
Abstract
A quantitative method using ultra-performance liquid chromatography coupled with tandem mass spectrometry (UPLC-MS/MS) was developed for the determination of 60 compounds, belonging to a variety of veterinary drug (VD) classes, in meat. The included analytes belong to the following VD classes: β-agonists, sulfanilamides, quinolones, macrolides, tetracyclines, β-lactams, nitroimidazoles, glucocorticoids, sex hormones, chloromycetins, sedatives, and olaquindox metabolite. The effective PRiME pass-through cleanup procedure was used to ensure high extraction efficiency and good sample cleanup after a simple liquid extraction of the meat samples with acetonitrile/water. The developed method was validated successfully. Mean recoveries for all analytes ranged from 80 to 116%, with the relative standard deviations (RSDs) ≤ 7.8%. Limits of quantification were in the range 0.05–3.0 μg kg−1 and limits of detection were in the range 0.1–10 μg kg−1. The matrix effect was evaluated for the different meat matrices and was found to be markedly different in different matrices. The validated method was used in a pilot study to analyze real samples of pork, beef, mutton, chicken, and pork liver, lambs’ liver, and chicken liver. Trace amounts of β-agonists, oxytetracycline, quinolones, chloromycetin sulfadimidine, and 3-methyl-quinoxaline-2-carboxylicacid were detected in these samples. In conclusion, this workflow can provide a simpler and more cost-effective alternative to conventional analytical methods and is compatible with processing large sample numbers in a short time period.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Veterinary drugs (VDs), which are widely used in agriculture and animal husbandry to prevent or cure diseases, to increase production or as growth promoters, may leave residues in food from treated animals, in animal excrement, and even in the environment (Paiga et al. 2017; Teglia et al. 2017; Li et al. 2017; Zhao et al. 2017b). Especially with overdosing and illegal use, VD residues and their metabolites pose a potential risk to both animal and human health since long-term exposure can cause cancer, anaphylactic shock, microbial resistance, and reproductive disorders (Lúcia Santos et al. 2016; Moreno-Gonzalez et al. 2017b; Qin et al. 2017).
To protect public health, pharmacologically active substances are strictly regulated and monitored in food stuffs of animal origin and several countries have established maximum residue limits (MRLs) or tolerances. Commission Regulation (EU) 37/2010 classified VDs in four annexes, on the basis of a scientific assessment of safety (Commission 2010). CAC/MRL 2-2015 recommends MRLs and provides risk management recommendations for residues of VDs, such as chloramphenicol, metronidazole, and dimetridazole, in food (Commission 2015). In China, announcement No. 235 of the Ministry of Agriculture also established MRLs for different substances in food of animal origin (China 2002). In all cases, different international regulations require that levels of residues in food should not harm consumers. Because of the high number of VDs that need to be controlled, it is important that regulatory monitoring laboratories have access to multi-class multi-residue analytical methods that permit analysis of a variety of drugs with a single procedure (Mainero Rocca et al. 2017; Wittenberg et al. 2017).
There have been several reports describing analysis of VDs in many products of animal origin, including bovine muscle, porcine muscle (Park et al. 2015; Zhang et al. 2016), chicken (Yoshikawa et al. 2017), eggs (Piatkowska et al. 2017), milk (Wittenberg et al. 2017), and honey (Jin et al. 2017). However, multi-class methods for analysis of VDs in more than two matrices are scarce because of the complexity of animal-origin samples and the large number of analytes (Rizzetti et al. 2017). Although multi-class methods are the most efficient way to maximize the number of analytes that may be simultaneously determined by a single analysis, most of the methodologies proposed in the literature can only be used for compounds from the same class (Matus and Boison 2016; Qin et al. 2017; Lu et al. 2016; Zhao et al. 2017a). There also is a growing trend to develop multi-class multi-residue methods for VD residues in food. Zanella et al. reported a simple extraction procedure, using acetonitrile and trichloroacetic acid for protein precipitation, coupled to liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) for the analysis of 20 VD residues in bovine kidney and liver (Rizzetti et al. 2017). Yoshikawa used a two-step extraction followed by a solid-supported liquid extraction cleanup for LC-MS/MS analysis of 37 VDs in seven different classes in two processed chicken foods (Yoshikawa et al. 2017). A dispersive solid-phase extraction and LC-MS/MS have been used to develop a multi-class method for determination of 30 VDs, belonging to three different classes in milk powder (Li and Wu 2017). The simultaneous detection of a large number of analytes at low concentrations requires the use of a highly sensitive and selective technique. LC–MS/MS has been widely used for this purpose and has enabled the simultaneous analysis of several classes of VDs at low concentrations, even in complex matrices such as bovine muscle (Dasenaki et al. 2016). Taking into account the wide variety of physicochemical properties of VDs and the complexity of meat matrices, an important and complex issue is how to simultaneously analyze multi-class VDs with generic extraction procedures (Yoshikawa et al. 2017). An effective sample extraction technique that can essentially eliminate matrices is necessary to achieve reliable results and maintain instrument performance (Tian et al. 2016). Several extraction approaches, such as the QuEChERS (Quick, Easy, Cheap, Effective, Rugged and Safe) approach (Guo et al. 2016; Jin et al. 2017; Paiga et al. 2017; Piatkowska et al. 2017; Turnipseed et al. 2017), solid phase extraction (SPE) (Jianfeng Wang et al. 2017; Turnipseed et al. 2017; Yoshikawa et al. 2017; Chen et al. 2016), matrix solid phase dispersion (Zhang et al. 2017), liquid–liquid extraction (Aguilera-Luiz et al. 2008; Pastor-Belda et al. 2017), and molecularly imprinted polymers (Doue et al. 2012; Samanidou et al. 2016) have been described. Because of the complex nature of the matrices, SPE is one of the most widely adopted cleanup steps for determination of VDs in food samples. However, because of the narrow selectivity of SPE, it is difficult to broaden the scope of the analytes and this method is not capable of determining a large number of compounds belonging to different classes (Guo et al. 2016). Waters Corporation has released a new Hydrophilic-Lipophilic Balance (HLB) sorbent called PRiME (process, robustness, improvements, matrix effects, ease of use), which we have found to be highly effective for the removal of both fats and phospholipids from meat extracts (Moreno-Gonzalez et al. 2017a). PRiME HLB sorbent has also been evaluated for the determination of different families of antibiotics in a complex matrix such as milk (Chen et al. 2016) and of multiclass VD residues in fish, shrimp, eel, and swine waste (Turnipseed et al. 2017; Li et al. 2017). This special cartridge does not require activation/equilibration or washing steps, and provides a better cleanup effect than Oasis HLB (Chen et al. 2016). The PRiME pass-through cleanup approach for different mechanisms of action is simple to operate, has high efficiency, and is suitable for high throughput analysis of analytes in complex samples.
In this study, different extraction solvents were investigated and modifications were made to accommodate a variety of target analytes. The efficiency of PRiME HLB was also compared with conventional HLB. The performance of this method was validated by estimating specificity, linearity, trueness, precision, analytical limits, and matrix effects. Finally, the method was successfully used for the analysis of VDs in several meat matrices.
Experimental
Chemicals, Reagents, and Solutions
HPLC grade methanol and acetonitrile were obtained from Fisher Scientific, Inc. (Pittsburgh, PA, USA). Formic acid of high-quality grade was purchased from Sigma-Aldrich (St. Louis, MO, USA), (Bedford, MA, USA). Oasis PRiME HLB extraction cartridges (200 mg, 6 mL) were supplied by Waters (Milford, MA, USA). The syringe filter was purchased from Agela (Tianjin, China).
Clenbuterol, salbutamol, ractopamine, mabuterol, cimbuterol, terbutaline, penbutolol, tulobuterol, fenoterol, clorprenaline, sulfamerazine, sulfamethoxazole, sulfadimidine, sulfadimethoxine, sulfamonomethoxine, sulfaquinoxaline, sulphacetamide, sulfapyridine, sulfisoxazole, sulfamethizole, sulfadoxine, sulfathizole, sulfaphenazole, sulfadiazine, sulfamethoxypyridazine, sulfameter, sulfachloropyridazine, enrofloxacin, ciprofloxacin, sarafloxacin, danofloxacin, flumequine, ofloxacin, norfloxacin, erythromycin, lincomycin, tetracycline, oxytetracycline, chlorotetracycline, doxycycline, amoxicillin, ampicillin, benzylpenicillin, cloxacillin, dimetridazole, metronidazole, betamethasone, dexamethasone, methyltestosterone, trenbolone, testosterone, mengestrol, chloromycetin, thiamphenicol, florfenicol, chlorpromazine, acetylpromazine, azaperol, phenylethanolamine A, and 3-methyl-quinoxaline-2-carboxylicacid were all purchased from Dr. Ehrenstorfer (Augsburg, Germany) or Sigma-Aldrich.
According to their classification and stability, the aforementioned compounds were divided into 12 different groups: β-agonists (10), sulfanilamides (17), quinolones (7), macrolides (2), tetracyclines (4), β-lactams (4), nitroimidazoles (2), glucocorticoids (2), sex hormones (4), chloromycetins (3), sedatives (4), and olaquindox metabolites (1).
Stock solutions of each compound (~ 1000 mg L−1) were prepared in different solvents and stored at − 20 °C in brown glass containers for a maximum period of 6 months. The analytes were dissolved and diluted with an appropriate solvent (acetonitrile, methanol, or water), based on their solubility properties.
Individual stock standard solutions were prepared by weighing approximately 10 mg of all compounds into 10-mL volumetric flasks. Depending on the specific solubility properties, the compounds were dissolved in acetonitrile or methanol. Multi-compound working standard solutions were prepared by diluting an appropriate amount of the standard solutions with methanol. The mixtures were stored at 4 °C in the dark and were stable for 2 weeks.
Instrumentation
A Waters Acquity™ ultra high-performance liquid chromatography system, equipped with a Waters Acquity autosampler and binary solvent management system, together with a Triple Quad™ 5500 mass spectrometer (AB Sciex, Framingham, USA), with an electrospray ionization source, were used for the analyses. A 3K-30 high speed refrigerated centrifuge (Sigma-Aldrich), MS3 basic vortex mixer (IKA, Germany), and KQ-250-E ultrasonic instrument (Kun Shan, China) were used for sample preparation.
Chromatographic separations were performed using Waters ACQUITY UPLC™ BEH C18 analytical columns (100 × 2.1 mm i.d., 1.7 μm particle size) maintained at 40 °C. Nitrogen (purity ≥ 99%), generated using a nitrogen generator (Peak Scientific, Billerica, MA, USA), was used in the ESI source. Chromatographic separation was carried out with gradient elution at a flow rate of 300 μL min−1, using water (eluent A) and methanol (eluent B), both containing 0.1% formic acid (v/v). The gradient elution was performed as follows, 95–80% A (0–3.0 min); 80–50% A (3.0–5.0 min); 50–0% A (5.0–8.0 min); 0% A (8.0–10.0 min); 0–95%A (10.0–10.1 min); and 95–95% A (10.1–12.0 min).
MS was performed using a Triple Quadrupole Mass Spectrometer, equipped with an ESI source working in positive ion mode (ESI+) as follows: ion spray (IS) voltage, 5500 V; curtain gas, 30 psi; nebulizer gas (GS1), 55 psi; auxiliary gas (GS2), 55 psi; and source temperature, 400 °C. When working in negative ion mode (ESI−), the same parameters were used, except the IS voltage, which was set at − 4500 V. Nitrogen was used for nebulization and collision. To optimize the mass spectrometer parameters for each compound, the compounds were infused separately using selective reaction monitoring (SRM) mode, with a continuous flow of 10 μL min−1 and a concentration of 1 mg L−1. The Analyst 1.6.2 software (AB Sciex) was used for data acquisition and processing. Optimal values of declustering potential (DP), entrance potential (EP), collision energy (CE), collision cell exit potential (CXP), and SRM for each analyte used for identification of VD residues are summarized in Table 1.
Meat Samples
Samples of pork, beef, mutton, chicken, pork liver, lambs’ liver, and chicken liver were purchased from a local supermarket. The samples were minced using a meat grinder and stored at − 18 °C until analysis.
Sample Preparation
A meat sample (2.50 ± 0.01 g) was weighed into a 50-mL conical centrifuge tube. Ten milliliters acetonitrile/water (80/20, v/v) was added and the tube was vortexed for 10 s. The sample was sonicated for about 30 min then centrifuged at 7000 rpm at 4 °C for 5 min. The resulting supernatant solution was slowly transferred into a 15-mL centrifuge tube. A Waters Oasis PRiME HLB cartridge (200 mg, 6 mL) was set up for pass-through filtration. The extraction mixture (5 mL) was passed through the cartridge under gravity and collected in a 15-mL centrifuge tube. The extract was concentrated to near dryness under nitrogen at 45 °C. The extract was then reconstituted with 20% acetonitrile-water (1 mL) and vortexed for 30 s. Finally, the extract was pressed through a 0.22-μm syringe filter into a vial and analyzed by UPLC-MS/MS.
Validation Procedure
The method was validated by evaluating linearity, the limits of detection (LODs), limits of quantification (LOQs), accuracy, repeatability and reproducibility, and matrix effect using UPLC-MS/MS. The linearity of the method was demonstrated using blank matrix-matched calibration standard for each analyte. A calibration curve was prepared for blank meat matrix with six different concentration ranges at levels of 0.1–1000 μg kg−1 for analytes, shown in Table 1. For each compound, the matrix-matched calibration curve was obtained by plotting the ratio of each analyte quantitative ion peak area (y) versus the corresponding mass concentrations (x). LODs and LOQs were estimated by fortifying blank samples with the 60 analytes and applying the extraction procedure before chromatographic determination. For each analyte, LODs and LOQs in meat samples were defined as the concentration giving a signal to noise ratio of 3 and 10, respectively.
The accuracy of the method was evaluated based on the recoveries of VD residues. The blank meat samples spiked with all analytes were prepared at three levels ((LOQ) (low), 20 × LOQ (medium), and 50 × LOQ (high)), then processed and measured under the experimental conditions. Repeatability and reproducibility was determined in six replicates at each level by measuring corresponding relative standard deviations (RSDs). Matrix effect (ME) was evaluated by comparing the response of each VD residue obtained from a standard solution in solvent and that from a matrix at the same concentration.
Results and Discussion
Optimization of Analytical Conditions
Various LC conditions, including different chromatography columns, mobile phases, modes of elution, flow rates, and column temperatures, were evaluated to obtain good response, peak shape, and separation. A comparison of acetonitrile and methanol as the organic solvent showed that higher intensities and better peak shapes were obtained for most analytes using methanol. Addition of 0.1% formic acid to both the aqueous phase and the organic phase enhanced the response of most analytes in positive ion mode and gave an outstanding response for chloromycetins (negative ion mode), with negligible suppression of the response. As a result of these preliminary studies, 0.1% formic acid in methanol and water using a BEH C18 column were chosen as the best LC conditions for further investigations.
The MS ESI parameters were optimized for both positive and negative ion modes by direct infusion of standard solutions of each target compound. For most compounds, the protonated ion [M + H]+ was present as the base peak and was selected as the precursor ion for quantification. Chloromycetins, however, gave the deprotonated ion [M-H]−. Cone voltage and collision energy were optimized to acquire two product ions and two transitions for each compound in SRM mode. The higher abundance transition with less interference was used for quantification and the other transition was used for identification. The MS parameters, cone voltage, collision energy, and SRM transition selected for each analyte are shown in Table 1.
Optimization of Sample Preparation
Sample Extraction
One of the greatest challenges faced was to develop extraction conditions suitable for a wide range of VD analytes in a single extraction procedure and to eliminate the complex interferences in meat tissue matrices. Acetonitrile, methanol, and water mixtures are frequently used in the extraction step in the analysis of VD residues. Acetonitrile gave less co-extraction of matrix and facilitated protein precipitation.
Various extraction solvent combinations were investigated in terms of the absolute recoveries obtained for each individual analyte. In the present study, we tested different acetonitrile/water ratios (50:50, 80:20, and 100:0, v/v) for the extraction step in the analysis of VD residues. Acetonitrile/water (80/20, v/v) extracted the most drug from the spiked samples, providing acceptable absolute recoveries of 80–110%. The recoveries obtained under the different test conditions are presented in Fig. 1. Recoveries were very low (< 10%) for tetracyclines and too high (> 120%) for β-lactams and glucocorticoids when 100% acetonitrile was used as the extraction solvent for our list of analytes. Increasing the amount of water in the extraction solvent gave more co-extraction of matrix. Acetonitrile/water (50/50, v/v) was not compatible with all the compound classes, and absolute recoveries were outside the acceptable range for certain β-lactams, nitroimidazoles, glucocorticoids, sex hormones, chloromycetins, and for olaquindox metabolite.

Recoveries for different classes of veterinary drugs with different extracting solution
Addition of 1 or 2% formic acid to the extraction solvent gave acceptable recoveries for most analytes, but low recoveries for β-agonists, macrolides, glucocorticoids, and olaquindox metabolite. Taking into account the different chemical properties of VD analytes, we decided not to use acid in the extractions.
The volume of the extraction solvent was then optimized by evaluating the difference between 5, 10, and 15 mL of 80% acetonitrile–water. When 5 mL of solvent was used, the recoveries of most analytes were very low since extraction was inadequate and the protein may not have precipitated completely. Although a slight improvement in recoveries was observed when the volume of the extraction solvent was increased, the improvement did not justify the higher amount of solvents needed and the increased time needed for the evaporation step. To avoid increasing the cost and decreasing the rapidity of the developed method, 10 mL was chosen as the best volume for the extraction solvent.
Selection of Procedures for Cleanup
The suitability of this pass-through SPE for analyzing multi-residue VDs was evaluated in various meat matrices. Several cleanup procedures, including no clean-up, MCX, HLB, and PRiME HLB, have previously been used in the determination of VDs. The performance of different purification technologies was evaluated by spiking meat samples with 50 μg kg−1 of VDs to choose the best eluting mixture. The recovery (%RE) was determined by comparing the response area by the spiked sample (Ab), the blank matrix (As), and theoretical addition (M) using Eq. 1. The PRiME HLB SPE cartridge was the most efficient sorbent for purifying the extracts for most VDs (Fig. 2). With the Oasis PRiME HLB, most compounds had recoveries between 80 and 116%, with all the relative standard deviations (RSDs) < 7.8%, shown in Table 2. Higher variability was seen with other cleanup methods. Without cleanup, the recovery of sex hormones was > 150%, which is unsatisfactory. With the Oasis MCX, low recoveries were obtained for almost all glucocorticoids, sex hormones, sedatives, and for olaquindox metabolite, although the β-agonists had better recoveries (85–116%). The common RP C18 SPE, Oasis HLB gave good recovery for most analytes, with the exception of glucocorticoids and sex hormones.

Comparison of extraction efficiencies of different cleanup strategies for all analytes
Method Validation
Matrix Effect
The ME is widely used to evaluate the performance of sample pretreatment methods. ME was investigated by comparing the peak areas of drugs in blank matrices (Am) to those of aqueous standards (Ae), using Eq. 2 (Yoshikawa et al. 2017):
An ME(%) value between − 20 and 20% indicates an acceptable matrix effect, a value <− 20% indicates ion suppression, and a value > 20% indicates an enhancement effect (Valese et al. 2017). MEs were evaluated for the different meat matrices (Fig. 3). It is noteworthy that ME is different in different matrices. All 60 compounds had an acceptable ME in pork, beef, mutton, and chicken whereas, in the liver samples, most compounds had a large ME because of the complex matrix. Some analytes that showed > 20% false negatives, especially sex hormones, exhibited apparent signal suppression, with ME values > 40%. Quinolones and chloromycetins showed matrix enhancement, ranging from 10 to 50%, and quinolones gave significant matrix enhancement (> 40%), which is in agreement with a report that all quinolones are subject to signal enhancement (Ortelli et al. 2009).

Matrix effects in HPLC-MS analysis of VDs in different matrix samples
Removal of Phospholipids
Because of their special physical and chemical properties, phospholipids co-extract with analytes, caused ionization suppression in the LC-ESI-MS analysis and an obvious matrix effect of analytes (José Ángel Salatti-Dorado et al. 2017). Since they are strongly retained on the chromatographic column, phospholipids can also produce shifts in retention time, increased baseline noise, and poor precision.
Both phosphatidylcholine (PC) and sphingomyelin (SM) have a phosphocholine head group and would be fragmented to generate a characteristic ion at m/z 184. The total amount of PC and SM in the sample can be determined by measuring the total precursor ion signals that generate product ion with m/z 184. This value can then be used in lipidomic studies (Chao et al. 2017). The removal of phospholipids was evaluated in a single injection by measuring the positive precursor ion scan at m/z 184 after sample extraction with no cleanup or cleanup using PRiME HLB. Large amounts of phospholipids were detected in positive ion mode in both muscle and yolk extracts, when no cleanup was carried out. A PRiME HLB cleanup step completely removed phospholipids (Fig. 4). PRiME HLB cartridges have a packed bed of a novel water-wettable reversed-phase SPE sorbent, designed for the gross level removal of phospholipids from biological tissue prior to UPLC-MS/MS analysis.

Parent scan mass spectrum at m/z 184 of pork blank with PRiME HLB (A) and no purification (B)
Accuracy, Repeatability, and Reproducibility
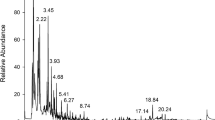
To evaluate the trueness of the proposed method, three concentrations, limits of quantification (LOQ) (low), 20 × LOQ (medium) and 50 × LOQ (high) for each analyte, were analyzed in seven meat samples. Each analysis was repeated six times in parallel. Repeatablility was evaluated at three levels as previously described, with six replicates at each level. The percentage recovery and precision results expressed as RSDs are given in Table 2. All of the compounds had recoveries between 80 and 116% in various meat and tissue samples. Although recoveries were slightly lower in the liver samples, these also met the quantitative requirements. The RSDs of all compounds, in all investigated matrices, was < 7.8%, indicating that the method can be carried out with a satisfactory level of precision. The results also indicate that good recoveries can be obtained using the developed method. Total ion current (TIC) chromatograms of blank and spiked pork samples were showed in Fig. 5.

Total ion current (TIC) chromatograms of blank and spiked pork samples
Linearity and Limit of Detection
Because of the difference in sensitivity of the analytes in the LC/MS, linearity of the analytical method was evaluated using matrix- matched calibration curves for each compound in blank meat samples at six different concentration levels, and for different compounds over different concentration ranges. All 60 compounds showed good linear regression, with R2 values > 0.995 (Table 1).
The LODs and LOQs were determined as explained in the “Validation Procedure” section. Concentration levels with signal to noise ≥ 3 were defined as LODs and concentration levels with signal to noise ≥ 10 were defined as LOQs. As shown in Table 1, the LODs and LOQs for 60 analytes ranged from 0.2 to 3 μg kg−1 and 0.5 to 10 μg kg−1, respectively, indicating the satisfactory sensitivity for the proposed analytical protocol.
Application to Real Samples
The developed method was used for the analysis of VDs in 280 meat samples, including pork, beef, mutton, pork liver, lambs’ liver, and chicken liver. Three β-agonists (clenbuterol, salbutamol, ractopamine) were identified in samples of mutton, beef, lambs’ liver, pork, and chicken with concentrations ranging from 0.90 to 410 μg kg−1. Oxytetracycline, enrofloxacin, ciprofloxacin, and chloromycetin were also found in chicken and chicken liver samples, with the concentrations < 222 μg kg−1. Sulfadimidine and 3-methyl-quinoxaline-2-carboxylicacid were detected in a pork and pork liver sample, at concentration levels of 607 and 2.73 μg kg−1, respectively. The typical SRM chromatograms of positive samples were presented in Fig. 6. These results are consistent with previous studies. Several analytical methods for the determination of different groups of VDs in meat samples have been developed by LC-MS/MS and applied in real meat samples. A method was developed for detecting sulfonamides, tilmicosin, and avermectins residues in animal samples, and sulfamethazine (sulfadimidine) was detected at concentration levels of 15 and 72 μg kg−1 in 2 bovine meat samples (Qin et al. 2017). LC-MS/MS had also been applied in 137 pilot meat samples and ractopamine had been identified with the concentration ranging from 1.8 to 6.3 μg kg−1 and 0.6 to 64 μg kg−1 in 12 bovine muscle and 15 bovine liver samples, respectively (Matus and Boison 2016). For all positive samples, the differences between values obtained using the new method and those obtained using the National Standard Method of China were < 7.78%, which are presented in Table 3.

SRM chromatograms of nine VDs in real samples (clenbuterol, salbutamol, ractopamine, oxytetracycline, enrofloxacin, ciprofloxacin, chloromycetin, sulfadimidine, and 3-methyl-quinoxaline-2-carboxylicacid)
Conclusion
The Oasis PRiME HLB UPLC-MS/MS method described here is a reliable procedure for the simultaneous determination of 60 VD residues in different meat samples. The new method was satisfactorily validated in terms of linearity, sensitivity, precision, repeatability, and MEs. The use of acetonitrile/water (80/20, v/v) and Oasis PRiME HLB for extraction and cleanup gave excellent recovery and purification. Oasis PRiME HLB, when used as a pass-through cleanup SPE, can remove most of the phospholipids from biological tissues prior to LC-MS analysis. Notably, the properties of the pass-through SPE technique mean that more compounds in more food matrices could easily be included in the future.
References
Aguilera-Luiz MM, Vidal JL, Romero-Gonzalez R, Frenich AG (2008) Multi-residue determination of veterinary drugs in milk by ultra-high-pressure liquid chromatography-tandem mass spectrometry. J Chromatogr A 1205:10–16. https://doi.org/10.1016/j.chroma.2008.07.066
Chao HC, Chen GY, Hsu LC, Liao HW, Yang SY, Wang SY, Li YL, Tang SC, Tseng YJ, Kuo CH (2017) Using precursor ion scan of 184 with liquid chromatography-electrospray ionization-tandem mass spectrometry for concentration normalization in cellular lipidomic studies. Anal Chim Acta 971:68–77
Chen ZY, Ying S, Liu JH, Zhan PP, Zhao YG (2016) PRiME pass-through cleanup for the fast determination of aflatoxins in human serum by using LC-MS/MS. Anal Methods 2016:1457–1462
China (2002) Announcement of the Ministry 450 of agriculture of the People’s Republic of China 235 th. http://www.moa.gov.cn/zwllm/tzgg/gg/200302/t20030226_59300.htm (03–26-2017)
Codex Alimentarius Commission (2015) Codex Maximum Residue Limit (MRLs) and risk management recommendations (RMRs) for residues of veterinary drugs in foods CAC/MRL 2–2015 Codex Alimentarius International Food Standards
Dasenaki ME, Michali CS, Thomaidis NS (2016) Analysis of 76 veterinary pharmaceuticals from 13 classes including aminoglycosides in bovine muscle by hydrophilic interaction liquid chromatography-tandem mass spectrometry. J Chromatogr A 1452:67–80. https://doi.org/10.1016/j.chroma.2016.05.031
Doue M, Bichon E, Dervilly-Pinel G, Pichon V, Chapuis-Hugon F, Lesellier E, West C, Monteau F, Le Bizec B (2012) Molecularly imprinted polymer applied to the selective isolation of urinary steroid hormones: an efficient tool in the control of natural steroid hormones abuse in cattle. J Chromatogr A 1270:51–61. https://doi.org/10.1016/j.chroma.2012.10.067
European Commission (2010) Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in food stuffs of animal origin. Off J Eur Union L15:1–72
Guo C, Wang M, Xiao H, Huai B, Wang F, Pan G, Liao X, Liu Y (2016) Development of a modified QuEChERS method for the determination of veterinary antibiotics in swine manure by liquid chromatography tandem mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci 1027:110–118. https://doi.org/10.1016/j.jchromb.2016.05.034
Jin Y, Zhang J, Zhao W, Zhang W, Wang L, Zhou J, Li Y (2017) Development and validation of a multiclass method for the quantification of veterinary drug residues in honey and royal jelly by liquid chromatography-tandem mass spectrometry. Food Chem 221:1298–1307. https://doi.org/10.1016/j.foodchem.2016.11.026
Li R, Wu M (2017) High-throughput determination of 30 veterinary drug residues in milk powder by dispersive solid-phase extraction coupled with ultra-high performance liquid chromatography tandem mass spectrometry. Food Anal Methods 2017:3753–3762
Li X, Guo P, Shan Y, Ke Y, Li H, Fu Q, Wang Y, Liu T, Xia X (2017) Determination of 82 veterinary drugs in swine waste lagoon sludge by ultra-high performance liquid chromatography-tandem mass spectrometry. J Chromatogr A 1499:57–64. https://doi.org/10.1016/j.chroma.2017.03.055
Lu Y, Cheng Z, Liu C, Cao X (2016) Determination of sulfonamides in fish using a modified QuEChERS extraction coupled with ultra performance liquid chromatography-tandem mass spectrometry. Food Anal Methods 2017:1857–1866
Mainero Rocca L, Gentili A, Perez-Fernandez V, Tomai P (2017) Veterinary drugs residues: a review of the latest analytical research on sample preparation and LC-MS based methods. Food Addit Contam A Chem Anal Control Expo Risk Assess 34:766–784. https://doi.org/10.1080/19440049.2017.1298846
Matus JL, Boison JO (2016) A multi-residue method for 17 anticoccidial drugs and ractopamine in animal tissues by liquid chromatography-tandem mass spectrometry and time-of-flight mass spectrometry. Drug Test Anal 8:465–476. https://doi.org/10.1002/dta.2019
Moreno-Gonzalez D, Hamed AM, Gilbert-Lopez B, Gamiz-Gracia L, Garcia-Campana AM (2017a) Evaluation of a multiresidue capillary electrophoresis-quadrupole-time-of-flight mass spectrometry method for the determination of antibiotics in milk samples. J Chromatogr A 1510:100–107. https://doi.org/10.1016/j.chroma.2017.06.055
Moreno-Gonzalez D, Rodriguez-Ramirez R, Del Olmo-Iruela M, Garcia-Campana AM (2017b) Validation of a new method based on salting-out assisted liquid-liquid extraction and UHPLC-MS/MS for the determination of betalactam antibiotics in infant dairy products. Talanta 167:493–498. https://doi.org/10.1016/j.talanta.2017.02.045
Ortelli D, Cognard E, Jan P, Edder P (2009) Comprehensive fast multiresidue screening of 150 veterinary drugs in milk by ultra-performance liquid chromatography coupled to time of flight mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci 877:2363–2374. https://doi.org/10.1016/j.jchromb.2009.03.006
Paiga P, Santos LH, Delerue-Matos C (2017) Development of a multi-residue method for the determination of human and veterinary pharmaceuticals and some of their metabolites in aqueous environmental matrices by SPE-UHPLC-MS/MS. J Pharm Biomed Anal 135:75–86. https://doi.org/10.1016/j.jpba.2016.12.013
Park JA, Zhang D, Kim DS, Kim SK, Cho KS, Jeong D, Shim JH, Kim JS, Abd EA, El-Aty AM, Shin HC (2015) Single-step multiresidue determination of ten multiclass veterinary drugs in pork, milk, and eggs using liquid chromatography with tandem mass spectrometry. J Sep Sci 38:2772–2780. https://doi.org/10.1002/jssc.201500501
Pastor-Belda M, Garrido I, Campillo N, Vinas P, Hellin P, Flores P, Fenoll J (2017) Combination of solvent extractants for dispersive liquid-liquid microextraction of fungicides from water and fruit samples by liquid chromatography with tandem mass spectrometry. Food Chem 233:69–76. https://doi.org/10.1016/j.foodchem.2017.04.094
Piatkowska M, Gbylik-Sikorska M, Gajda A, Jedziniak P, Bladek T, Zmudzki J, Posyniak A (2017) Multiresidue determination of veterinary medicines in lyophilized egg albumen with subsequent consumer exposure evaluation. Food Chem 229:646–652. https://doi.org/10.1016/j.foodchem.2017.02.147
Qin Y, Jatamunua F, Zhang J, Li Y, Han Y, Zou N, Shan J, Jiang Y, Pan C (2017) Analysis of sulfonamides, tilmicosin and avermectins residues in typical animal matrices with multi-plug filtration cleanup by liquid chromatography-tandem mass spectrometry detection. J Chromatogr B Anal Technol Biomed Life Sci 1053:27–33. https://doi.org/10.1016/j.jchromb.2017.04.006
Rizzetti TM, Souza MP, Prestes OD, Adaime MB, Zanella R (2017) A simple and fast method for the determination of 20 veterinary drug residues in bovine kidney and liver by ultra-high-performance liquid chromatography tandem mass spectrometry. Food Anal Methods 2017:854–864
Salatti-Dorado JA, Caballero-Casero N, Sicilia MD, Lunar ML, Rubio S (2017) The use of a restricted access volatile supramolecular solvent for the LC/MS-MS assay of bisphenol A in urine with a significant reduction of phospholipid-based matrix effects. Anal Chim Acta 950:71–79
Samanidou V, Kehagia M, Kabir A, Furton KG (2016) Matrix molecularly imprinted mesoporous sol-gel sorbent for efficient solid-phase extraction of chloramphenicol from milk. Anal Chim Acta 914:62–74. https://doi.org/10.1016/j.aca.2016.02.003
Santos L, Soares B, Rosa J, Freitas A, Leston S, Barbosa J, Ramos F (2016) Detection and quantification of 41 antibiotic residues in Gilthead Sea Bream (Sparus aurata) from aquaculture origin, using a multiclass and multi-residue UHPLC-MS/MS method. Food Anal Methods 2016:2749–2753
Teglia CM, Peltzer PM, Seib SN, Lajmanovich RC, Culzoni MJ, Goicoechea HC (2017) Simultaneous multi-residue determination of twenty one veterinary drugs in poultry litter by modeling three-way liquid chromatography with fluorescence and absorption detection data. Talanta 167:442–452. https://doi.org/10.1016/j.talanta.2017.02.030
Tian H, Wang J, Zhang Y, Li S, Jiang J, Tao D, Zheng N (2016) Quantitative multiresidue analysis of antibiotics in milk and milk powder by ultra-performance liquid chromatography coupled to tandem quadrupole mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci 1033-1034:172–179. https://doi.org/10.1016/j.jchromb.2016.08.023
Turnipseed SB, Storey JM, Lohne JJ, Andersen WC, Burger R, Johnson AS, Madson MR (2017) Wide-scope screening method for multiclass veterinary drug residues in fish, shrimp, and eel using liquid chromatography-quadrupole high-resolution mass spectrometry. J Agric Food Chem 65:7252–7267. https://doi.org/10.1021/acs.jafc.6b04717
Valese AC, Molognoni L, de Souza NC, de Sa Ploencio LA, Costa ACO, Barreto F, Daguer H (2017) Development, validation and different approaches for the measurement uncertainty of a multi-class veterinary drugs residues LC-MS method for feeds. Journal of chromatography B. Analytical technologies in the biomedical and Life Sci 1053:48–59. https://doi.org/10.1016/j.jchromb.2017.03.026
Wang J, Fan X, Liu Y, Du Z, Feng Y, Jia L, Zhang J (2017) Extraction optimization of sixteen cephalosporins in milk by filtered solid phase extraction and ultra high pressure liquid chromatography coupled to tandem mass spectrometry. Anal Methods 2017:1282–1289
Wittenberg JB, Simon KA, Wong JW (2017) Targeted multiresidue analysis of veterinary drugs in milk-based powders using liquid chromatography-tandem mass spectrometry (LC-MS/MS). J Agric Food Chem 65:7288–7293. https://doi.org/10.1021/acs.jafc.6b05263
Yoshikawa S, Nagano C, Kanda M, Hayashi H, Matsushima Y, Nakajima T, Tsuruoka Y, Nagata M, Koike H, Sekimura K, Hashimoto T, Takano I, Shindo T (2017) Simultaneous determination of multi-class veterinary drugs in chicken processed foods and muscle using solid-supported liquid extraction clean-up. Journal of chromatography B. Analytical technologies in the biomedical and Life Sci 1057:15–23. https://doi.org/10.1016/j.jchromb.2017.04.041
Zhang D, Park ZY, Park JA, Kim SK, Jeong D, Cho SH, Shim JH, Kim JS, Abd El-Aty AM, Shin HC (2016) A combined liquid chromatography-triple-quadrupole mass spectrometry method for the residual detection of veterinary drugs in porcine muscle, milk, and eggs. Environ Monit Assess 188:348. https://doi.org/10.1007/s10661-016-5344-x
Zhang Z, Wu Y, Li X, Wang Y, Li H, Fu Q, Shan Y, Liu T, Xia X (2017) Multi-class method for the determination of nitroimidazoles, nitrofurans, and chloramphenicol in chicken muscle and egg by dispersive-solid phase extraction and ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem 217:182–190. https://doi.org/10.1016/j.foodchem.2016.08.097
Zhao Y, Yue T, Tao T, Wang X, Huang L, Xie S, Pan Y, Peng D, Chen D (2017a) Simultaneous determination of quinoxalines in animal feeds by a modified QuEChERS method with MWCNTs as the sorbent followed by high-performance liquid chromatography. Food Anal Methods 2017:2085–2091
Zhao H, Zulkoski J, Mastovska K (2017b) Development and validation of a multiclass, multiresidue method for veterinary drug analysis in infant formula and related ingredients using UHPLC-MS/MS. J Agric Food Chem 65:7268–7287. https://doi.org/10.1021/acs.jafc.7b00271
Acknowledgements
The authors wish to thank Dr. Perry G. Wang and Prof. Shulin Peng for their scientific advices. Funding for this work was financially supported by the National Key Research and Development Program of China (No. 2017YFC1601600).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Yanxia Zhang declares no conflict of interest. Xia Xue declares no conflict of interest. Shufang Su declares no conflict of interest. Zhimou Guo declares no conflict of interest. Jun Wang declares no conflict of interest. Lisheng Ding declares no conflict of interest. Yanming Liu declares no conflict of interest. Jianhua Zhu declares no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Informed consent is not applicable for this study.
Electronic supplementary material
ESM 1
(DOC 2688 kb)
Rights and permissions
About this article

Cite this article
Zhang, Y., Xue, X., Su, S. et al. A Multi-Class, Multi-Residue Method for Detection of Veterinary Drugs in Multiple Meat Using a Pass-Through Cleanup SPE Technique and UPLC-MS/MS Analysis. Food Anal. Methods 11, 2865–2884 (2018). https://doi.org/10.1007/s12161-018-1244-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1244-8