
Abstract
Although glycosyl inositol phosphoryl ceramides (GIPCs) represent the most abundant class of sphingolipids in plants, they still remain poorly characterized in terms of structure and biodiversity. More than 50 years after their discovery, little is known about their subcellular distribution and their exact roles in membrane structure and biological functions. This review is focused on extraction and characterization methods of GIPCs occurring in plants and fungi. Global methods for characterizing ceramide moieties of GIPCs revealed the structures of long-chain bases (LCBs) and fatty acids (FAs): LCBs are dominated by tri-hydroxylated molecules such as monounsaturated and saturated phytosphingosine (t18:1 and t18:0, respectively) in plants and mainly phytosphingosine (t18:0 and t20:0) in fungi; FA are generally 14–26 carbon atoms long in plants and 16–26 carbon atoms long in fungi, these chains being often hydroxylated in position 2. Mass spectrometry plays a pivotal role in the assessment of GIPC diversity and the characterization of their structures. Indeed, it allowed to determine that the core structure of GIPC polar heads in plants is Hex(R1)-HexA-IPC, with R1 being a hydroxyl, an amine, or a N-acetylamine group, whereas the core structure in fungi is Man-IPC. Notably, information gained from tandem mass spectrometry spectra was most useful to describe the huge variety of structures encountered in plants and fungi and reveal GIPCs with yet uncharacterized polar head structures, such as hexose–inositol phosphoceramide in Chondracanthus acicularis and (hexuronic acid)4–inositol phosphoceramide and hexose–(hexuronic acid)3–inositol phosphoceramide in Ulva lactuca.

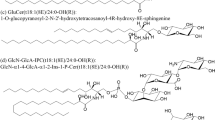
Example of GIPC with its three building blocks (fatty acid, FA; long chain base, LCB; polar head) where R1 could be a hydroxyl, an amine or a N-acetylamine group
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipids having sphingoid backbones, namely sphingolipids, are ubiquitous and essential components of biological membranes. They can represent up to 30 % of total lipids in the plasma membrane of cells [1, 2]. Detected for the first time in 1870 in brain samples, their name comes from the Sphinx, because of their enigmatic and mysterious roles at the time of their discovery. The complex structural diversity of sphingolipids arises from the possible occurrence of three building blocks connected to the sphingoid backbone: a polar head, a fatty acyl chain (FA) linked by an amide bond (to form a ceramide) to a long-chain amino-alcohol called long-chain base (LCB) (Fig. 1). A systematic nomenclature of sphingolipids has been proposed in which d and t designate di- and tri-hydroxylation, respectively, and two colon-delineated numbers designate the numbers of carbon atoms and unsaturation(s).

Structure of sphingolipids: a 4-hydroxysphig-8-enine, b ceramide, c sphingomyelin, d example of GIPC with its three building blocks (FA, LCB, polar head) and where R1 could be a hydroxyl, an amine or a N-acetylamine group
Sphingomyelin, i.e., a ceramide molecule (Fig. 1b) to which a phosphocholine head group is attached (Fig. 1c), is a highly representative structure of animal sphingolipids. However, plants and fungi do not possess these sphingolipids. Instead, they contain complex glycosphingolipids known as glycosyl inositol phosphoryl ceramides (GIPC) [3]. The core structure of GIPCs consists of a ceramide moiety linked to an inositol–glucuronic acid unit via a phosphodiester bond. Many diverse saccharides can be added to this core structure, forming compounds such as Gal-Glc(R1)-GlcA-inositol-1-phosphoceramide in plants or Man-inositol-1-phosphoceramide in fungi as shown in Fig. 1d, [1, 4, 5] (where Gal refers to galactose, GlcA to glucuronic acid and Glc to glucose, with R1 being a hydroxyl, an amine or a N-acetylamine group).
The work of Carter et al. [6] described the first isolation process of GIPCs, also providing insights into the structure of polar heads and ceramide moieties of these plant lipids. However, more than 50 years later, little is known about the diversity of structures encountered for GIPCs from plant and fungal origin, whereas these compounds are believed to be involved in diverse cellular functions such as structuring of membrane microdomains, membrane integrity, response to abiotic stresses and regulation of programmed cell death associated with resistance to pathogens [4].
This situation might be partly attributed to the physico-chemical properties of GIPCs. Indeed, the solubility of GIPCs in usual organic solvents is rather limited due to their large hydrophilic polar head. This property has been a major obstacle for their isolation and characterization. Indeed, most lipid extraction methods relied on phase partition in chloroform/methanol/water mixtures; this method was largely inefficient to extract GIPCs that remained insoluble for the most part, or were lost in the aqueous phase and interphase [3, 7].
Global methods for characterizing ceramide moieties of GIPCs revealed the structures of LCBs and FAs: LCBs are dominated by tri-hydroxylated molecules such as monounsaturated and saturated phytosphingosine (t18:1 and t18:0, respectively) in plants and mainly phytosphingosine (t18:0 and t20:0) in fungi, and FA are generally 14–26 carbon atoms long in plants and 16–26 carbon atoms long in fungi, these chains being often hydroxylated in position 2 [1]. Further, the characterization of polar heads encountered in GIPC structures is still incomplete: the exact nature of glycans and their branching have not yet been completely identified in plants. This situation hampers our understanding of the overall GIPC functions in cell physiology as well as the role of specific GIPC molecular species in peculiar molecular mechanisms.
The purpose of this review is to provide a state-of-the-art description of purification and separation methods of plant and fungal GIPCs, followed by their structure characterization, essentially by means of mass spectrometry. The biodiversity of GIPCs will also be presented, with a compendium of structures encountered in plants and fungi.
Extraction and purification of GIPCs
The first GIPC purification method took advantage of the difference in solubility between GIPCs and glycerolipids at low pH in 70 % ethanol, GIPCs being insoluble in this solvent. Carter et al. [6] were the first ones who described a method dedicated to the purification of phytoglycolipids (the previous designation of GIPCs) from Phaseolus vulgaris (bean) leaves. The extraction was performed by blending the plant leaves in acidic solution and refluxing the residue in aqueous acidic 70 % ethanol for 20 min. This step was followed by several washes (cold acidic ethanol, acetone and diethyl ether). The residue was then dissolved in butanol/acetic acid/water (4/1/5, v/v/v) before purification by ion exchange chromatography (diethyl-amino-ethyl- (DEAE) cellulose column) to eliminate proteins and peptides. A final desalting step was performed by size exclusion chromatography (Sephadex G-25 column). Kaul and Lester [8, 9] have reported on a similar procedure for the isolation of GIPCs from tobacco leaves, except that they raised the pH of the extraction solvent up to 8.5 before reflux, and sample washing was carried out using only acetone. Prior to purification, the residue was dissolved in chloroform/methanol/water (16/16/5, v/v/v). A separation between N-acetyl and amine classes of phytosphingolipids was achieved by means of a Porasil column and a second separation with an acetate form of DE52-cellulose column allowed concentrating each species.
More recently, Markham et al. [7] reported four different extraction methods from frozen plant tissues (Arabidopsis thaliana, tomato and soybean leaves): (1) addition of methanol/chloroform/water (2/1/0.35, v/v/v), extraction with chloroform and back-extraction in the presence of KCl; (2) extraction with 2-propanol and then with chloroform/2-propanol (1/1, v/v); (3) three rounds of extraction with ethanol/water/diethylether/pyridine/ammonia (15/15/5/1/0.018, v/v/v/v/v) at 60 °C; (3) extraction three times with 2-propanol /hexane/water (55/20/25, v/v/v) at 60 °C. The latter solvent combination was preferred for its relatively innocuous character and for the capacity of 2-propanol to inhibit lipases. Moreover, adequate solubilization of GIPCs was observed at 60 °C, a temperature that did not affect their composition. Neutral (ceramide, 2-hydroxy-ceramide, monohexosylceramide) and anionic (GIPC) sphingolipids were separated by normal-phase liquid chromatography. In another study, Markham and Jaworski [10] extracted sphingolipids from frozen A. thaliana leaves with the same 2-propanol/hexane/water solvent mixture at 60 °C, but the extract was dissolved in THF/methanol/water (2/1/2, v/v/v) containing 0.1 % formic acid prior to analysis by liquid chromatography–tandem mass spectrometry (LC-MS/MS).
More recently, we have slightly modified the original protocol from Carter and Koob [11] for GIPC extraction and purification [12]. Protocol optimizations merely consisted in (1) getting rid of proteins and (2) finding a solvent mixture that was ensuring total GIPC solubility and compatibility with MALDI- and electrospray ionization (ESI)-MS experiments. A two-phase partition step with water/butan-1-ol (1:1, v/v) was found to be optimum for GIPC recovery without any significant protein contamination. The extraction residue was further dissolved in THF/methanol/water (4/4/1, v/v/v) containing 0.1 % formic acid before analysis by mass spectrometry.
In most instances, GIPC extraction from fungi relied on similar procedures. Toledo et al. [13] extracted glycosphingolipids from frozen fungi (Aspergillus fumigatus) six times with each of the following solvent mixtures: isopropanol/hexane/water (55/20/25, v/v/v) and chloroform/methanol (2/1, v/v). Then, the residue was partitioned with water/1-butanol (1/1, v/v). The sample dissolved in chloroform/methanol/water (30/60/8, v/v/v) was loaded on top of a DEAE-Sephadex A-25 column (acetate form) to separate acidic glycosphingolipids from neutral ones. GIPCs from A. fumigatus cultures were extracted in a similar way by Simenel et al. [14] using a chloroform/methanol/water (10/10/3, v/v/v) solvent combination. The final separation step of the various GIPC species was achieved by normal phase chromatography (silica 60). Heise et al. [15] extracted GIPC from frozen Cryptococcus neoformans fungi with aqueous phenol at 75 °C. After injection on a Bio-Gel P-6DG column, excluded material was extracted three times with chloroform/methanol/water (10/10/3, v/v/v) and separated on a Florisil column. This method was also used by Loureiro y Penha et al. [16] and Gutierrez et al. [17]. Jennemann et al. [18, 19] extracted GIPCs three times from frozen Agaricus bisporus and Agaricus campestris fungi with chloroform/methanol/water (60/35/8, v/v/v) at 50 °C. GIPCs were separated by ion exchange chromatography (DEAE-Sephadex A-25 column, acetate form).
Analysis of fatty acids released from GIPCs
Hydrolysis of FA chains has been widely used for structure characterization of GIPCs. Fatty acid chains were released and trans-methyl-esterified with hot acidic methanol [8, 9, 12, 13, 20] or, less currently, diazomethane [11]. Most often, the resulting free fatty acid methyl esters (FAMEs) were trimethyl-silylated to facilitate their analysis by gas chromatography. Sometimes, FA chains of GIPCs were analyzed after methylamine treatment, in order to eliminate contaminating ester bond-containing lipids such as galactolipids and glycerophospholipids [10, 12]. Identification and quantitative analysis of FAMEs were performed by GC and/or GC-MS (Table 1).
Wells et al. [21] saponified samples before HPLC, and FA chains were identified by comparison with standard molecules.
Jennemann et al. [19] liberated the ceramide moiety by mineral acid hydrolysis. Fatty acid chains were identified by ESI-MS/MS in the negative product ion mode giving the [RCO2]− ion, R corresponding to the chain with (n − 1) carbon atoms. Buré et al. [12] identified some fatty acids by ESI-MS/MS in the negative mode on the basis of the occurrence of a new fragment ion named Fa in their publication.
Fatty acids were also characterized without hydrolysis, using ESI-MS and ESI-MS/MS. Levery et al. [5] and Toledo et al. [13] used the fact that Li+ cationization increased sensitivity and fragmentation of the pseudo-molecular ion [M + Li]+, compared to [M + H]+ or [M + Na]+ ions, to characterize the ceramide moiety. Markham et al. [7] exploited ESI-MS3 fragmentation of [M + Na]+ precursor ions for identification of the FA and LCB chains. In the same way, Markham et al. [10] and Salas et al. [22] used ESI-multiple reaction monitoring (MRM) to analyze the transition [M + H]+/Z0 ions permitting the identification of FA chains and LCBs (see below and Fig. 2).

CID fragmentation pattern of [M + Li]+ GIPC precursors in the positive mode, adapted from Levery et al. [5]
Analysis of long-chain bases released from GIPCs
Hydrolysis was also used for the characterization of LCBs released from GIPCs. After hydrolysis of GIPCs by methanolic HCl, LCBs were identified by TLC using reference molecules, but most often were trimethyl-silylated [8, 9, 11, 13, 14, 20, 23–25]. Identification and quantification were then performed by GC and/or GC-MS (Table 1).
Alternatively, hydrolysis with methanolic HCl could be followed by a derivatization step to convert LCBs into their N-biphenylcarbonyl derivatives [21] prior to a separation by liquid chromatography. The LCBs were then identified by comparison with standards.
Another way leading to the characterization of LCBs was followed by Cacas et al. [2, 12] who established a protocol consisting in (1) release of the LCB from sphingolipids by treatment with barium hydroxide at 110 °C; (2) their oxidation into their corresponding aldehydes by sodium periodate, and (3) the quantification of these aldehydes by GC-MS.
Jennemann et al. [19] liberated the ceramide moiety by mineral acid hydrolysis. The long-chain base was identified by ESI-MS/MS in the positive product ion mode giving m/z 318, 300, 282, and 264 ions for t18:0 phytosphingosine.
As in the case of FAs, LCBs have also been identified by ESI-MS and ESI-MS/MS without the use of degradation chemistry. As for FA analysis, Levery et al. [5] and Toledo et al. [13] characterized the ceramide moiety by fragmenting the pseudo-molecular ion [M + Li]+. ESI-MS3 and ESI-MRM were used in the positive ion mode for the identification of FA and LCB chains [7, 10, 22], whereas Buré et al. [12] characterized the ceramide moiety by ESI-MS/MS using the fragmentation of the pseudo-molecular ion [M-2H]2−.
Characterization of the glycan moiety
Characterization of glycan moieties of GIPC polar heads was often accomplished after hydrolysis. In some instances, released oligosaccharides were analyzed without derivatization [18, 19], but they were most often submitted to permethylation [15–17, 23, 24], or trimethyl-silylation [8, 14]. Structure characterization was achieved by TLC and comparison with standards [11], GC and/or GC-MS or by ESI-MS/MS [15] (Table 1). In another approach, Hsieh et al. [26] removed the phosphoceramide moiety by a treatment with a phosphatase. Free oligosaccharides were acetylated (acetic anhydride/pyridine), separated by normal and reverse HPLC, purified by TLC and identified by GC-MS.
Alternatively, spectral information obtained by NMR analysis allows establishing the nature of the saccharide units engaged in the GIPC polar heads, thus differentiating species that have the same mass, i.e., mannose, glucose, and galactose. NMR also allows establishing the conformation of the bonds linking these units [13–20, 24, 25, 27] and the type of bond (linear or branched) between saccharides [15, 17], i.e., if there is a unique chain of saccharide molecules, or several chains that create a branching point.
Tandem mass spectrometry (ESI-MS/MS) was used for the partial characterization of the glycan moiety in the positive [7] and negative ion modes [12]. As for the FA and LCB analysis, Levery et al. [5] and Toledo et al. [13, 20] characterized the saccharide moiety by the fragmentation of the pseudo-molecular ion [M + Li]+.
MALDI-MS analysis
MALDI ionization leads to singly charged ions of GIPCs, and thus provides rather simple spectra that greatly facilitate fast screening. Owing to the structure of GIPCs, several options were tried for the selection of proper matrices and associated ionization modes, such as: 2,5-dihydroxybenzoic acid and positive mode [13], norharmane [15–17] and 7-amino-4-methylcoumarin [23, 24] in negative mode, or indole-2-carboxylic acid [18]. Plant GIPC extracts were analyzed in the positive and negative ion modes with several matrices (alpha-cyano-4-hydroxy-cinnamic acid, 2,5-dihydroxybenzoic acid, 2,4,6-trihydroxy-acetophenone, 3-hydroxy-picolinic acid and 2,6-dihydroxy-acetophenone) [12]. In the latter work, the best result in terms of sensitivity was obtained with 2,6-dihydroxy-acetophenone in the negative ion mode, with the clear advantage of the observation of the sole [M-H]− pseudo-molecular species (see above). This latter approach revealed at a glance the occurrence of highly glycosylated GIPCs for A. thaliana and N. tabacum BY-2 cells samples [12].
The comparison of full-scan MALDI mass spectra and global structure assignment of GIPC isolated from N. tabacum and A. thaliana (cell culture and leaf samples) led to the identification of 6 GIPC clusters having mass differences of either 162 or 132 Da, corresponding to additional hexose or pentose units. The series with the lowest masses (m/z 1,200 to 1,340) contained two saccharides, whereas the series with the highest masses (m/z 1,930 to 2,040) contained seven saccharides [12].
Since pure reference GIPCs were not available at this stage of the work, relative abundances of GIPC series could not be safely assessed on the basis of full-scan MALDI mass spectra, and should be taken as a rough indication of real GIPC distributions according to their number of glycans.
ESI-MS analysis
In plants, ESI-MS full-scan spectra of GIPCs were usually dominated by doubly charged pseudo-molecular ions, with preferential sites of ionization likely located on the phosphate group and on the carboxyl group of hexuronic acid [12]. In contrast, in fungi, spectra were dominated by single charged pseudo-molecular ions since hexuronic acid was not found in fungal GIPC structures and the preferential site of ionization became the sole phosphate group.
Tandem mass spectrometry in ESI and MALDI ionization modes
Full-scan MALDI-MS measurements can be achieved with a mass accuracy of a few parts per million with a time of flight (ToF) analyzer, providing a quick overview of GIPC structures, but are not sufficient for a safe assignment of all detected species. Fragment ions produced by tandem mass spectrometry using collision-induced dissociation (CID) provide most of the information required for the description of GIPC structures. The nomenclature of fragmentations was proposed by Costello et al. [28] and Levery et al. [5].
In the positive ion mode, pseudo-molecular ions observed for GIPC ions were mainly due to ammonium, sodium or potassium adducts [7]; alternatively, the addition of LiI was used to produce lithium adducts [5, 13]. In this case, the pseudo-molecular ions [M + Na]+, [M + K]+, [M + Li]+, [M + NH4]+, more generally named [M + Cat]+ (with Cat being the cation Na, K, Li, or NH4), were observed in mass spectra instead of [M + H]+. CID mass spectra of [M + Cat]+ precursor ions showed fragment ions characteristic of glycosylinositol, [Bn + Cat]+ and [Cn + Cat]+ and their complementary fragment ions [Yn + Cat]+ and [Zn + Cat]+ (Fig. 2). It can be noticed that the fragment ions kept the cation in their structure. Other major fragment ions were glycosylinositol phosphate ions [BnPO3 + Cat]+ and [CnPO3 + Cat]+, ceramide phosphate ions [Y0PO3 + Cat]+ and [Z0PO3 + Cat]+, and ceramide ions [Y0 + Cat]+ and [Z0 + Cat]+. Internal fragment ions were also observed as [BnPO3-Bm + Cat]+, [BnPO3-Cm + Cat]+, [CnPO3-Bm + Cat]+ and [CnPO3-Cm + Cat]+ [7]. Using CID of [M(Cat) + Cat]+ precursor ions, Levery et al. [5, 13] reported more complex positive ion spectra, since fragments with two cations could also be observed. In this study, the ions [BnPO3(Cat) + Cat]+, [CnPO3(Cat) + Cat]+, [Y0PO3(Cat) + Cat]+ and [Z0PO3(Cat) + Cat]+ were also found in addition to the ions mentioned above. In this paper, internal fragment ions were called [Ym/Cn + Cat]+, [Ym/Bn + Cat]+, [Ym/CnPO3(Cat) + Cat]+, [Ym/BnPO3(Cat) + Cat]+, [Zm/BnPO3(Cat) + Cat]+, and [Zm/Bn + Cat]+.
Tandem mass spectrometry experiments were conducted in the negative ion mode on ESI-Q-IT and MALDI-Q-ToF instruments by Buré et al. [12]. The Q-ToF instrument provided a narrow isolation width of precursor ions, and accurate mass measurements for both precursor and fragment ions, two features that were particularly useful for structure assignment of plant GIPCs by means of MALDI-MS/MS. Fragmentation patterns obtained by MALDI-MS/MS and ESI-MS/MS were similar.
Nevertheless, complementary information was derived from these two ionization modes, since the C3PO3 ion representative of the carbohydrate moiety was only obtained by MALDI-MS/MS and the Fa ion, representative of the ceramide moiety, was only obtained by ESI-MS/MS (Fig. 3). When present, the Fa ion allowed identifying the fatty acid chain of the ceramide moiety. Furthermore, ions at m/z 373 and 355 observed in the negative ion mode represent a useful signature of the polar head when the arrangement of saccharides is linear (Fig. 3).

CID fragmentation pattern of GIPC precursor ions in the negative mode, adapted from Buré et al. [12]
Tandem mass spectrometry allowed distinguishing isobaric GIPC species. For instance, as encountered in the case of BY-2 cell extracts, the R1 substituent could be either an amine or an acetylamine, and the corresponding 42 Da mass difference could be balanced by a difference of three carbon atoms in the length of the fatty acid chain (Figs. 1 and 3). In this particular case, such isobaric species occurring at m/z 1,259.76 in MALDI-MS (and doubly charged ion at m/z 629.38 in ESI-MS), could be differentiated and attributed to hexose(NH2)–hexuronic acid–inositol phosphoceramide (t18:1h24:0) and hexose(NAc)–hexuronic acid–inositol phosphoceramide (t18:1h21:0) [12].
HPLC-ESI-MS/MS
To our knowledge, studies concerning the characterization of GIPC species by HPLC-ESI-MS/MS were carried out only in plants: A. thaliana [10, 29], Helianthus annuus (sunflower seeds) [22, 29], Spinacia oleracea (spinach leaves), Brassica oleracea (white cabbage), Glycine max (soybean), and Lycopersicon esculentum (tomato leaves) [29]. All these experiments were achieved in reversed-phase liquid chromatography with solvents A and B containing THF/methanol/5 mM ammonium formate or ammonium sulfate + 0.1 % formic acid in different proportions. In this mode, the various GIPC species were separated as a function of their chain length and desaturation. Mass spectrometric parameters were established by infusing each purified samples. The characterization of GIPCs obtained by infusion was used to choose precursor/product ions transitions for MRM experiments, i.e. from [M + H]+ to Z0 ions, the Z0 ion being characteristic of the ceramide moiety. Low pH HPLC eluents were found to favor positively charged GIPC species that were detectable with great sensitivity. The positive ion mode was required to detect the positively charged ceramide fragment. Combined HPLC-ESI-MS/MS advantageously provides a third identification level with reference to the retention time, in addition to the precursor mass and the MRM transitions. As no commercial GIPC standard was available, the quantification of GIPC species proposed by Markham et al. [10] was based on a commercial ovine ganglioside standard, GM1. This analytical method seemed to be influenced by some matrix effects. In their approach, Blaas and Humpf [29] used sphingosyl phosphoinositol (Lyso-IPC, commercially available) as a standard for semi-quantitative HPLC-ESI-MS/MS of GIPCs, and C17-IPC (synthesized for this purpose) as ionization standard, to minimize matrix effects.
Diversity of GIPC structures in plants and fungi
As indicated by the information on GIPC structures assembled in Table 1, GIPC composition was found to be slightly less complex in fungi than in plants. The simple distribution reported for A. thaliana GIPCs containing mostly two glycosylated residues [7] seems rather to be a special case in the plant kingdom.
In plants, the core structure of GIPC polar heads is Hex(R1)-HexA-IPC with R1 being a hydroxyl, an amine or a N-acetylamine group, whereas the core structure in fungi is Man-IPC. Additional saccharides linked to inositol were arabinose, galactose, mannose and fucose in plants, and mannose, galactose, xylose, fucose, GlcN, and GlcNAc in fungi. In plants, the ceramide moiety consisted in h16:0 to h26:1, 20:0 to 28:0, and t18:1, t18:0, d18:1, d18:0, d18:2 for FA and LCB chains, respectively; in fungi, the composition was rather h22:0 to h26:0, 2h24:0 to 2h25:0 and 16:0 to 24:0 and t18:0, t20:0 for FA and LCB chains, respectively (Table 1).
Markham et al. found that sphingolipids from A. thaliana differed by 28 Da, indicating fatty acid chains with even carbon number only [7, 10]. In A. thaliana (cells and leaves) and BY-2 cells, our group found evidence for fatty acid chains with odd and even carbon numbers. The occurrence of fatty acid chains with odd carbon numbers has been confirmed by GC-MS analysis of FAMEs, and is consistent with fatty acid chain distributions reported for A. fumigatus and Nicotiana tabacum [1, 14].
The number of saccharides linked to inositol is rather large in GIPC structures: up to 14 saccharides were found in tobacco leaves [8] and up to 20 saccharides in Candida albicans [27]. The arrangement of these saccharides is thought to be linear in most cases, but Hsieh et al. [26] have found, by a combination of HPLC and GC-MS, a partially atypical structure with two saccharides linked to the inositol instead of one, GlcNAc-GlcA-Ins(Man)-P-Cer.
ESI-MS/MS analysis provided evidence for new GIPC structures in algae. In Chondracanthus acicularis, an unusual GIPC structure lacking hexuronic acid was identified by ESI-MS/MS as hexose–inositol phosphoceramide. These unusual GIPCs constitute a new GIPC series with a single saccharide. In Ulva lactuca, other new structures were identified as (hexuronic acid)4–inositol phosphoceramide and hexose–(hexuronic acid)3–inositol phosphoceramide.
Thus, the exact molecular structure of GIPCs still leaves us a certain number of enigmas to resolve.
Conclusion
By combining selective extraction, LC-MS/MS, MALDI-MS/MS, and NMR, all methodologies allowing for complete structure characterization of GIPCs from plant and fungal origins are currently available. Evidence for a broad diversity of GIPC structures has been found by applying these methodologies, but leaves open a whole bunch of questions related to the understanding of GIPC functions [4]: What are the exact structures of the polar heads of GIPCs? How many sugars are engaged in this polar head? Where are GIPCs localized? In which membrane leaflet? How are they structured in membranes? Do they influence membrane thickness? Are they structuring elements for microdomains formation? What are their roles in plant physiology? Are they involved in host-pathogen relationships?
These questions give a clear indication for future work: adequate answers will require molecular tools that are still missing at this moment. Indeed, there are no commercially available GIPC standards that are needed for absolute quantitation by MRM. Further, besides molecular tools required for quantitative mass spectrometry, there are neither antibodies against GIPCs, nor fluorescently labeled GIPCs to allow imaging.
Abbreviations
- CID:
-
Collision-induced dissociation
- DHA:
-
2,6-dihydroxy-acetophenone
- ESI:
-
Electrospray ionization
- FA:
-
Fatty acid
- FAME:
-
Fatty acid methyl ester
- Gal:
-
Galactose
- GIPC:
-
Glycosyl inositol phosphoryl ceramide
- GlcA:
-
Glucuronic acid
- GlcN:
-
Glucosamine
- GlcNAc:
-
N-acetyl glucosamine
- Hex:
-
Hexose
- IPC:
-
Inositol phosphoryl ceramide
- IT:
-
Ion trap
- LCB:
-
Long-chain base
- MALDI:
-
Matrix-assisted laser desorption ionization
- Man:
-
Mannose
- MRM:
-
Multiple reaction monitoring
- MS/MS:
-
Tandem mass spectrometry
- NAc:
-
N-acetyl
- Q:
-
Quadrupole
- TIC:
-
Total ion current
- ToF:
-
Time of flight
References
Pata MO, Hannun YA, Ng CKY (2010) New Phytol 185:611–630
Cacas JL, Melser S, Domergue F, Joubès J, Bourdenx B, Schmitter JM, Mongrand S (2012) Anal Bioanal Chem 403:2745–2755
Sperling P, Franke S, Luthje S, Heinz E (2005) Plant Physiol Biochem 43:1031–1038
Markham JE, Lynch DV, Napier JA, Dunn TM, Cahoon EB (2013) Curr Opin Plant Biol DOI: 10.1016/j.pbi.2013.02.009
Levery SB, Toledo MS, Straus AH, Takahashi HK (2001) Rapid Commun Mass Spectrom 15:2240–2258
Carter HE, Gigg RH, Law JH, Nakayama T, Weber E (1958) J Biol Chem 233:1309–1314
Markham JE, Li J, Cahoon EB, Jaworski JG (2006) J Biol Chem 281:22684–22694
Kaul K, Lester RL (1975) Plant Physiol 55:120–129
Kaul K, Lester RL (1978) Biochemistry 17:3569–3575
Markham JE, Jaworski JG (2007) Rapid Commun Mass Spectrom 21:1304–1314
Carter HE, Koob JL (1969) J Lipid Res 10:363–369
Buré C, Cacas JL, Wang F, Gaudin K, Domergue F, Mongrand S, Schmitter JM (2011) Rapid Commun Mass Spectrom 25:3131–3145
Toledo MS, Levery SB, Bennion B, Guimaraes LL, Castle SA, Lindsey R, Momany M, Park C, Straus AH, Takahashi HK (2007) J Lipid Res 48:1801–1824
Simenel C, Coddeville B, Delepierre M, Latgé JP, Fontaine T (2008) Glycobiology 18:84–96
Heise N, Gutierrez ALS, Mattos KA, Jones C, Wait R, Previato JO, Mendonça-Previato L (2002) Glycobiology 12:409–420
Penha CV L y, Todeschini AR, Lopes-Bezerra LM, Wait R, Jones C, Mattos KA, Heise N, Mendonça-Previato L, Previato JO (2001) Eur J Biochem 268:4243–4250
Gutierrez ALS, Farage L, Melo MN, Mohana-Borges RS, Guerardel Y, Coddeville B, Wieruszeski JM, Mendonça-Previato L, Previato JO (2007) Glycobiology 17:1C–11C
Jennemann R, Bauer BL, Bertalanffy H, Geyer R, Gschwind RM, Selmer T (1999) Wiegandt H 259:331–338
Jennemann R, Geyer R, Sandhoff R, Gschwind RM, Levery SB, Grone HJ, Wiegandt H (2001) Eur J Biochem 268:1190–1205
Toledo MS, Levery SB, Straus AH, Takahashi HK (2001) FEBS Lett 493:50–56
Wells GB, Dickson RC, Lester RL (1996) J Bact 178:6223–6226
Salas JJ, Markham JE, Martinez-Force E, Garces R (2011) J Agric Food Chem 59:12486–12492
Uchiyama R, Aoki K, Sugimoto H, Taka N, Katayama T, Itonori S, Sugita M, Che FS, Kumagai H, Yamamoto K (2009) Biosci Biotechnol Biochem 73:74–78
Aoki K, Uchiyama R, Itonori S, Sugita M, Che FS, Isogai A, Hada N, Hada J, Takeda T, Kumagai H, Yamamoto K (2004) Biochem J 378:461–472
Levery SB, Toledo MS, Straus AH, Takahashi HK (1998) Biochemistry 37:8764–8775
Hsieh TC, Lester RL, Laine RA (1981) J Biol Chem 256:7747–7755
Trinel PA, Maes E, Zanetta JP, Delplace F, Coddeville B, Jouault T, Strecker G, Poulain D (2002) J Biol Chem 277:37260–37271
Costello CE, Vath JE (1990) Meth Enzymol 193:738–768
Blaas N, Humpf HU (2013) J Agric Food Chem DOI: 10.1021/jf4001499
Barr K, Lester RL (1984) Biochemistry 23:5581–5588
Carter HE, Kisic A (1969) J Lipid Res 10:356–362
Acknowledgments
The work was supported by the French Agence Nationale pour la Recherche (contract no. NT09_517917 PANACEA).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue Analytical Science in France with guest editors Christian Rolando and Philippe Garrigues.
Rights and permissions
About this article
Cite this article
Buré, C., Cacas, JL., Mongrand, S. et al. Characterization of glycosyl inositol phosphoryl ceramides from plants and fungi by mass spectrometry. Anal Bioanal Chem 406, 995–1010 (2014). https://doi.org/10.1007/s00216-013-7130-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-7130-8