisation methods) and the size of the spectral library available rather than the chromatography.
Discussion and perspectives
Data processing aspects, HRMS–MS
The participation of many institutes in analysing the one sample reveals that non-target screening methods have come a long way in a short time and that a comprehensive target screening complemented with suspect screening forms an essential prerequisite to non-target identification. Most substances reported were identified through target or suspect screening of data acquired with non-targeted acquisition. Tentatively identified non-target compounds were reported by a few participants, even though non-target identification is still a very time-intense process. Many participants included MS–MS and retention time information where possible to support the proposed identity of suspect compounds, addressing the disadvantages of performing suspect screening on the basis of exact mass only. Suspect screening proved a useful step to propose tentative identifications for several compounds and ease the burden of identification. The large overlap between target and suspect compounds proves that this was successful in many cases. The selection of the peaks of interest for non-target identification remains very subjective; it is clear that not all of the up to several thousand unknown peaks can be identified. One option is the prioritisation of masses occurring in several samples (e.g. Refs. [3, 9], and although only one sample was available for the trial, several participants only considered peaks when they occurred in at least two repeated analytical runs, to reduce the effect of noise or other interferences on the results. In terms of non-target identification, whereas some participants determined the molecular formula first, followed by elucidation on the basis of the one or several possible molecular formulas, other participants went straight to identification with an exact mass. The latter strategy, although resulting in more candidates, has been revealed recently to be more effective from a data processing perspective [72] (it is for example easier to select candidates with one or some formula(s) from an exact mass search than to merge the results of several separate calculations).
A new trend revealed during the trial is the move towards finding related substances, with several strategies used. These included searching for common fragments or neutral losses, suspect screening for homologues, and even homologue series detection. Other strategies that have been used elsewhere included selection by mass defect to avoid matrix interferences in sediments [23], and although the Kendrick mass defect is often used for natural organic matter or petroleum analysis (e.g. Refs. [73, 74]), this was not mentioned by any of the participants and it remains to be seen whether this may have an application to environmental samples. As discussed above, challenges still remain with regard to the reporting of substances that cannot be fully characterised structurally using the information available (e.g. long chain surfactants, lipid species with undefined branching and/or double bond locations) and also the level of confirmation required before being able to report these substances in investigations. As discussed in the Introduction, a combination of a revised version of the EU IP system (to, e.g., better reflect the availability of instruments with much higher resolution than the 10,000 in the original guideline [6]) with the level system shown in Fig. 1 could be a way to move forward. A further improvement would be better processing of data-independent acquisition data, such that fragments are available for all substances, not just selected precursors. Another interesting approach could be to speed up non-target identification using alternative databases, for example those including natural products, to assist in finding background natural compounds that are not necessarily of interest for non-target identification of potentially hazardous anthropogenic substances.
The number of targets and suspects detected only once indicates that the results are highly dependent on the institute knowledge and focus area and on the target and suspect lists used. This reinforces the need for additional evidence before reporting suspect identification and for an improved representation of identification confidence, for example updated IPs. It is clear from the number of different software packages and approaches used that the data processing techniques are very different, with vendor versus open source approaches and many different sources of information used by the participants. The need to harmonise information sources was expressed and, as a result of the recognition that one participant’s target is another participant’s suspect or unknown, an agreement was made to exchange target and suspect lists in a centralised platform, where the development of search criteria and prioritisation methods for these lists will be essential to enable application of the “screen smart” approach. Additionally, the results of this study have been provided as supporting information for use in future investigations. Enhancing this with the upload of mass spectra of target compounds to an open access database, for example the NORMAN MassBank [43], would help improve the success of target, suspect, and even non-target screening immensely. Another need clearly expressed by the participants was for better data processing of complementary ionisation modes and methods (e.g. positive or negative, APCI, and APPI) and for data-independent acquisition techniques, such that fragmentation can be obtained for all substances rather than selected precursor masses, and a better integration of retention information.
Lessons learned from the trial
The first collaborative non-target screening trial run by the NORMAN Association was a very ambitious endeavour coinciding with the Third Joint Danube Survey in response to the need expressed by members in late 2012. The response from participants was very positive and the objective of discussing ways to harmonise non-target screening of environmental samples within Europe was fulfilled with a series of three well-attended workshops [75–77]. The analytical methods are already reasonably well harmonised, with the main differences resulting from instrument-specific settings that participants cannot reasonably be expected to change. Although the data processing workflows were not as harmonised, with many participants expressing the need for better integration of information, workflows, and software, a great deal can be learnt by reflecting on the results of the other participants. Participants were given a lot of freedom for this collaborative trial, and there was also interest in a more tightly-defined interlaboratory comparison in the future, with an additional spiked sample and a more comprehensive suspect list from greater comparability.
All participants provided the raw data of their measurements for this trial, such that a retrospective analysis of the results will be possible, and finding a suitable environmental repository for this data will be the subject of future efforts. Many participants were keen to investigate their results further, using the information gained during the trial, to perform retrospective identification. The trend for retrospective analysis, used already during the evaluation of these results, is increasing and the importance of this in the future should not be underestimated.
References
Krauss M, Singer H, Hollender J (2010) LC-high resolution MS in environmental analysis: from target screening to the identification of unknowns. Anal Bioanal Chem 397(3):943–951
Hernandez F, Pozo OJ, Sancho JV, Lopez FJ, Marin JM, Ibanez M (2005) Strategies for quantification and confirmation of multi-class polar pesticides and transformation products in water by LC-MS2 using triple quadrupole and hybrid quadrupole time-of-flight analyzers. TraC Trends Anal Chem 24(7):596–612
Schymanski EL, Singer HP, Longree P, Loos M, Ruff M, Stravs MA, Ripolles Vidal C, Hollender J (2014) Strategies to Characterize Polar Organic Contamination in Wastewater: Exploring the Capability of High Resolution Mass Spectrometry. Environ Sci Technol 48(3):1811–1819
Zedda M, Zwiener C (2012) Is nontarget screening of emerging contaminants by LC-HRMS successful? A plea for compound libraries and computer tools. Anal Bioanal Chem 403(9):2493–2502
Hug C, Ulrich N, Schulze T, Brack W, Krauss M (2014) Identification of novel micropollutants in wastewater by a combination of suspect and nontarget screening. Environ Pollut 184:25–32
European Commission (2002) Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Official Journal of the European Communities L221:29 http://old.eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2002:221:0008:0036:EN:PDF. Accessed 30 Jan 2015
Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, Hollender J (2014) Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ Sci Technol 48(4):2097–2098
Creek DJ, Dunn WB, Fiehn O, Griffin JL, Hall RD, Lei ZT, Mistrik R, Neumann S, Schymanski EL, Sumner LW, Trengove R, Wolfender JL (2014) Metabolite identification: are you sure? And how do your peers gauge your confidence? Metabolomics 10(3):350–353
Letzel T, Lucke T, Schulz W, Sengl M, Letzel M (2014) OMI (Organic Molecule Identification) in water using LC-MS(/MS): Steps from “unknown” to “identified”: a contribution to the discussion. Lab More Int 4:24–28
Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR (2007) Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3(3):211–221
NIST/EPA/NIH (2011) NIST Mass Spectral Library, various editions. National Institute of Standards and Technology. US Secretary of Commerce, USA
McLafferty FW (2000) Wiley Registry of Mass Spectral Data, 7th edn, ISBN-10: 0471440981
Stein S (2012) Mass Spectral Reference Libraries: An Ever-Expanding Resource for Chemical Identification. Anal Chem 84(17):7274–7282. doi:10.1021/ac301205z
Kind T, Fiehn O (2010) Advances in structure elucidation of small molecules using mass spectrometry. Bioanal Rev 2(1–4):23–60
Scheubert K, Hufsky F, Bocker S (2013) Computational mass spectrometry for small molecules. J Cheminform 5(12). doi:10.1186/1758-2946-5-12
Portoles T, Mol JGJ, Sancho JV, Hernandez F (2014) Use of electron ionization and atmospheric pressure chemical ionization in gas chromatography coupled to time-of-flight mass spectrometry for screening and identification of organic pollutants in waters. J Chromatogr A 1339:145–153
Hernandez F, Ibanez M, Portoles T, Cervera MI, Sancho JV, Lopez FJ (2015) Advancing towards universal screening for organic pollutants in waters. J Hazard Mater 282:86–95
Helbling DE, Hollender J, Kohler HPE, Singer H, Fenner K (2010) High-Throughput Identification of Microbial Transformation Products of Organic Micropollutants. Environ Sci Technol 44(17):6621–6627
Kern S, Fenner K, Singer HP, Schwarzenbach RP, Hollender J (2009) Identification of Transformation Products of Organic Contaminants in Natural Waters by Computer-Aided Prediction and High-Resolution Mass Spectrometry. Environ Sci Technol 43(18):7039–7046
Huntscha S, Hofstetter TB, Schymanski EL, Spahr S, Hollender J (2014) Biotransformation of benzotriazoles: insights from transformation product identification and compound-specific isotope analysis. Environ Sci Technol 48(8):4435–4443
Moschet C, Piazzoli A, Singer H, Hollender J (2013) Alleviating the Reference Standard Dilemma Using a Systematic Exact Mass Suspect Screening Approach with Liquid Chromatography-High Resolution Mass Spectrometry. Anal Chem 85(21):10312–10320
Thurman EM, Ferrer I, Blotevogel J, Borch T (2014) Analysis of Hydraulic Fracturing Flowback and Produced Waters Using Accurate Mass: Identification of Ethoxylated Surfactants. Anal Chem 86(19):9653–9661
Chiaia-Hernandez AC, Schymanski EL, Kumar P, Singer HP, Hollender J (2014) Suspect and nontarget screening approaches to identify organic contaminant records in lake sediments. Anal Bioanal Chem 406(28):7323–7335
Howard PH, Muir DCG (2010) Identifying New Persistent and Bioaccumulative Organics Among Chemicals in Commerce. Environ Sci Technol 44(7):2277–2285
Howard PH, Muir DCG (2011) Identifying New Persistent and Bioaccumulative Organics Among Chemicals in Commerce II: Pharmaceuticals. Environ Sci Technol 45(16):6938–6946
Howard PH, Muir DCG (2013) Identifying New Persistent and Bioaccumulative Organics Among Chemicals in Commerce. III: Byproducts, Impurities, and Transformation Products. Environ Sci Technol 47(10):5259–5266
Ulrich N, Schuurmann G, Brack W (2011) Linear Solvation Energy Relationships as classifiers in non-target analysis-A capillary liquid chromatography approach. J Chromatogr A 1218(45):8192–8196
Wolf S, Schmidt S, Muller-Hannemann M, Neumann S (2010) In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinf 11:148
HighChem (2014) Mass Frontier, 7th edn. HighChem Ltd./Thermo Scientific, Bratislava
Little JL, Cleven CD, Brown SD (2011) Identification of "Known Unknowns" Utilizing Accurate Mass Data and Chemical Abstracts Service Databases. J Am Soc Mass Spectrom 22(2):348–359
Little JL, Williams AJ, Pshenichnov A, Tkachenko V (2012) Identification of "Known Unknowns" Utilizing Accurate Mass Data and ChemSpider. J Am Soc Mass Spectrom 23(1):179–185
RSC (2014) ChemSpider www.chemspider.com. Royal Society of Chemistry. Accessed 17 Dec 2014
ICPDR (2015) Joint Danube Survey 3 www.danubesurvey.org. ICPDR - International Commission for the Protection of the Danube River. Accessed 23 Jan 2015
Liska I, Wagner F, Deutsch K, Sengl M, Slobodnik J (2015) Joint Danube Survey 3 Final Scientific Report (in print). Vienna, Austria
Schulze T, Krauss M, Bahlmann A, Hug C, Walz K-H, Brack W (2014) Onsite large volume solid phase extraction – how to get 1000 litres of water into the laboratory? Society for Environmental Toxicology and Chemistry (SETAC) Europe 24th Annual Meeting, Basel, Switzerland, 11–15 May, 2014
ChemAxon (2015) MarvinSketch Calculator Plugins 15.1.12.0 http://www.chemaxon.com. Accessed 30 Jan 2015
Greco G, Grosse S, Letzel T (2013) Serial coupling of reversed-phase and zwitterionic hydrophilic interaction LC/MS for the analysis of polar and nonpolar phenols in wine. J Sep Sci 36(8):1379–1388
LW/HSWT (2014) DAIOS Substance Database http://www.daios-online.de/daios/. Zweckverband Landeswasserversorgung, Langenau, Germany. Accessed 18 Dec 2014 (login only)
Muller A, Schulz W, Ruck WKL, Weber WH (2011) A new approach to data evaluation in the non-target screening of organic trace substances in water analysis. Chemosphere 85(8):1211–1219
NCBI (2014) PubChem (https://pubchem.ncbi.nlm.nih.gov/). National Center for Biotechnology Information. Accessed 17/12/2014
LfU/LW/HSWT/TUM (2014) STOFF-IDENT Substance Database http://bb-x-stoffident.hswt.de/stoffidentjpa/app Environmental Agency of Bayern, Germany (Bayerisches Landesamt für Umwelt). Accessed 1 Apr 2015 (login only)
MassBank (2014) MassBank www.massbank.jp. Accessed 17 Dec 2014
MassBank (2014) NORMAN MassBank www.massbank.eu. Accessed 17 Dec 2014
Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, Oda Y, Kakazu Y, Kusano M, Tohge T, Matsuda F, Sawada Y, Hirai MY, Nakanishi H, Ikeda K, Akimoto N, Maoka T, Takahashi H, Ara T, Sakurai N, Suzuki H, Shibata D, Neumann S, Iida T, Funatsu K, Matsuura F, Soga T, Taguchi R, Saito K, Nishioka T (2010) MassBank: a public repository for sharing mass spectral data for life sciences. J Mass Spectrom 45(7):703–714
HighChem (2014) mzCloud https://www.mzcloud.org/. HighChem Ltd., Bratislava, Slovakia. Accessed 17 Dec 2014
SIS (2014) The NIST 14 Mass Spectral Library http://www.sisweb.com/software/ms/nist.htm. Scientific Instrument Services. Accessed 17 Dec 2014
Wiley (2013) Wiley Registry of Mass Spectral Data, 10th Edn. Wiley, ISBN: 978-0-470-52037-6
Broecker S, Herre S, Wust B, Zweigenbaum J, Pragst F (2011) Development and practical application of a library of CID accurate mass spectra of more than 2,500 toxic compounds for systematic toxicological analysis by LC-QTOF-MS with data-dependent acquisition. Anal Bioanal Chem 400(1):101–117
Agilent (2015) Broecker, Herre, and Pragst Accurate Mass Personal Compound Database and Library (PCDL) for Forensics and Toxicology. Agilent Technologies, Santa Clara
Agilent (2015) Accurate Mass Personal Compound Database and Library (PCDL) for Pesticides. Agilent Technologies, Santa Clara
Scripps (2015) METLIN: Metabolite and Tandem MS Database http://metlin.scripps.edu/. Scripps Center for Metabolomics. Accessed 23/3/2015
Smith CA, O'Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, Siuzdak G (2005) METLIN: a metabolite mass spectral database. Ther Drug Monit 27(6):747–751
Bruker (2014) Bruker ToxScreenerTM - A Comprehensive Screening Solution for Forensic Toxicology. Bruker Daltronics, Inc., Bremen, Germany
Sciex (2015) iMethod Application - LC/MS/MS Meta Library Version 1.0 for Cliquid Software http://sciex.myshopify.com/products/imethod-application-lcmsms-meta-library-version-10-for-cliquid-software. AB Sciex. Accessed 23/3/2015
Gerlich M, Neumann S (2013) MetFusion: integration of compound identication strategies. J Mass Spectrom 48:291–298
Waters (2014) MassFragment (version 1.3) http://www.waters.com/webassets/cms/library/docs/720004823en.pdf?locale=en_US. Accessed 27 Jan 2015
Tellstroem V, Dunsbach R (2014) Technical Note TN-26: SmartFormula 3D – the new Dimension in Substance Identification – From Mass Spectrum to Chemical Formula. Bruker Daltronics, Inc., Bremen, Germany
Hill AW, Mortishire-Smith RJ (2005) Automated assignment of high-resolution collisionally activated dissociation mass spectra using a systematic bond disconnection approach. Rapid Commun Mass Spectrom 19:3111–3118
Agilent (2011) Agilent MassHunter Molecular Structure Correlator (MSC) Software, Revision A. Agilent Technologies, Santa Clara
NIST (2005) Automated Mass Spectral Deconvolution and Identification System (AMDIS), 26th edn. National Institute of Standards and Technology (NIST), US Department of Defense, USA
Daylight (2012) SMILES- A Simplified Chemical Language http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html. Daylight Chemical Information Systems Inc. Accessed 30 Jan 2015
IUPAC (2012) The IUPAC International Chemical Identifier http://www.iupac.org/inchi/. International Union of Pure and Applied Chemitstry. Accessed 30 Jan 2015
O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: An open chemical toolbox. J Cheminform 3:33
Liska I, Wagner F, Slobodnik J (2008) Joint Danube Survey 2: Final Scientific Report. Editors: Liska I, Wagner F, Slobodnik J ICPDR – International Commission for the Protection of the Danube River, Vienna, Austria
Kind T, Fiehn O (2007) Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinf 8:105
Weiss S, Jakobs J, Reemtsma T (2006) Discharge of three benzotriazole corrosion inhibitors with municipal wastewater and improvements by membrane bioreactor treatment and ozonation. Environ Sci Technol 40(23):7193–7199
Rasche F, Svatos A, Maddula RK, Bottcher C, Bocker S (2011) Computing Fragmentation Trees from Tandem Mass Spectrometry Data. Anal Chem 83(4):1243–1251
Meringer M, Reinker S, Zhang JA, Muller A (2011) MS/MS Data Improves Automated Determination of Molecular Formulas by Mass Spectrometry. Match-Commun Math Co 65(2):259–290
Pluskal T, Uehara T, Yanagida M (2012) Highly Accurate Chemical Formula Prediction Tool Utilizing High-Resolution Mass Spectra, MS/MS Fragmentation, Heuristic Rules, and Isotope Pattern Matching. Anal Chem 84(10):4396–4403
Schymanski EL, Gallampois CMJ, Krauss M, Meringer M, Neumann S, Schulze T, Wolf S, Brack W (2012) Consensus Structure Elucidation Combining GC/EI-MS, Structure Generation, and Calculated Properties. Anal Chem 84(7):3287–3295
Eckel WP, Kind T (2003) Use of boiling point-Lee retention index correlation for rapid review of gas chromatography-mass spectrometry data. Anal Chim Acta 494(1–2):235–243
Schymanski EL, Gerlich M, Ruttkies C, Neumann S (2014) Solving CASMI 2013 with MetFrag, MetFusion and MOLGEN-MS/MS. Mass Spectrometry 3 (Special Issue 2):S0036
Reemtsma T (2009) Determination of molecular formulas of natural organic matter molecules by (ultra-) high-resolution mass spectrometry Status and needs. J Chromatogr A 1216(18):3687–3701
Reemtsma T (2010) The carbon versus mass diagram to visualize and exploit FTICR-MS data of natural organic matter. J Mass Spectrom 45(4):382–390
NORMAN Association (2015) Non-target screening techniques for environmental monitoring http://www.norman-network.net/?q=node/190. NORMAN Association. Accessed 27 Jan 2015
NORMAN Association (2015) Workshop on Non-Target Screening: http://www.norman-network.net/?q=node/162. NORMAN Association. Accessed 27 Jan 2015
NORMAN Association (2015) NORMAN MassBank Workshop http://www.norman-network.net/?q=node/163. NORMAN Association. Accessed 27 Jan 2015
Acknowledgments
The authors gratefully acknowledge the contributions of many others who helped during the analysis, reporting and discussion of the results presented here: Philipp Longree, Martin Loos, Matthias Ruff and Jennifer Schollee (Eawag), Margit Petre, Anett Kloß and Werner Brack (UFZ), Reza Aalizadeh, Nikiforos Alygizakis and Pablo Gago Ferrero (University of Athens), Sylvain Merel (University of Tübingen), Alexander van Nuijs, Alin Ionas and Adrian Covaci (University of Antwerp), Martijn Pijnappels (Rijkswaterstaat), Christelle Margoum (Irstea), Claudio Bortolini (University of Padua), Marie-Hélène Devier and Hélène Budzinski (University of Bordeaux, France), Christophe Tondelier and Mathilde Chachignon (VERI), Tobias Bader and Thomas Lucke (Zweckverband Landeswasserversorgung), Michael Schlüsener (BfG), Mar Esperanza (Suez Environnement), Siniša Repec (Croatian Waters), Peter Tarabek (Slovak National Water Reference Laboratory), Christoph Ruttkies (IPB, Halle, Germany), the ChemSpider support team, and Thierry Faye from Agilent. The water sample of the Danube was kindly provided by the International Commission for the Protection of the Danube River (IPCDR) as part of the Joint Danube Survey 3. Author contributions: ELS, HPS, JS, IMI, PO, MK, TS, PH, TL, SG, NST, CZ, and JH were involved in the core team preparing the trial, evaluation, workshop, and publication; MI, TP, RdB, MR, MO, UK, WS, AG, NN, GL, PB, SB, DS, and PR participated in the trial and publication efforts.
This trial was initiated by the NORMAN Association with in-kind contributions from participants; no fee was charged for participation. This work was supported in part by the SOLUTIONS project, which received funding from the European Union’s Seventh Framework Programme for research, technological development and demonstration under Grant Agreement No. 603437.
Compliance with ethical standards
ᅟ
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
No violation of human or animal rights occurred during this investigation.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published in the topical collection High-Resolution Mass Spectrometry in Food and Environmental Analysis with guest editor Aldo Laganà.
Rights and permissions
About this article

Cite this article
Schymanski, E.L., Singer, H.P., Slobodnik, J. et al. Non-target screening with high-resolution mass spectrometry: critical review using a collaborative trial on water analysis. Anal Bioanal Chem 407, 6237–6255 (2015). https://doi.org/10.1007/s00216-015-8681-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8681-7