
Abstract
Xanthine oxidase (XOD) catalyzes the metabolism of hypoxanthine and xanthine to uric acid, the overproduction of which could cause hyperuricemia, a risk factor for gout. Inhibition of XOD is a major treatment for gout, and biflavonoids have been found to act as XOD-inhibitory compounds. In this study, ultrafiltration liquid chromatography with photodiode-array detection coupled to electrospray-ionization tandem mass spectrometry (UF-LC-PDA–ESI-MS) was used to screen and identify XOD inhibitors from S. tamariscina. High-performance counter-current chromatography (HPCCC) was used to separate and isolate the active constituents of these XOD inhibitors. Furthermore, ultrahigh-performance liquid chromatography (UPLC) and triple-quadrupole mass spectrometry (TQ-MS) was used to determine the XOD-inhibitory activity of the obtained XOD inhibitors, and enzyme kinetics was performed with Lineweaver–Burk (LB) plots using xanthine as the substrate. As a result, two compounds in S. tamariscina were screened as XOD inhibitors: 65.31 mg amentoflavone and 0.76 mg robustaflavone were isolated from approximately 2.5 g S. tamariscina by use of HPCCC. The purities of the two compounds obtained were over 98 % and 95 %, respectively, as determined by high-performance liquid chromatography (HPLC). Lineweaver–Burk plot analysis indicated that amentoflavone and robustaflavone were non-competitive inhibitors of XOD, and the IC 50 values of amentoflavone and robustaflavone for XOD inhibition were 16.26 μg mL−1 (30.22 μmol L−1) and 11.98 μg mL−1 (22.27 μmol L−1), respectively. The IC 50 value of allopurinol, used as the standard, was 7.49 μg mL−1 (46.23 μmol L−1). The results reveal that the method for systematic screening, identification, and isolation of bioactive components in S. tamariscina and for detecting their inhibitory activity using ultrafiltration LC–ESI-MS, HPCCC, and UPLC–TQ-MS is feasible and efficient, and could be expected to extend to screening and separation of other enzyme inhibitors.

ᅟ
Similar content being viewed by others
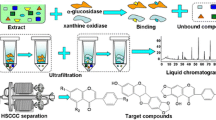
Avoid common mistakes on your manuscript.
Introduction
Hyperuricemia (excess uric acid in blood plasma) and its complications are commonly accepted as the primary risk factor for chronic gout, and also believed to be a risk factor of cardiovascular disease and metabolic syndrome [1, 2]. Previous reports revealed that a high intake of purine-rich foods and drinks could increase uric-acid levels [3–5]. In purine metabolism, xanthine and hypoxanthine are oxidized to uric acid by xanthine oxidase (XOD), which is strongly expressed in the liver and intestine [6]. Overproduction of uric acid can lead to painful inflammation, caused by deposition of uric acid in the joints [7]. Consequently, XOD is believed to be an important enzyme causing gout and many other diseases related to hyperuricemia.
XOD inhibitors can block the synthesis of uric acid and reduce the generation of superoxide anions [8]. Allopurinol, a competitive inhibitor of XOD, has been widely used for clinical applications in the treatment of gout and hyperuricemia. However, it has potentially serious side effects including skin rash and hypersensitivity syndrome [6, 9]. Therefore, it is necessary to find alternative, safe inhibitors from natural sources. Biflavonoids, which are flavonoid dimers connected with a C–C or a C–O–C bond, as a class of flavonoids have been found to act as XOD-inhibitory compounds [10]. In addition, sometimes the bioactivity of a biflavonoid is greater than that of the corresponding monomers [11]. In searching for inhibitors from natural products, much effort is being made to establish sensitive and simple methods for screening the XOD inhibitors [12]. In recent years, mass-spectrometry-based methods have been successfully used as a sensitive, rapid, and accurate tool for screening and analysis of enzyme inhibitors [13, 14]. Ultrafiltration-LC–ESI-MS, being an effective strategy to screen and identify the active compounds from complex mixtures, has been used to separate biologically active compounds from botanical extracts. The ultrafiltration facilitates the separation of biflavonoid–receptor complexes from unbound compounds, and the LC–ESI-MS step identifies the biflavonoids. Spectrophotometry methods have been often reported for evaluating the XOD-inhibitory activity of compounds or mixtures [10, 15]. In our previous study, UPLC–TQ-MS was developed to determine the XOD-inhibitory activity and superoxide-scavenging activity of some herbal extracts by accurately quantifying the amount of xanthine and WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium sodium salt) [8]. Compared with the commonly used spectroscopy method, the UPLC–TQ-MS method is not only sensitive, accurate, and fast, but also able to eliminate false-positive and false-negative results. With minor modification, UPLC–TQ-MS was used in this study to evaluate the XOD-inhibitory activity of compounds by directly quantifying uric acid of enzymatic reaction.
Selaginella tamariscina (Beauv) Spring belongs to the family Selaginellaceae, and is a traditional medicine in China. Investigation of the phytochemical constituents of S. tamariscina revealed that the major constituents are flavonoids and that it is an especially rich source of biflavonoids, which have a variety of biological activity including cytotoxic, antiallergenic, anti-inflammatory, antiviral, antioxidant, inhibiting nuclear factor-κB activation, hypoglycemic, hypolipidemic, and more [16–19]. However, it is yet to be fully exploited, because most of the biflavonoids had not yet been found in nature or else had not been synthesized [20]. High-performance counter-current chromatography (HPCCC), a continuous liquid–liquid partition chromatography without a support matrix, has become established as an effective technique for separating bioactive compounds from plant extracts [21–23]. HPLC–ESI-MS was usually used to identify the active ingredients of complex mixtures, and a previous report had analyzed the compounds in S. tamariscina in positive mode [24]. However, it was reported that there was higher sensitivity for the flavonoid signal in negative-ion mode than positive, and our preliminary LC–ESI-MS studies confirmed that negative-ion mode ESI-MS flavonoid data provided more structural information.
In this work, ultrafiltration LC-PDA–ESI-MS was used for rapid screening and identification of the two bioactive compounds from extract of S. tamariscina, and HPCCC was used to separate the XOD inhibitors from the crude extract. Biflavonoids obtained were of over 95 % purity. Then UPLC–TQ-MS was used to determine the XOD-inhibitory activity of the obtained XOD inhibitors. This might be the first report on this system for screening, isolation, and activity detection of XOD inhibitors from S. tamariscina.
Experimental
Chemicals and reagents
Xanthine oxidase (E.C. 1.17.3.2) from bovine milk and trizma base were obtained from Sigma Aldrich (St. Louis, MO, USA); xanthine, uric acid, and allopurinol were acquired from Alfa Aesar Company and used directly; 5-fluorouracil was obtained from Aladdin industrial Co., Ltd. (Shanghai, China); amentoflavone was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). The ultrafiltration chamber used was Microcon YM-100 (Millipore, Bedford, MA), and the molecular weight cutoff was 100,000 Da. The solvents for HPLC analysis were of LC grade and purchased from Fisher Chemicals (Pittsburg, PA, USA). The other solvents used for the preparation of crude extract of S. tamariscina and for fractionation, ultrafiltration, and isolation were all of analytical grade, and were obtained from Beijing Chemical Engineering Company (Beijing, China). All aqueous solutions were prepared with pure water produced by a Milli-Q water (18.2 MΩ) system (Millipore, Bedford, MA, USA). The dried material of S. tamariscina was taken from Hebei Kangpai Medicinal Store (Hebei, China), and was identified by Professor Shumin Wang, Chang Chun University of Chinese Medicine, Jilin Province.
Preparation of samples
Dry S. tamariscina was ground before being extracted, and 5.002 g powder was extracted three times by reflux with 50 mL 70 % ethanol for 0.5 h. After filtration, the combined extracts were concentrated under reduced pressure and re-dissolved in 10 mL methanol (crude-extract concentration 500 mg mL−1). The extract was stored at 4 °C before ultrafiltration-LC-PDA–ESI-MS and HPCCC separation.
Ultrafiltration HPLC-PDA–ESI-MS
Ultrafiltration LC-PDA–ESI-MS screening based on MS includes three steps: incubation, ultrafiltration, and analysis. First 20 μL xanthine oxidase (E.C. 1.17.3.2) (0.5 U mL−1, dissolved in 50 mmol L−1 Tris–HCl buffer, pH 8.7) and 20 μL S. tamariscina extract (250 mg mL−1, dissolved in methanol) were added to 100 μL 50 mmol L−1 Tris–HCI buffer (pH 8.7), and then the mixture was incubated at 37 °C for 40 min. Next, the mixture was ultrafiltrated using a Sigma 1-14 centrifuge (Sigma, Germany) with ultramembrane filter (Microcon YM-100, Millipore, Bedford, MA) at 10,000 rpm for 10 min at room temperature. The filter was washed three times by centrifugation with 100 μL aliquots Tris–HCI buffer (pH 8.7) to separate the unbound low-mass molecules. The bound biflavonoids were released by adding 100 μL methanol–water solution (50:50, v/v) (pH 3.1), followed by centrifugation at 10,000 rpm for 10 min, which was repeated three times. The control experiment without enzyme was also performed before each screening experiment. The ultrafiltrate was injected directly into the HPLC–ESI-MS system for analysis.
The mass-spectrometry experiments were performed on a Thermo Scientific LCQ Fleet mass spectrometer with an electrospray-ionization (ESI) interface (ThermoFisher, USA). Chromatographic analysis was performed in an Agilent Extend-C18 column (150 × 4.6 mm, 5 μm). The binary mobile phase consisted of methanol (solvent A) and water containing 0.2 % acetic acid (solvent B). The sample injection volume was 5 μL. The flow was maintained at 0.5 mL min−1 with a total run time of 25 min. The column temperature was 30 °C. The system was run with the gradient program: 0–2 min: 50 % B; 2–12 min: 50 % B to 25 % B; 12–22 min: 25 % B to 10 % B; 22–25 min: 10 % B to 50 % B. The detection wavelength was 337 nm. The sheath-gas and auxiliary-flow rates were set at 40 and 10 (arbitrary unit), respectively. The capillary voltage was set at 4.5 kV, and its temperature was maintained at 350 °C. The isolation width for ESI-MSn was 2.0 Da, and a source-CID experiment was performed to obtain the detailed structural information; the collision energy was 20–25 %. Full scan of ions ranged from 100 to 1500 molecular weights in the negative-ion mode.
HPCCC-separation procedure
The HPCCC was performed in a Midi HPCCC (Dynamic Extractions, England). The apparatus had a coil of 22 mL. The HPCCC system was equipped with two check pumps (Smartline Pump 100, KNAUER, Germany), a 2500 sensitive UV detector (KNAUER, Germany), and a sample-injection valve with a 1 mL sample loop for analytical separation.
A mixture of hexane–ethyl acetate–methanol–water (2.2:2.8:2:3, v/v) was shaken vigorously in a separatory funnel and separated at room temperature (<30 min). The two phases were used in the HPCCC after the system reached equilibrium. The 22 mL coil was used, and the apparatus was set in the reverse-phase mode. The entire coiled column was first filled with the upper phase, which served as the stationary phase, and then the lower layer (as the mobile phase) was pumped into the head-end of the column at a flow of 1.0 mL min−1. The rotation speed was set at 1200 rpm. The temperature was held at 30 °C, and 0.5 mL 500 mg mL−1 extract was concentrated to dryness under reduced pressure, dissolved in 2 mL hexane–ethyl acetate–methanol–water mixture (2.2:2.8:2:3, v/v), and then continuously loaded into the injection valve after the system reached hydrodynamic equilibrium. The retention of stationary phase in the coil was approximately 77.3 %. The analysis time was recorded to be the same as the sample-injection time. The effluent from the outlet of the column was continuously monitored by a UV detector at 337 nm and collected into the test tubes, with a collecting time of 1 min for each tube. Those fractions from the HPCCC with one single peak the same as that obtained by use of HPLC were combined.
HPLC analysis
An Agilent 1100 HPLC equipped with a DAD detector was used for the analysis of biflavonoids of S. tamariscina. The separation was performed using an Agilent Extend-C18 column (250 × 4.6 mm, 5 μm) with a C18 guard column. The binary mobile phase consisted of methanol (solvent A) and water containing 0.2 % acetic acid (solvent B). The sample injection volume was 10 μL. The flow was maintained at 1.0 mL min−1 with a total run time of 25 min. The column temperature was 30 °C. The system was run with a gradient program, 0–2 min: 50 % B; 2–12 min: 50 % B to 25 % B; 12–22 min: 25 % B to 10 % B; 22–25 min: 10 % B to 50 % B. The detection wavelength was 337 nm.
Determination of XOD-inhibitory activity by UPLC–TQ-MS
The XOD-inhibitory activity was determined by the methods used by Shu Liu et al. (2012) with minor modifications. The formation of uric acid was determined directly by measuring the content of uric acid using the UPLC–TQ-MS method. The XOD reaction was performed in a total volume of 200 μL containing 0.05 mol L−1 Tris–HCl (pH 8.7) and 20 mU mL−1 XOD, and initiated by adding xanthine at an appropriate concentration; it was then left for 5 min at 37 °C, and then halted by adding 0.8 mL methanol. Then 10 μL 5-FU was added as the internal standard (I.S.) to give a final concentration of 0.1 μg mL−1. A series of concentrations of standards (1, 5, 10, 50, 100, 150, and 200 μg μL−1) were prepared for determining the calibration curve of uric acid. The assay was performed in triplicate and IC 50 values were calculated from the percentage inhibition.
Aliquots (5 μL each) of each enzymatic reaction solution were analyzed using an Acquity UPLC system (Waters, Milford, MA, USA) with a thermostated autosampler at 4 °C. The separation was performed on an Acquity UPLC BEH C18 column (50 mm × 2.1 mm i.d., 1.7 μm; Waters, Milford, MA, USA) maintained at 25 °C. Mobile-phase component A was methanol, component B was water with 0.1 % formic acid, and the flow was set at 0.2 mL min−1. Isocratic elution was used at 10 % A and 90 % B for 2 min. The injection volume was 5 μL, and partial-loop mode was used for sample injection. Mass-spectrometry detection was performed on a Xevo TQ mass spectrometer (Waters, Milford, MA, USA) with an electrospray-ionization (ESI) source. The ESI source was operated in negative-ionization mode. Quantification analysis was performed in multiple reaction monitoring (MRM). The following settings were used for MRM: capillary voltage 2.0 kV; source temperature 350 °C; cone gas flow 50 L h−1; desolvation gas flow 600 L h−1. The cone and collision energy are presented in Table 1.
Enzyme-inhibition mechanism
To determine the mode of inhibition, kinetic studies were performed in the absence and presence of allopurinol and individual constituents of S. tamariscina with varying concentrations of xanthine as substrate. Different concentrations of allopurinol (up to 5.0 μg mL−1) and isolated compounds (up to 12.5 μg mL−1) of S. tamariscina were pre-incubated with xanthine for approximately 2 min. Afterwards the reaction was started by adding xanthine oxidase. Kinetic measurements were performed using at least three different inhibitor concentrations and two or three different xanthine concentrations (20, 50, 100 μmol L−1). The data obtained from the kinetic studies were transformed into Lineweaver–Burk (LB) plots.
Results and discussion
Screening of XOD inhibitors by ultrafiltration LC-PDA–ESI-MS
The ultrafiltration-LC-PDA–ESI-MS method can be used for rapid screening of bioactive compounds from complex mixtures, particularly for natural products with a minimum of sample preparation [25]. When the extract of S. tamariscina was incubated with XOD, active ligands could bind with XOD, and then unbound and small molecules could be separated from the ligand–XOD complexes or XOD by use of an ultrafiltration membrane. In this manner, the ultrafiltration chamber functions as a solution-phase extraction device, retaining ligands that can tightly bind to a receptor, whereas unbound compounds will be washed away. Then the ligand–receptor complex is disrupted by addition of organic solvent or by changing pH value, and the released ligands can be identified by LC-PDA–ESI-MS analysis. On the basis of this, the degree of activity of three compounds could be analyzed directly by comparing the HPLC peak areas of the control (extract without XOD) and the sample (extract + XOD) experiment. Degree of binding activity = (As − Ac)/Ac, where As and Ac are the areas of each compound from the sample experiment and control experiment, respectively. The results revealed that compound 2 had the greatest degree of binding activity (80.6 %), followed by compound 1 (59.6 %), and that compound 3 (−54.6 %) did not bind with XOD, which might be caused by the structures of the compounds. Figure 1 shows the ultrafiltration-HPLC analysis of S. tamariscina.

LC-PDA chromatograms of the interaction of the extract of S. tamariscina XOD ((a), extract with XOD) and of the control experiment ((b), extract without XOD). Experimental conditions: column, Agilent Extend-C18 column (250 × 4.6 mm, 5 μm) and a C18 guard column; mobile phase, methanol (phase component A) and water containing 0.2 % acetic acid (phase component B); gradient program, 0–2 min: 50 % B; 2–12 min: 50 % B to 25 % B; 12–22 min: 25 % B to 10 % B; 22–25 min: 10 % B to 50 % B; fl B, 1.0 mL min−1; column temperature 30 °C; detection wavelength 337 nm
Identification of XOD inhibitors by LC-PDA–ESI-MS
To identify the biflavonoids screened from S. tamariscina, LC-PDA–ESI-MS analysis was performed. From the LC-PDA–ESI-MS chromatogram of the crude extract from S. tamariscina (Fig. 1), it can be seen that a good separation was achieved within 25 min. Three major peaks were separated and detected with retention times of 15.64, 19.23, and 21.72 min. The retention times, values of the mass-to-charge ratios (m/z) for the [M−H]− ions, and the CID fragments in the negative-ion mass spectra (NI-MS) of compounds 1 and 2 are listed in Table 2. The molecular weights of compounds 1 and 2 were confirmed to be 538. By comparing retention time (t R), UV spectra, and ESI-MS fragmentation patterns with those of the corresponding reference compound and the literature, the compounds were identified; the detailed compound identification is discussed below.
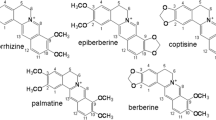
The LC-PDA–ESI-MS data for peak 1 is shown in Table 2. From this table we can see that the deprotonated molecular ion [M−H]− at m/z 537 and its CID spectrum of the ions at m/z 443, 417, 375, and 331 could be found by NI-MS. On the basis of the LC peaks, through comparing the retention time with that of the standard, detection of the molecular ions and the fragment ions of the molecular ion obtained by using CID experiments, and referring to the literature, the compound corresponding to peak 1 was identified as amentoflavone [23, 24]. Amentoflavone is a biflavonoid, which consists of two flavonoids connected by IC3′–IIC8″. The first fragmentation route might be that two γ-H of the A-ring in flavonoid part I underwent McLafferty rearrangement to lose C3O2, followed by the cleavage of the C-ring in flavonoid part I at positions 0 and 4 (0,4A), and loss of C2H2 to give the ion at m/z 443. The second fragmentation route involved the cleavage of the C-ring in flavonoid part II at positions 0 and 2 (0,2A), and the loss of C7H5O2 to produce a fragment ion at m/z 417. A low-abundance fragment ion at m/z 331 was also observed, which could be obtained from two γ-H of the part I A-ring in m/z 417 through McLafferty rearrangement, loss of C3O2, and loss of a H2O molecule via intramolecular dehydration between −OH on the IB4′ and IIA7″. The third fragmentation route produced a fragment ion at m/z 375, which corresponded to the loss of a C9H6O3 unit from the cleavage of the C-ring in flavonoid part II at positions 0 and 4 in the parent ion m/z 537, which is similar to the biflavonoids mass spectra of a previous report by Shi et al. [26, 27]. Usually, fragmentation routes involving the cleavage of the C-ring in flavonoid part II at positions 0 and 2 and 0 and 4 were the primary pathways of amentoflavone-type biflavonoids in our experiment. The fragmentation pathways of the amentoflavone (peak 1) are summarized in Scheme 1a.

Proposed fragmentation pathway of amentoflavone (a) and robustaflavone (b)
The LC–MS of peak 2 also gave m/z 537 as the deprotonated molecular ion [M−H]−, which confirmed the molecular mass also to be 538. Further experiments in the form of CID of the m/z 537 ([M−H]−) produced three main fragment ions at m/z 469, 387, and 305 (Table 2). On the basis of the fragmentation pattern and molecular weight, the compound corresponding to peak 2 was identified as robustaflavone [23, 24]. Two flavonoids of robustaflavone are linked by IC3′–IIC6″; for these one fragmentation route, involving two γ-H of the A-ring in flavonoid part I losing C3O2 through McLafferty rearrangement, generated the fragment ions at m/z 469. The fragment ions at m/z 387 corresponded to the loss of a C10H6O3 unit from the cleavage of the C-ring in flavonoid part II at positions 0 and 3 in the parent ion m/z 537. The ESI-MS fragmentation routes of robustaflavone-type biflavonoids were different from those of amentoflavone-type biflavonoids. Two flavonoid parts in robustaflavone are linked via IC3′–IIC8″, from which it is easy to lose a H2O molecule via intramolecular dehydration between −OH on the IB4′ and IIB5″ (IIB7″) because of the spatial configuration; thus the cleavage of the C-ring in flavonoid part II at positions 0 and 2 resulted in loss of C7H5O2. As two γ-H of the A-ring in flavonoid part I underwent McLafferty rearrangement to lose C3O2, the IC-ring simultaneously cracked at positions 0 and 4 to lose C2H2; this generated the fragment ion at m/z 305, but with relatively low abundance. The fragmentation pathways of robustaflavone (peak 2) are summarized in Scheme 1b.
HPCCC separation and analysis of compounds
A successful HPCCC separation depends on a suitable two-phase solvent system that provides an apposite K of approximately 1 for target compounds. Such a biphasic system should also yield a reasonably short settling time. In our experiment the crude extract mainly contained biflavonoids, and their structures are very similar, so the separation was not an easy task. We selected five series of solvent systems on the basis of the solubility of the target compounds. The K values of these compounds were calculated from the HPLC data as aforementioned, and are summarized in Table 3.
The first two systems, containing hexane–ethyl acetate–ethanol–water (2:3:0.1:5, v/v) and hexane–ethyl acetate–ethanol–water (1:1:0.2:2, v/v), had poor separation performances because the settling times were quite short and the K values were quite low for the two compounds, causing the eluted peaks to be close together and too near the front. On modifying the ratios to ethyl acetate–ethanol–water (1:0.2:1, v/v), the modified system obtained long settling times and higher K values for compound 2. When ethanol was replaced by methanol and the solvents were set at the ratios hexane–ethyl acetate–methanol–water (2.2:2.8:2:3, v/v) and hexane–ethyl acetate–methanol–water (2:3:1:5, v/v) the settling time improved significantly, but separation performance for the latter solvent system was poor because of the high K value of compound 2 (see Table 3). However, the system of hexane–ethyl acetate–methanol–water (2.2:2.8:2:3, v/v) provided good separation of the compounds. The K values for compounds 1 and 2 in the former systems were 0.56 and 1.12, respectively. When this solvent system was used in HPCCC separation with a flow of 1 mL min−1, two XOD inhibitors were separated within 90 min. The HPCCC separation is shown in Fig. 2.

HPCCC chromatograms of crude extract of S. tamariscina (1, amentoflavone; 2, robustaflavone). Experimental conditions: two-phase solvent system, hexane–ethyl acetate–methanol–water (2.2:2.8:2:3, v/v); the other HPCCC conditions were as follows: stationary phase, upper phase; mobile phase, lower phase; rotation speed, 1200 rpm; injection volume, 2 mL; detection wavelength, 377 nm; flow, 1.0 mL min−1; separation temperature, 30 °C
The crude extract of S. tamariscina and the fractions corresponding to each peak isolated by HPCCC were analyzed by HPLC, and the results are given in Fig. 3. The XOD inhibitors 1 and 2 were sequentially separated and detected. The purities of the two compounds (65.31 mg amentoflavone and 0.76 mg robustaflavone) as determined by HPLC were 98 % and 95 %, respectively.

HPLC profiles of the extract of S. tamariscina (a) and of compounds 1 and 2 in HPCCC separation (b, c). Experimental conditions: column, Agilent Extend-C18 column (250 × 4.6 mm, 5 μm) and a C18 guard column; mobile phase, methanol (phase component A) and water containing 0.2 % acetic acid (phase component B); gradient program, 0–5 min: 70 % B to 40 % B; 5–10 min: 40 % B to 20 % B; 10–18 min: 20 % B to 10 % B; 18–20 min: 10 % B to 70 % B; flow, 1.0 L min−1; column temperature 30 °C; detection wavelength 337 nm
To further investigate the structures of two compounds, an ESI-MSn experiment was performed. Compounds related to peaks 1 and 2 in HPCCC both had intense deprotonated molecular ions [M−H]− at m/z 537 in the negative-ion mode. Comparing the fragment ions with those of the corresponding reference compound and the literature, peak 1 and 2 were assigned to be amentoflavone and robustaflavone, respectively.
Optimization of UPLC–TQ-MS
Ultrafiltration-LC-PDA–ESI-MS screening of the plant extract revealed that amentoflavone and robustaflavone could bind with XOD. Xanthine is oxidized to uric acid by xanthine oxidase, and inhibition of xanthine oxidase resulted in reduced production of uric acid, which was measured by UPLC–TQ-MS. The uric acid and 5-FU were detected by direct full-scan mass-spectrometry in both positive and negative-ionization modes. The results revealed that the analyte signals obtained from the negative-ion ESI mode had higher sensitivity and intensity, permitting quantitative measurement. The negative-ion ESI-MS of uric acid and 5-FU produced abundant deprotonated molecular ions [M−H]− at m/z 166.5 and 129.2, respectively. To develop a sensitive mass-spectrometry method for quantification of uric acid and 5-FU, we should select at least two precursor–product ion pairs for each analyte. On the basis of the analytic results, the most sensitive ion pairs for each analyte were selected for the quantitative ion pairs: the precursor–product ion pairs 166.5–124.09 for uric acid and 129.2–86.03 for 5-FU. The effect of different mobile phases on the separation of uric acid and 5-FU was also investigated. When water and acetonitrile were used as mobile phase, uric acid and 5-FU were not well separated, and the peaks had tailing when water and methanol were used as the mobile phase. However, a good separation of the two compounds was achieved when water with 0.1 % formic acid and methanol were used as mobile phase.
XOD-inhibitory activity and mode of the inhibitors
The XOD-inhibitory capacity of obtained compounds and allopurinol was evaluated by UPLC–TQ-MS. The amount of uric acid created was calculated as the calibration curve of standard regression equations: y = 0.1184x + 0.4096. The standard curves had good linearity, with coefficient of correlation (r) of 0.999, and the linear range was 1–200 μg μL−1. XOD inhibition was calculated as: ((A − B)/A) × 100 %, where A is the uric-acid concentration of the sample without inhibitor and B is the uric-acid concentration of the sample with inhibitor.
The evaluation results are shown in Table 4. In the XOD-inhibitory activity experiment, amentoflavone and robustaflavone caused a dose-dependent inhibition of xanthine-oxidase-enzyme activity. The known synthetic XOD-inhibitor allopurinol had the IC 50 value 7.49 ± 0.14 μg mL−1. Of the two obtained biflavonoids, robustaflavone (IC 50 = 11.98 ± 0.12 μg mL−1) had a higher XO-inhibitory capacity than amentoflavone (IC 50 = 16.26 ± 0.15 μg mL−1); this result is similar to that of an earlier report [28]. In the ultrafiltration LC-PDA–ESI-MS, the degree of binding activity of the two inhibitors was 80.6 % for robustaflavone and 59.6 % for amentoflavone. Therefore, the results of the two methods revealed that the binding activity and inhibitory activity of robustaflavone was greater than amentoflavone. This distinction might be caused by their structure (Scheme 1): both are dimers of apigenin but the two flavonoid parts of robustaflavone are linked via IC3′–IIC6″, whereas the two flavonoid parts of amentoflavone are linked via IC3′–IIC8″. The former link may be responsible for the higher XOD-inhibitory activity.
Kinetic studies were performed and an LB plot was drawn to determine the mode of XO inhibition of the two biflavonoids and allopurinol (see Electronic Supplementary Material (ESM) Fig. S1, Fig. S2, Fig. S3). In the LB plot robustaflavone and amentoflavone revealed a linear non-competitive inhibitory activity, whereas allopurinol had a competitive XOD-inhibitory activity; this is in agreement with an earlier report [15]. According to previous reports of structure–function correlation for XOD inhibition, hydroxyl groups at C5 and C7 positions and CO at the C4 position in flavonoids facilitate XOD inhibition [15], and robustaflavone and amentoflavone meet these conditions. Their high molecular weight (Mol. Wt. 538), higher number of hydroxyl groups (six), and non-planarity could be attributed to the non-competitive nature of XOD inhibition.
Conclusions
Two biflavonoids, amentoflavone and robustaflavone, with XOD-inhibiting activity were successively isolated from S. tamariscina by HPCCC methods under the guidance of ultrafiltration-based biflavonoid screening, and then the XOD-inhibitory activity of the two obtained compounds and of allopurinol (a complex XOD inhibitor) was determined by UPLC–TQ-MS. The ultrafiltration-LC-PDA–ESI-MS method is an effective tool for screening and identifying active compounds from S. tamariscina, the analysis could be finished within 25 min, and this may be the first time the fragmentation pathways in negative-ion mode for the two biflavonoids have been discussed in detail. HPCCC was successfully used for the isolation and purification of the XOD inhibitors: two biflavonoids, 65.31 mg amentoflavone and 0.76 mg robustaflavone, were obtained from approximately 2.5 g S. tamariscina, and their purities were over 98 % and 95 %, respectively. Furthermore, from the results of the Lineweaver–Burk plots study, both amentoflavone and robustaflavone were non-competitive XO inhibitors, and their IC 50 was 16.26 ± 0.12 μg mL−1 and 11.98 ± 0.03 μg mL−1, respectively. Therefore, the method described in this research is a rapid and feasible means for systematic screening, purification, and evaluation of the active compounds of S. tamariscina and other crude extracts.
References
Dawson J, Quinn T, Walters M (2007) Uric acid reduction: a new paradigm in the management of cardiovascular risk. Curr Med Chem 14:1879–1886
Lin KC, Lin HY, Chou P (2000) The interaction between uric acid level and other risk factors on the development of gout among asymptomatic hyperuricemic men in a prospective study. J Rheumatol 27:1501–1505
Gao X, Qi L, Qiao N, Choi HK, Curhan G, Tucker KL, Ascherio A (2007) Intake of added sugar and sugar-sweetened drink and serum uric acid concentration in US men and women. Hypertension 50:306–312
Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera-Acosta J, Patel JM, Johnson RJ (2006) A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol 290:625–631
Kondo M, Hirano Y, Nishio M, Furuya Y, Nakamura H, Watanabe T (2013) Xanthine oxidase inhibitory activity and hypouricemic effect of aspalathin from unfermented rooibos. J Food Sci 78:1935–1939
Borges F, Fernandes E, Roleira F (2002) Progress towards the discovery of xanthine oxidase inhibitors. Curr Med Chem 9:195–217
Harris MD, Siegel LB, Alloway JA (1999) Gout and hyperuricemia. Am Fam Physician 59:925–934
Liu S, Xing JP, Zheng Z, Song FR, Liu ZQ, Liu SY (2012) Ultrahigh performance liquid chromatography–triple quadrupole mass spectrometry inhibitors fishing assay: a novel method for simultaneously screening of xanthine oxidase inhibitor and superoxide anion scavenger in a single analysis. Anal Chim Acta 715:64–70
Tsai TF, Yeh TY (2010) Allopurinol in dermatology. Am J Clin Dermatol 11:225–232
Arimboor R, Rangan M, Aravind SG, Arumughan C (2011) Tetrahydroamentoflavone (THA) from Semecarpus anacardium as a potent inhibitor of xanthine oxidase. J Ethnopharmacol 133:1117–1120
Kim HP, Park H, Son KH, Chang HW, Kang SS (2008) Biochemical pharmacology of biflavonoids: implications for anti-inflammatory action. Pharm Res 31:265–273
Li DQ, Li SP, Zhao J (2014) Screening of xanthine oxidase inhibitors in complex mixtures using online HPLC coupled with postcolumn fluorescencebased biochemical detection. J Sep Sci 37:338–344
Zhou H, Xing JP, Liu S, Song FR, Cai ZW, Pi ZF, Liu ZQ, Liu SY (2012) Screening and determination for potential xanthine oxidase inhibitors from leaves of Acanthopanax senticosus harms by using UF-LC/MS and ESI-MSn. Phytochem Anal 24:315–323
Yang ZZ, Zhang YF, Sun LJ, Wang Y, Gao XM, Cheng YY (2012) An ultrafiltration high-performance liquid chromatography coupled with diode array detector and mass spectrometry approach for screening and characterizing tyrosinase inhibitors from mulberry leaves. Anal Chim Acta 719:87–95
Lin CM, Chen CS, Chen CT, Liang YC, Lin JK (2002) Molecular modeling of flavonoids that inhibits xanthine oxidase. Biochem Biophys Res Commun 294:167–172
Mahjoub MA, Ammar S, Edziri H, Mighri N, Bouraoui A, Mighri Z (2010)Anti-inflammatory and antioxidant activities of some extracts and pure natural products isolated from Rhus tripartitum (Ucria). Med Chem Res 19:271–282
Adaramoye OA, Medeiros IAJ (2009) Endothelium independent vasodilation induced by kolaviron, a biflavonoid complex from Garcinia kola seeds, in rat superior mesenteric arteries. Smooth Muscle Res 45:39–53
Woo ER, Pokharel YR, Yang JW, Lee SY (2006) Inhibition of nuclear factor-k B activation by 2′,8″-biapigenin. Biol Pharm Bull 29:976–980
Zheng XK, Li YJ, Zhang L, Feng WS, Zhang X (2011) Antihyperglycemic activity of Selaginella tamariscina (Beauv.) Spring. J Ethnopharmacol 133:531–537
Rahman M, Riaz M, Desai UR (2007) Synthesis of biologically relevant biflavanoids–a review. Chem Biodivers 4:2495–2527
Wang J, Bai HL, Liu CM, Li L (2010) Isolation and purification of ginsenosides from plant extract of Panax quinquefolium L. by high performance centrifugal partition chromatography coupled with evaporative light scattering detection. Chromatographia 71:267–271
Yuan ED, Liu BG, Ning ZX, Chen CG (2009) Preparative separation of flavonoids in Adinandra nitida leaves by high-speed counter-current chromatography and their effects on human epidermal carcinoma cancer cells. Food Chem 115:1158–1163
Yuan Y, Wang BQ, Chen LJ, Luo HD, Fisher D, Sutherland IA, Wei YQ (2008) How to realize the linear scale-up process for rapid purification using high-performance counter-current chromatography. J Chromatogr A 1194:192–198
Zhang YX, Li QY, Yan LL, Shi Y (2011) Structural characterization and identification of biflavones in Selaginella tamariscina by liquid chromatography-diode-arraydetection/electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom 25:2173–2186
Shi SY, Peng MJ, Zhang YP, Peng S (2013) Combination of preparative HPLC and HSCCC methods to separate phosphodiesterase inhibitors from Eucommia ulmoides bark guided by ultrafiltration-based ligand screening. Anal Bioanal Chem 405:4213–4223
Shi SY, Zhou HH, Huang KL (2008) Hyphenated HSCCC-DPPH for rapid preparative isolation and screening of antioxidants from Selaginella moellendorffii. Chromatographia 68:173–178
Romani A, Galardi C, Pinelli P (2002) HPLC quantification of flavonoids and Biflavonoids in Cupressaceae leaves. Chromatographia 56:469–474
Tan WJ, Xu JC, Li L, Chen KL (2009) Bioactive compounds of inhibiting xanthine oxidase from Selaginella labordei. Nat Prod Res 23:393–398
Acknowledgments
This work was supported by the National Natural Science Foundation of China (nos 21175128, 81303280, 31370374, 31373899) and the Natural Science Foundation of Jilin Province (No. 20130413043GH, 20130521013ZH, [2013]253, [2013]254, [2012]224).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 29 kb)
Rights and permissions
About this article

Cite this article
Wang, J., Liu, S., Ma, B. et al. Rapid screening and detection of XOD inhibitors from S. tamariscina by ultrafiltration LC-PDA–ESI-MS combined with HPCCC. Anal Bioanal Chem 406, 7379–7387 (2014). https://doi.org/10.1007/s00216-014-8132-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8132-x