
Abstract
In this work, the development and optimization of a new methodology to analyze grape seed procyanidins based on the application of two-dimensional comprehensive LC is presented. This two-dimensional method involves the use of a microbore column containing a diol stationary phase in the first dimension coupled to either a C18 partially porous short column or a C18 monolithic column in the second dimension. The orthogonal hydrophilic interaction × reversed phase liquid chromatography (HILIC×RP-LC) system is interfaced through a ten-port two-position switching valve. The optimized HILIC×RP-LC separation followed by diode array and tandem mass spectrometry detection (HILIC×RP-LC-DAD-MS/MS) made possible the direct analysis of a complex grape seed extract and allowed the tentative identification of 43 flavan-3-ols, including monomers and procyanidin oligomers till a polymerization degree of 7 units with different galloylation degrees. To the best of our knowledge, this is the first time that this powerful analytical technique is employed to characterize complex procyanidin samples. This work successfully demonstrates the great capabilities of the HILIC×RP-LC-DAD-MS/MS coupling for the direct analysis of very complex natural samples like grape seeds.

Two-dimensional HILIC x RP plot (280 nm) of grape seed procyanidins.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Flavan-3-ol polymers, more frequently known as proanthocyanidins or condensed tannins, are a complex group of phenolic compounds widely distributed in the plant kingdom and the second most abundant group of natural plant phenolic compounds after lignin [1]. This wide group of compounds is divided into different smaller categories depending on the monomers included in their composition [2]. The main group belongs to procyanidins, which are basically composed by catechin and epicatechin units, which are the largest class of proanthocyanidins [3]. Besides, monomeric forms esterified with gallic acid are also found in nature, such as catechin, epicatechin, gallocatechin, and epigallocatechin gallates. This fact makes even more complex the number of possible combinations for the different proanthocyanidins forms, increasing considerably the chemical diversity of these components and, therefore, the complexity of their analysis. Procyanidins are among the most common phenolic compounds in grapes, which can be found both in skins and seeds [4]. In fact, grape seeds are an important natural source of these components, although they exist in other important food sources, such as cocoa, apples, peanuts, or berries [5]. Vitis vinifera grape seed procyanidins are particularly interesting because of their unlimited structural diversity based on the combination of only three elemental units, i.e., catechin, epicatechin, and epigallocatechin (Fig 1). This is due to the stereochemistry of the asymmetric carbons C2 and C3 of the flavan skeleton, the type of interflavan bond (C4-C8 and C4-C6, B-type procyanidins), the length of the polymer chain (degree of polymerization), the degree of galloylation, and the position of the gallic acid ester [6].

Chemical structure of the main flavan-3-ols forming part of grape seed procyanidins
Grape procyanidins have a critical importance in winemaking as these compounds have been demonstrated to have a great contribution to the astringency of wines as well as to other organoleptic properties, such as improving color stability of red wines [7]. Besides, it has been suggested that the galloylation degree of the contained procyanidins could increase wine coarseness [8]. Nevertheless, the increasing interest on this class of compounds nowadays is mainly due to their important functional and bioactive activities, such as antioxidant, antibacterial [9], anti-inflammatory [10], or anti-cancer [11] effects. These important technological and biological properties have raised interest in developing new methods to analyze these compounds.
However, due to their huge chemical complexity and diversity, the analysis of procyanidins is very difficult and typically requires the use of a purification step before chromatographic separation followed by mass spectrometry (MS) to identify the multiple compounds present. Different strategies have been explored for their separation, being the most commonly applied reversed phase high-performance liquid chromatography (RP-LC). In this case, mainly C18 columns are employed, although the complete resolution of all the possible components present on this type of complex profiles is quite difficult to achieve, even using very lengthy gradient elutions (from 60 to 120 min) [12,13]. Besides, the elution order does not correspond to their polymerization degree [5]. Although this RP-LC mode could be well suited for monomers and oligomers, the separation of procyanidins with a degree of polymerization (DP) higher than 4 is usually not possible. In general, polymers with higher DP cannot be separated and coelute in a large unresolved peak. In order to partially solve this problem, other separation modes, such as normal phase LC, have been also employed. In this case, procyanidins can be separated according to their polymerization degree [5]. This approach has been also followed to separate procyanidins from grapes [14], among other food samples [15]. The NP approach has been shown to be useful to separate up to DP 10, for instance, to analyze procyanidins from cocoa and chocolate products [16,17] or different berries [18,19]. However, when the sample is more complex, as grape seeds, it is not possible to have proper resolution between peaks, due to the huge number of isomers and because the degree of galloylation of the polymers varies. Besides, all the compounds which present the same DP coelute. As an evolution of these latter methods, hydrophilic interaction LC (HILIC) has been also employed in order to overcome the problems related to the solvents needed in NP. HILIC acceptance is increasing in the last years because it enables to achieve separation similar to those obtained in NP using solvents compatible with RP [20]. The use of water in the mobile phase allows the formation of a water-enriched layer partially immobilized on the polar stationary phase. Subsequently, the separation is achieved, mainly, by partitioning between the organic-rich mobile phase and the water-enriched layer, although other interactions might also occur [21]. However, despite the development of different methods based on the use of these separation methods, the complete analysis of procyanidins still remains unresolved.
To avoid the lack of resolution provided by the mentioned monodimensional separation strategies, bidimensional approaches might be utilized. Comprehensive two-dimensional LC (LC × LC) is characterized by an enhanced resolving power, which can be very useful for the analysis of complex samples [22]. This technique has been already applied for the separation and characterization of different food-related complex matrices [23,24]. In LC × LC, the sample is subjected to two independent separation processes, so that different fractions from the first dimension are continuously transferred to the second dimension for further separation. This approach is far from being straightforward because multiple parameters have to be optimized [25–27]. For instance, very fast analyses are needed in the second dimension to be able to handle all the fractions coming from the first one, and consequently, very high flow rates should be employed. In order to keep the system backpressure under attainable conditions, two types of columns are typically employed in the second dimension in comprehensive two-dimensional LC systems: monolithic columns and partially porous columns. Monolithic columns are formed by a porous continuous gel with a porosity typically 15 % higher than a conventional packed columns. On the other hand, partially porous columns are packed with particles with 2.7 μm of diameter containing a solid core of 1.7 μm and a superficial porous section of 0.5 μm. These particles provide a decrease on analyte diffusivity into the column compared to conventional fully porous particles, allowing higher mass transfer rates and higher flow rates without compromising the column efficiency and generating significantly less backpressure. Two-dimensional LC in off-line mode has already been applied to the separation of procyanidins from apple and cocoa [28]. In that work, a first dimension HILIC separation was coupled to a RP-based separation in the second dimension. However, in the mentioned work, fractions eluting from the first dimension were collected and, later on, injected in a second dimension making the procedure lengthy and laborious.
The aim of the present work is to develop a new LC × LC method to automatically analyze grape seed procyanidins without any previous pretreatment. To do that, the performance of different HILIC and RP columns is tested and compared in both dimensions, and all the parameters involved in this challenging coupling closely studied. By using this novel LC × LC-DAD-MS/MS approach, it is possible to separate and identify 46 different phenolic compounds, mostly, grape seed procyanidins, in a single chromatographic run.
Experimental
Samples and chemicals
Grape seeds (V. vinifera L., cv. Malvar) were from the El Encín plantation (IMIDRA, Madrid, Spain). All the solvents employed (acetonitrile, methanol, and 2-propanol) were of HPLC-grade and acquired from Lab-Scan (Dublin, Ireland). Formic acid was supplied by Sigma-Aldrich (Madrid, Spain), whereas acetic acid was purchased from Scharlab (Barcelona, Spain). Water employed was Milli-Q grade obtained from a Millipore system (Billerica, MA). (+)-Catechin, (−)-epicatechin, ethyl gallate, and procyanidin B1 reference samples were acquired from Extrasynthèse (Genay, France) and gallic acid from Scharlab (Barcelona, Spain).
Sample preparation
A fast and simple extraction protocol was followed for the extraction of proanthocyanidins from grape seeds [29]: briefly, 80 g of finely ground grape seeds was weighted and added to 90 mL of extraction solvent (methanol/water 80:20, v/v). The solution was sonicated for 15 min. Afterwards, the solution was left to stand protected from light for 2 h, and then, it was again sonicated for 15 min. The extract was centrifuged at 8,000 rpm for 20 min. The supernatant was recovered and filtered through 0.45-μm nylon syringe filters (Symta, Madrid, Spain). Lastly, after evaporation of methanol in a Rotavapor R-210 (Buchi Labortechnik AG, Flawil, Switzerland), the extract was lyophilized using a freeze-dryer (Labconco Corporation, MO). The extraction yield obtained after extraction was 2.75 % (dry weight basis).
For the LC × LC analysis of procyanidins, a 50-mg/mL solution of the dried extract was prepared in methanol and filtered (0.45 μm). Then, 300 μL of this solution was added to 700 μL ACN to obtain a 15-mg/ml solution which was filtered again (0.20 μm, Symta) and finally injected.
LC × LC instrumentation
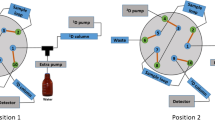
Comprehensive two-dimensional LC analyses were carried out on an Agilent 1200 series liquid chromatograph (Agilent Technologies, Santa Clara, CA) equipped with a diode array detector and an autosampler. In order to have robust and reproducible low flow rates and gradients in the first dimension, a Protecol flow splitter (SGE Analytical Science, Milton Keynes, UK) was placed between the first dimension pumps and the autosampler. Besides, an additional LC pump (Agilent 1290 Infinity) was coupled to this instrument to perform the second dimension separation through an electronic controlled two-position ten-port switching valve. In addition, an Agilent 6320 Ion Trap mass spectrometer equipped with an electrospray interface was coupled on-line and operated in negative ionization mode using the following conditions: dry temperature, 350 °C; mass range, m/z 90–2,200 Da; dry gas flow rate, 12 L/min; and nebulization pressure, 40 psi. The LC data were elaborated and visualized in two and three dimensions using LC Image software (version 1.0, Zoex Corp., Houston, TX).
LC × LC separation conditions
Different columns and conditions were tested for the optimization of the HILIC × RP separation of grape seed procyanidins. In the first dimension, a Syncronis HILIC column (250 × 2.1 mm, 5 μm d.p., Thermo Scientific, Waltham, MA) and a Lichrospher diol-5 (150 × 1.0 mm, 5 μm d.p., HiChrom, Reading, UK) column were tested. Under the optimum conditions, the diol column with a precolumn with the same stationary phase was employed using 15 μl/min as flow rate. The mobile phases employed were (A) acetonitrile/acetic acid (98:2, v/v) and (B) methanol/water/acetic acid (95:3:2, v/v/v) eluted according to the following gradient: 0 min, 0 % B; 5 min 20 % B; 10 min, 30 % B; 30 min, 50 % B; 40 min, 50 % B; 50 min, 80 % B; 75 min, 100 % B; 85 min, 100 % B. The injection volume was 20 μl.
In the second dimension separation, two different columns were tested. The optimum separation conditions were optimized separately. In first place, a partially porous column Ascentis Express C18 (50 × 4.6 mm, 2.7 μm d.p., Supelco, Bellefonte, CA) was employed. During the whole LC × LC separation, 1.3-min repetitive second dimension gradients were employed, being also 1.3 min the modulation time programmed in the switching valve. Water (0.1 % formic acid, A) and acetonitrile/methanol (50:50, v/v, B) were the mobile phases, using a repetitive gradient consisting of: 0 min, 0 % B; 0.1 min, 15 % B; 0.3 min, 25 % B; 1 min, 45 % B; 1.01 min, 0 % B. The flow rate was 3 mL/min.
The second column tested in the second dimension was a C18 monolithic column (100 × 4.6 mm, Onyx C18, Phenomenex, Torrance, CA), and it was also eluted using 1.3-min repetitive gradients. Two different gradient profiles were employed throughout the analysis. During the first 52 min, the mobile phase employed consisted of water (0.1 % formic acid, A) and methanol (B) eluted according to the following gradient: 0 min, 0 % B; 0.1 min, 15 % B; 0.3 min, 25 % B; 1.0 min, 45 % B; 1.01 min, 0 % B. From minute 52 till the end of the analysis, the mobile phase composition was changed to water (0.1 % formic acid, A) and acetonitrile/methanol (B) using the following program: 0 min, 0 % B; 0.1 min, 15 % B; 0.3 min, 25 % B; 0.8 min, 45 % B; 0.9 min, 90 % B; 1.0 min, 0 % B. In this case, the flow rate was 4 mL/min.
In all cases, 280 nm was the wavelength used to monitor the separations, although UV–vis spectra were collected from 190–550 nm using a sampling rate of 20 Hz in the diode array detector. The MS was operated under negative ESI mode. The flow eluting from the second dimension column was splitted before the MS instrument, introducing approximately 600 μL/min into the MS detector.
Results and discussion
The coupling of a HILIC column in the first dimension to a RP-LC column in the second dimension to separate grape procyanidins by LC × LC is, theoretically, a promising alternative. However, this coupling is far from being straightforward due to numerous technical difficulties that arise during the method development [30]. For this reason, an initial individual optimization of the two intended dimensions was carried out. Once the preliminary conditions were independently obtained for both columns, their coupling was set up and fine-tuned, as well as the electrospray conditions prior to MS analysis. Data from DAD and MS were combined to chemically characterize the grape seed sample studied.
Optimization of the first dimension separation
To achieve a proper LC × LC separation of procyanidins, it was decided to employ a HILIC column in the first dimension in order to obtain a first distribution of the grape seed components according to their increasing DP. This novel approach would allow the coupling to a RP-based second dimension avoiding problems related to solvent immiscibility. Nevertheless, as in every comprehensive multidimensional system, some requirements should be met in order to have a good coupling depending on the chosen configuration. One of the most successful multidimensional approaches is to use a switching valve as interface between dimensions equipped with two identical sample loops to transfer the fractions collected from the first dimension to the second dimension [24]. This setup was chosen in this work. In this kind of systems, the first dimension separation has to be carried out at very low flow rates so that enough time is allowed to fill one of the loops while the fraction previously collected in the other loop is being analyzed in a fast second dimension. For this reason, the optimization of the first dimension separation implies great difficulties, mainly related to the proper distribution of wide peaks throughout the chromatogram and the effective translation of the gradient program to the column at those very low flow rates. In this sense, obviously, the nature of the column employed is of great importance as it will influence how the components of the sample will interact with the whole chromatography system during the analysis. To achieve the separation of the procyanidins present in the grape seed sample studied, two different columns were tested from the wide group of stationary phases available to carry out HILIC separations [31], a silica column and a bonded diol column. Different mobile phases and elution gradients were studied, always keeping a small proportion of water during the separation. In any case, the retention obtained in the silica column was stronger making very difficult the correct separation of the procyanidins under the desired conditions. The diol column offered better capabilities in this application, allowing a better separation and distribution of the different procyanidin oligomers during the first dimension analysis. The particular conditions were optimized by changing the mobile phases employed as well as the gradients and flow rates and also studying the influence of the sample solvent. This factor was shown to have a critical importance on the separation. In fact, at the beginning of the optimization, the sample was injected dissolved in methanol, which is a good solvent to dissolve the procyanidins’ oligomers and polymers. However, considering the narrow dimensions of the column employed (150 × 1.0 mm), the introduction of the sample in methanol, with higher eluotropic power than the acetonitrile-rich initial mobile phase (under HILIC conditions), prevented the separation as the components of the sample did not have enough time to interact with the stationary phase and eluted practically unretained. For this reason, the sample was firstly prepared more concentrated in methanol and then diluted in acetonitrile (to maintain the same concentration). This change modified completely the behavior of the chromatographic process allowing the separation of the different components. In Fig 2, a typical first dimension chromatogram obtained under optimum conditions is shown. As it can be observed, flavan-3-ols were grouped and distributed along the analysis according to their different DP, as it was expected. In fact, this behavior has been previously observed in other applications in which this type of stationary phase was used just in one dimension [1,17,19].The flow rate employed was 15 μl/min. This flow rate was selected as it was low enough to permit the transfer of a relatively low volume of the first dimension eluate to the second dimension separation.

HILIC chromatogram (280 nm) of a grape seed procyanidins’ extract obtained under the optimum first dimension separation conditions using a microbore column (diol stationary phase, 150 × 1.0 mm, 5 μm). DP degree of polymerization
Optimization of the second dimension separation
The second dimension separation conditions should allow the fast separation of the components present on each fraction of the first dimension transferred. The length of the second dimension analysis directly influences the volume being transferred in each fraction as each first dimension fraction cannot be transferred until the second dimension analysis is not finished and the second dimension column is prepared. For this reason, 20 μl was the fraction volume targeted from the beginning of the second dimension optimization, to keep a reasonable injection volume in these analyses. This fact is interesting because even if the solvents employed in both dimensions of a HILIC × RP coupling are compatible, the strength of the solvents relative to the two different stationary phases brings about an important problem. Indeed, the stronger solvent in the first dimension will be the weaker one in the second dimension and vice versa. Considering the flow rate employed in the first dimension (15 μl/min), ca. 1.3 min was available for the separation and re-equilibration of the second dimension column after injecting the mentioned 20 μl from the first dimension. Thus, very fast analyses are needed, and consequently, very high flow rates should be employed. In order to keep the system backpressure under attainable conditions, two types of columns have been mainly employed in the second dimension in comprehensive LC systems: monolithic columns and partially porous columns. In this work, both types of columns with C18 stationary phases were studied carrying out a preliminary optimization of the separation conditions for each column injecting 20 μl of the whole sample. Different mobile phases (acetonitrile, methanol, acetonitrile/methanol 80:20, v/v, and acetonitrile/2-propanol 80:20, v/v), flow rates (2–4 ml/min), gradients, and temperatures (30–55 °C) were tested. According to the obtained results, mobile phases composed by (A) water (0.1 % formic acid) and (B) acetonitrile/methanol eluted at 3 ml/min were selected for the partially porous column, whereas (A) water (0.1 % formic acid) and (B) methanol eluted at 4 ml/min were chosen for the monolithic column. The gradients employed are described in the “Experimental” section. The separation temperature did not influence significantly the separation, and only slight decreases on the system backpressure were observed. For this reason, 30 °C was selected as the separation temperature. At the optimum analysis conditions, the backpressures observed were under the instrument working interval, ranging from 150 to 320 bar for the monolithic and partially porous columns, respectively. Using both sets of conditions, it was possible to accomplish the second dimension separation in 1.3 min, including the time for column reconditioning. Therefore, this time could be selected as modulation time. To this time corresponds the transfer of a volume of 19.5 μl of eluate from the first dimension. In this regard, it is important to note that although the sample solvent has great importance on the analysis obtained in the second dimension, as it can be observed in Fig 3a, in LC × LC the second dimension injection solvent cannot be selected as it is imposed by the fractions eluting from the first dimension. Consequently, in the present HILIC × RP approach, 19.5 μl of a strong solvent is injected in the second dimension, which deteriorates the separation obtained. To partially solve this problem, the use of loops in the switching valve with different internal diameter was studied. The idea was to take advantage of the miscibility of the solvents employed in HILIC and RP modes to dissolve the first dimension fraction in the second dimension mobile phase in order to avoid the problems generated by solvent incompatibility. As it is shown in Fig 3b, loops with internal volumes of 20, 30, and 50 μl were tested. It was observed that by using a 30-μl loop, the separation was significantly improved, and at the same time, the volume of injection in the second dimension was kept as low as possible.

a Effect of the sample solvent on a 2D separation using the partially porous column, and b effect of the use of loops with different internal volume installed in the switching valve maintaining 19.5 μl as first dimension transfer volume
Overall HILIC × RPLC-DAD-MS optimization
Under the conditions optimized for each dimension, the negative effect of the differential HILIC and RP solvent strength was successfully solved. Thus, HILIC × RP-LC separations of grape seed procyanidins were carried out using the best conditions for the combinations diol × partially porous columns and diol × monolithic columns. In both cases, the modulation time was 1.3 min corresponding, as mentioned, to 19.5 μl of first dimension eluate being transferred each time and diluted in the second dimension mobile phase up to 30 μl that corresponds to the internal volume of the loop installed on the switching valve. As it can be observed in Figs. 4 and 5, good separations of most of the compounds included in this complex sample were attained. In order to try to improve the separation and ionization yield in the electrospray of the studied compounds (mainly focusing on those eluting at the end of the first dimension separation, i.e., procyanidins with higher DP), different gradients and mobile phases were tested. In the case of the partially porous column, no significant improvement was observed when the gradient was modified from minute 52. On the other hand, a slight improvement was attained for the monolithic column changing the composition of the mobile phases in this last part of the analysis, above all, regarding the ionization of these components. Thus, after this optimization, when using the monolithic column in the second dimension, water (0.1 % formic acid) and methanol were employed as mobile phases during the first 52 min, while water (0.1 % formic acid) and acetonitrile/methanol were used for the rest of the analysis. Besides, the gradient was also slightly modified, as can be observed in Fig. 4.

Two-dimensional HILIC × RP plot (280 nm) of the separation of grape seed procyanidins using a monolithic column in the second dimension. Scheme of the gradients employed during the analysis in both dimensions. D1, (A) acetonitrile/acetic acid (98:2, v/v) and (B) methanol/water/acetic acid (95:3:2, v/v/v); D2 purple line, (A) water (0.1 % formic acid) and (B) methanol; D2 red line, (A) water (0.1 % formic acid) and (B) acetonitrile/methanol (50:50, v/v)

Two-dimensional HILIC × RP plot (280 nm) of grape seed procyanidins separated using a partially porous column in the second dimension. For peak identification, see Table 1
Concerning the MS detection of these components, negative ESI ionization was employed as this mode is widely considered as the most suitable to obtain a high response for flavan-3-ols [5]. The particular ionization parameters, namely, dry temperature, dry gas flow rate, and nebulization pressure, were adapted to the second dimension flow rate splitted and introduced into the MS.
It is important to note that the procyanidin polymers with higher DP were not completely resolved. In this sense, although those polymers were eluted at the end of the first dimension analysis, the extremely fast second dimension analyses needed to perform LC × LC did not provide the conditions required to have these compounds properly separated. Nevertheless, it has to be also remarked that procyanidin polymers with DP> 4 have not been fully resolved by monodimensional RP [32]. On the other hand, compared to NP procyanidin separations, polymers up to DP 10 could be separated using similar analysis times, although no separation and identification of the compounds possessing the same DP could be obtained using that approach [17]. Instead, compounds are just grouped by DP. Thus, the application of the methodology developed in this work implies a clear and significant improvement over the previously published analytical methods either based on the application of NP or RP.
To proceed with the exhaustive characterization of the natural food sample, the use of the partially porous column in the second dimension was selected as this set of columns allowed using less amount of solvents compared to the set of columns including the monolithic column. Using the optimized method, the peak capacity of the system was studied according to Neue [33]:
where, n is the number of components employed to make the calculations, t g is the gradient time, and w is the average peak width. Theoretical peak capacity of the separation, assuming that the total peak capacity of the two-dimensional system, n c2D, will be the product of the two independent peak capacities values (n c2D = 1 n c × 2 n c), is 1,435, considering that 1 n c = 41 and 2 n c = 35. As usual, this value assumes that the entire two-dimensional plane might be occupied by peaks, which is not really the case. For this reason, other approaches have been developed to obtain more realistic peak capacity values. Following the equation developed by Li et al. [34], a value of effective peak capacity equal to 875 is obtained:
This equation takes into account the second dimension time cycle (2 t c, equal to modulation time in this case, 1.3 min) as well as the influence of undersampling of the first dimension.
In any case, this value clearly shows the extremely high potential and enhanced separation power of the developed method and its application to complex real samples. Besides, it is important to remark the low correlation obtained between the retention mechanisms employed in both dimensions, as it can be clearly appreciated in Figs 4 and 5, thus, assuring high orthogonality.
Characterization of grape seed procyanidins by HILIC × RP-LC-DAD-MS/MS
The development of this HILIC × RP method allowed the comprehensive two-dimensional analysis of procyanidins for the first time. The tentative identification of the separated compounds was carried out combining the information from the two detectors employed, DAD and MS detectors. In Table 1, the data corresponding to the separated compounds as well as their tentative identification are presented. In Fig 5, the two-dimensional chromatogram obtained for the grape seed procyanidins analyzed under optimum conditions is also shown. As it can be observed, the compounds were mainly grouped according to their molecular mass, firstly eluting the monomers and then the different oligomers according to their increasing DP and increasing degree of galoylation (for the same DP) with respect to the first dimension separation.
Although two different detectors were employed, it is important to remark that the UV–vis spectra of the different grape seeds procyanidins were quite similar, thus, not allowing the differentiation among them. Therefore, the information from the MS detector was decisive in order to provide a tentative identification of each separated compound. The first identified compounds were the monomers catechin and epicatechin. Both compounds provided molecular ions with m/z 289 ([M-H]−). The fragmentation of those ions generated ions at m/z 245 as a result of loss of CO2. By comparing with commercial standards, peaks 2 and 3 could be identified as catechin and epicatechin, respectively. Other compounds were detected in the area in which these monomers eluted; peak 1 produced an ion at m/z 418.2 ([M-H]−) whose fragmentation generated ions at m/z 373, 289, and 127. Peak 4 was tentatively identified as gallic acid ethyl ester, according to its UV–vis spectrum (very similar to that of gallic acid) and the molecular ion detected (m/z 197.0, [M-H]−). Different ions at m/z 577 ([M-H]−) were detected corresponding to peaks 6, 7, and 8. The fragmentation pattern of these three compounds was the same (see Table 1) giving prominent fragments at m/z 469 and 425. These latter fragments would be theoretically formed through a retro-Diels-Alder mechanism ([M-H-152]−) [32] from a procyanidin dimer structure. Thus, according to this information, these peaks were assigned to procyanidin dimers.
Ions with m/z of 865 (peaks 9, 15, 16, and 18) were assigned to procyanidins trimers. The fragments derived from these ions consisted on m/z 739 and 577, corresponding probably to the loss of a phloroglucinol unit and to the loss of a (epi)catechin molecule, respectively. These fragments have been previously observed in procyanidin trimers [5]. On the other hand, compounds with m/z 729 ([M-H]−) generated fragments at m/z 577, 559, 441, 407, and 289. The fragments with m/z 577 corresponded to the loss of the galloyl group, whereas the fragments at m/z 559 would be formed as a result of the subsequent loss of a water molecule. The fragments with m/z 441 and 289 were detected as a result of the loss of a (epi)catechin unit and its detection, respectively. The ion at m/z 407 would correspond to the loss of water from a fragment formed through a retro-Diels-Alder mechanism from the m/z 577 ion. Consequently, these components (peaks 10–13) were tentatively identified as procyanidin dimer monogallates. Besides, a single peak eluting among these components with m/z 881.2 ([M-H]−) was detected (peak 14). This compound was assigned to a procyanidin dimer digallate according to the fragment at m/z 729 detected after its MS/MS analysis.
Peaks 17, 19, and 22 were tentatively assigned to procyanidin trimer monogallates. The detection of molecular ions ([M-H]−) at m/z 1,017 together with the presence of fragments at m/z 729 (loss of a (epi)catechin unit), 865 (loss of galloyl group), 577 (loss of a (epi)catechin unit with a galloyl group), and 891 allowed the identification. These fragment ions have been described as common fragments resulting from these compounds [5]. In Fig. 6b, a typical fragmentation pattern for these compounds is shown. Besides, a doubly-charged ion ([M-2H]2−) with m/z 584.5 was also detected. The MS/MS analysis of this compound (peak 21) allowed the detection of fragment ions closely related to loses of galloyl moieties (m/z 1,017 and 999), (epi)catechin units (m/z 880) and both (m/z 729). Consequently, peak 21 was assigned to a procyanidin trimer digallate.

UV–vis and MS spectra of a procyanidin hexamer monogallate (a) and a procyanidin trimer monogallate (b). Main detected fragments at m/z 940.7 ([M-2H]2−): m/z 1,593 ([M-H-(epi)catechin]−), m/z 1,305 ([M-H-2(epicatechin)]−), m/z 1,153 ([M-H-2(epicatechin)-phloroglucinol]−), m/z 577 (dimer), m/z 289 ((epi)catechin). Main detected fragments of m/z 1,017.2 ([M-H]−): m/z 865 (([M-H-phloroglucinol]−), m/z 729 (([M-H-(epi)catechin]−), m/z 577 (dimer)
Several procyanidin tetramers and procyanidin tetramer monogallates were found. Procyanidin tetramers were detected either as single-charged ions ([M-H]−) m/z 1,153 (peaks 20, 23, 25, and 26) or doubly-charged ions (m/z 576.7, peak 24). In any case, the fragmentation pattern was quite similar producing fragments at m/z 1,027 (loss of phloroglucinol unit), 865 (loss of (epi)catechin), 575 (loss of two (epi)catechin units) as well as other typical fragments (m/z 739, 403). Likewise, procyanidin tetramer monogallates (peaks 27–31) were mostly detected as doubly-charged ions ([M-2H]2−) at m/z 652, although the detection of singly-charged ions was also possible with m/z 1,305.4 ([M-H]−). Moreover, a procyanidin tetramer digallate was also detected as [M-2H]2− with m/z 728.5 (peak 36). The fragments found corresponding to losses similar to those already described confirmed the identification of all these procyanidin tetramers.
The procyanidins with DP 5 detected included pentamers (peaks 32–35, 37, and 39), a procyanidin pentamer monogallate (peak 38) and procyanidin pentamer digallates (peaks 40–42). All these components were detected as doubly-charged ions, with the exception of two procyanidin pentamers that were detected as [M-H]− with m/z 1,141.5. Concerning the procyanidin pentamers’ fragmentation pattern, several fragment ions which confirmed the identification were detected; among them, fragments with m/z 1,315 corresponding to a phloroglucinol unit loss, m/z 1,153 (matching with a (epi)catechin loss), m/z 1,027 (equivalent to a (epi)catechin with a phoroglucinol unit loss), m/z 863 (corresponding to the loss of two (epi)catechin units), and m/z 577 (matching the loss of three (epi)catechin units). Ions with m/z 796.4 and 872.5 were assigned to procyanidin pentamer monogallates and digallates ([M-2H]2−) and presented typical procyanidin oligomer fragments that confirmed the identification.
Lastly, the highest DP compounds identified corresponded to procyanidin hexamer (peak 44, m/z 864.7), procyanidin hexamer monogallates (peaks 43 and 45, m/z 940.8, see Fig 6) and procyanidin heptamer (peak 46, m/z 1008.4) which were detected as doubly-charged ions. Their fragmentation patterns were consistent with those typical of procyanidin oligomers.
Thus, the application of this new two-dimensional comprehensive HILICxRP-LC-DAD-MS method to the analysis of procyanidins from grape seeds allowed the separation and tentative identification of 46 compounds, including gallic acid ethyl ester, catechin, epicatechin and 43 procyanidin oligomers with degree of polymerization of up to 7 and degree of galloylation of up to 2. Although the separation and identification of procyanidin dimers and some trimers is already established in the chromatographic practice, it is not the same with respect to the vast number of galloylated procyanidin oligomers; in fact, one of the most important contributions of the application of the developed two-dimensional method is the separation and tentative identification of 20 non-galloylated procyanidins with DP up to 7, 11 monogalloylated procyanidins with DP from 2 to 6 and 5 digalloylated procyanidins with DP 2 to 5 in a single chromatographic run. According to the best of our knowledge, just as much as 7 monogalloylated and one digalloylated procyanidin dimers and 3 monogalloylated and a digalloylated procyanidin trimers have been previously identified using a single one-dimensional RP-LC method [29,35].
Conclusions
In this work, the development of a new two-dimensional comprehensive HILIC × RP-LC-DAD-MS/MS method to analyze grape seed procyanidins is presented. This application included the use of a microbore column with diol stationary phase for the HILIC separation in the first dimension and either a C18 partially porous column or a C18 monolithic column to carry out the RP-LC separations in the second dimension. These combinations provided high orthogonality to the multidimensional system. The optimization of this methodology allowed the separation and tentative identification of 46 phenolic compounds including flavan-3-ol monomers (catechin and epicatechin) and procyanidin oligomers up to DP 7. To the best of our knowledge, this is the first time that an on-line two-dimensional comprehensive LC is applied to the separation of procyanidins in such a complex natural sample, like grape seeds. This work also opens new possibilities to the analysis of such complex compounds as proanthocyanidins in other food and natural complex sources.
References
Kelm MA, Johnson JC, Robbins RJ, Hammerstone JF, Schmitz HH (2006) High performance liquid chromatography separation and purification of cacao (Theobroma cacao L.) procyanidins according to degree of polymerization using diol stationary phase. J Agric Food Chem 54:1571–1576
Aron PM, Kennedy JA (2008) Flavan-3-ols: nature, occurrence and biological activity. Mol Nutr Food Res 52:79–104
Sarnoski PJ, Johnson JV, Reed KA, Tanko JM, O’Keefe SF (2012) Separation and characterization of proanthocyanidins in Virginia type peanut skins by LC-MSn. Food Chem 131:927–939
Hayasaka Y, Waters EJ, Cheynier V, Herderich MJ, Vidal S (2003) Characterization of proanthocyanidins in grape seeds using electrospray mass spectrometry. Rapid Commun Mass Spectrom 17:9–16
Valls J, Millan S, Marti MP, Borras E, Arola L (2009) Advanced separation methods of food anthocyanins, isoflavones and flavonols. J Chromatogr A 1216:7143–7172
Santos-Buelga C, García-Viguera C, Tomás-Barberán FA (2003) In: Santos-Buelga C, Williamson G (eds) Methods in polyphenol analysis. The Royal Society of Chemistry, Cambridge
Monagas M, Gomez-Cordoves C, Bartolome B, Laureano O, Da Silva JMR (2003) Monomeric, oligomeric and polymeric flavan-3-ol composition of wines and grapes from Vitis vinifera L. Cv. Graciano, Tempranillo and Cabernet Sauvignon. J Agric Food Chem 51:6475–6481
Vidal S, Francis L, Guyot S, Marnet N, Kwiatkowski M, Gawel R, Cheynier V, Waters EJ (2003) The mouth-feel properties of grape and apple proanthocyanidins in a wine-like medium. J Sci Food Agric 83:564–573
Yu J, Ahmedna M, Goktepe I (2010) Potential of peanut skin phenolic extract as antioxidative and antibacterial agent in cooked and raw ground beef. Int J Food Sci Technol 45:1337–1344
Pallares V, Calay D, Cedo L, Castell-Auvi A, Raes M, Pinent M, Ardevol A, Arola L, Blay M (2012) Additive, antagonistic and synergistic effects of procyanidins and polyunsaturated fatty acids over inflammation in RAW 264.7 macrophages activated by lipopolysaccharide. Nutrition 28:447–457
Dinicola S, Cucina A, Pasqualato A, D’Anselmi F, Proietti S, Lisi E, Pasqua G, Antonacci D, Bizzarri M (2012) Antiproliferative and apoptocic effects triggered by grape seed extracts (GSE) versus epigallocatechin and procyanidins on colon cancer cell lines. Int J Mol Sci 13:651–664
Bartolomé B, Hernandez T, Bengoechea ML, Quesada C, Gomez-Cordovés C, Estrella I (1996) Determination of some structural features of procyanidins and related compounds by photodiode-array detection. J Chromatogr A 723:19–26
Peng Z, Hayasaka Y, Iland PC, Sefton M, Høj P, Waters EJ (2001) Quantitative analysis of polymeric procyanidins (tannins) from grape (Vitis vinifera) seeds by reverse phase high-performance liquid chromatography. J Agric Food Chem 49:26–31
Hellstrom JK, Mattila PH (2008) HPLC determination of extractable and unextractable proanthocyanidins in plant materials. J Agric Food Chem 56:7617–7624
Counet C, Ouwerx C, Rosoux D, Collin S (2004) Relationships between procyanidin and flavor contents of cocoa liquor from different origins. J Agric Food Chem 52:6243–6249
Robbins R, Leonczak J, Li J, Johnson JC, Collins T, Kwik-Uribe C, Schmitz HH (2012) Determination of flavanol and procyanidin (by degree of polymerization 1–10) content of chocolate, cocoa liquors, powders and cocoa flavanol extracts by normal phase high-performance liquid chromatography: collaborative study. J AOAC Int 95:1153–1160
Robbins R, Leonczak J, Johnson JC, Li J, Kwik-Uribe C, Prior RL, Gu L (2009) Method performance and multi-laboratory assessment of a normal phase high pressure liquid chromatography–fluorescence detection method for the quantitation of flavanols and procyanidins in cocoa and chocolate containing samples. J Chromatogr A 1216:4831–4840
Prior RL, Lazarus SA, Gao G, Muccitelli H, Hammerstone JF (2001) Identification of procyanidins and anthocyanins in blueberries and cranberries (Vaccinium spp.) using high-performance liquid chromatography/mass spectrometry. J Agric Food Chem 49:1270–1276
Khanal RC, Howard LR, Prior RL (2009) Procyanidin composition of selected fruits and fruit by-products affected by extraction method and variety. J Agric Food Chem 57:8839–8843
Buszewski B, Noga S (2012) Hydrophilic interaction liquid chromatography (HILIC)—a powerful separation technique. Anal Bioanal Chem 402:231–237
Greco G, Crosse S, Letzel T (2011) Study of the retention behavior in zwitterionic hydrophilic interaction chromatography of isomeric hydroxyl- and aminobenzoic acids. J Chromatogr A 1235:60–67
Stoll DR (2010) Recent progress in online, comprehensive two-dimensional high-performance liquid chromatography for non-proteomic applications. Anal Bioanal Chem 397:979–986
Herrero M, Ibañez E, Cifuentes A, Bernal J (2009) Multidimensional chromatography in food analysis. J Chromatogr A 1216:7110–7129
Dugo P, Cacciola F, Kumm T, Dugo G, Mondello L (2008) Comprehensive multidimensional liquid chromatography: theory and applications. J Chromatogr A 1184:353–368
Li X, Carr PW (2011) Effects of first dimension eluent composition in two-dimensional liquid chromatography. J Chromatogr A 1218:2214–2221
Groskreutz SR, Swenson MM, Secor LB, Stoll DR (2012) Selective comprehensive multi-dimensional separation for resolution enhancement in high performance liquid chromatography. Part I—principles and instrumentation. J Chromatogr A 1228:31–40
Donato P, Cacciola F, Tranchida PQ, Dugo P, Mondello L (2012) Mass spectrometry detection in comprehensive liquid chromatography: basic concepts, instrumental aspects, applications and trends. Mass Spectrom Rev 31:523–559
Kalili KM, de Villiers A (2009) Off-line comprehensive 2-dimensional hydrophilic interaction × reversed phase liquid chromatography analysis of procyanidins. J Chromatogr A 1216:6274–6284
Prodanov M, Vacas V, Hernández T, Estrella I (2010) In Polyphenols communications. A. Ageorges, V. Cheynier, P. Lefer and P. Sarni-Machado (eds.) vol. 2., pp. 592–593.
Bedani F, Schoenmakers PJ, Janssen HG (2012) Theories to support method development in comprehensive two-dimensional liquid chromatography—a review. J Sep Sci 35:1697–1711
Jandera P (2008) Stationary phases for hydrophilic interaction chromatography, their characterization and implementation into multidimensional chromatography concepts. J Sep Sci 31:1421–1437
Rockenbach II, Jungfer E, Ritter C, Santiago-Schübel B, Thiele B, Fett R, Galensa R (2012) Characterization of flavan-3-ols in seeds of grape pomace by CE, HPLC-DAD-MSn and LC-ESI-FTICR-MS. Food Res Int 48:848–855
Neue UD (2005) Theory of peak capacity in gradient elution. J Chromatogr A 1079:153–161
Li X, Stoll DR, Carr PW (2009) Equation for peak capacity estimation in two-dimensional liquid chromatography. Anal Chem 81:845–850
Santos-Buelga C, Francia-Aricha EM, Escribano-Bailón MT (1995) Comparative flavan-3-ol composition of seeds from different grape varieties. Food Chem 53:197–201
Acknowledgments
M.H. would like to thank MICINN for his “Ramón y Cajal” research contract. The authors want to thank Projects AGL2011-29857-C03-01 and CONSOLIDER INGENIO 2010 CSD2007-00063 FUN-C-FOOD (Ministerio de Educación y Ciencia) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Nutraceuticals and Separations with guest editor Luigi Mondello.
Rights and permissions
About this article
Cite this article
Montero, L., Herrero, M., Prodanov, M. et al. Characterization of grape seed procyanidins by comprehensive two-dimensional hydrophilic interaction × reversed phase liquid chromatography coupled to diode array detection and tandem mass spectrometry. Anal Bioanal Chem 405, 4627–4638 (2013). https://doi.org/10.1007/s00216-012-6567-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6567-5