
Abstract
Capillary electrophoresis coupled with time-of-flight mass spectrometry was used to explore new serum biomarkers with high sensitivity and specificity for diabetic nephropathy (DN) diagnosis, through comprehensive analysis of serum metabolites with 78 diabetic patients. Multivariate analyses were used for identification of marker candidates and development of discriminative models. Of the 289 profiled metabolites, orthogonal partial least-squares discriminant analysis identified 19 metabolites that could distinguish between DN with macroalbuminuria and diabetic patients without albuminuria. These identified metabolites included creatinine, aspartic acid, γ-butyrobetaine, citrulline, symmetric dimethylarginine (SDMA), kynurenine, azelaic acid, and galactaric acid. Significant correlations between all these metabolites and urinary albumin-to-creatinine ratios (p < 0.009, Spearman’s rank test) were observed. When five metabolites (including γ-butyrobetaine, SDMA, azelaic acid and two unknowns) were selected from 19 metabolites and applied for multiple logistic regression model, AUC value for diagnosing DN was 0.927 using the whole dataset, and 0.880 in a cross-validation test. In addition, when four known metabolites (aspartic acid, SDMA, azelaic acid and galactaric acid) were applied, the resulting AUC was still high at 0.844 with the whole dataset and 0.792 with cross-validation. Combination of serum metabolomics with multivariate analyses enabled accurate discrimination of DN patients. The results suggest that capillary electrophoresis-mass spectrometry based metabolome analysis could be used for DN diagnosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic nephropathy (DN) is one of the major complications of diabetes mellitus (DM) and has become the most prevalent cause of end-stage renal disease worldwide [1]. DN is also one of the most significant long-term diseases in terms of morbidity and mortality for individuals with diabetes [2]. Recent studies have shown that several interventions can slow the progression of DN, and their impact is greater if they are started at an early stage of the development of nephropathy [3]. Although renal biopsy is the most accurate diagnostic method for DN, routine renal biopsies are not acceptable in current clinical practice because of their invasiveness. Microalbuminuria is an alternative, non-invasive marker that can be used for DN risk assessment, and the urinary albumin-to-creatinine ratio (UACR) on first-void urine sample is recommended for DN screening. However, large prospective studies have revealed poor accuracy of this marker, even though urine samples are collected two or three times a day to normalize day-to-day variation [4]. Therefore, identifying reliable and versatile biomarkers for risk assessment of DN is important.
Mass spectrometry-based urinary proteomics is used for biomarker discoveries of DN. Dihazi et al. used surface-enhanced laser desorption/ionization time-of-flight mass spectrometry to identify three urinary proteins that differentiated patients with DN from patients with type 2 DM without nephropathy, patients with type 2 DM with micro- or macroalbuminuria, patients with proteinuria caused by non-diabetic renal disease, and healthy controls [5]. Mischak et al. profiled urinary polypeptides using capillary electrophoresis-mass spectrometry (CE-MS) and found that the MS patterns could be used to differentiate type 2 DM from healthy controls [6]. However, urinary protein markers sometimes show a wider variation than blood samples. Thus, it is necessary to discover biomarkers with small diurnal variations.
Metabolomics is the comprehensive analysis of low weight molecules in a sample, and has become a powerful tool in the biomarker discovery field. Nuclear magnetic resonance [7], gas chromatography-mass spectrometry [8], liquid chromatography-mass spectrometry (LC-MS; [9]), and CE-MS [10–12] are currently used for metabolomics. Targeted profiling, that is, detection of only a few sets of metabolites, has been used to discover biomarkers for DN. Xia et al. analyzed six intermediate metabolites of the purine degradation pathway in plasma from patients with non-DN and DN using LC with or without MS [13]. They found that adenosine, inosine, uric acid, and xanthine were useful biomarkers for monitoring DM progression. Jiang et al. used LC-tandem mass spectrometry to simultaneously quantify eight aminothiols in the homocysteine metabolic cycle in plasma and found two sulfur-containing metabolites, S-adenosylmethionine and S-adenosylhomocysteine, as potential biomarkers for DM and DN [14].
Compared to targeted profiling, comprehensive metabolome analysis of all metabolites in the given sample is a more powerful technique. Zhang et al. used non-targeted LC-MS to detect potential biomarker candidates of DN and type 2 DM, and observed significant differences in the serum levels of leucine, dihydrosphingosine, and phytosphingosine [15]. However, there are few published comprehensive metabolome profiles of DN.
Recently, we developed a non-targeted CE-MS-based metabolome profiling technique [11, 16] and applied it to biomarker discovery for acetaminophen-induced hepatotoxicity in mice [11] and several types of cancer-specific profiles in human saliva [12]. In the present study, we used CE-MS to identify serum metabolite biomarkers for DN diagnosis. Furthermore, classification models incorporating multiple biomarkers were constructed for discriminating DN from non-DN.
Materials and methods
Sample collection and metabolite extraction
All experiments were conducted in accordance with study protocol approved by the Institutional Ethics Committee of Chubu Rosai Hospital. Informed consent was obtained from all patients according to the Declaration of Helsinki as revised in 2000. Serum samples from 78 type 2 DM patients were collected and classified into the following three groups: DM group without nephropathy and albuminuria (non-DN, UACR < 30 mg/g, n = 20), early DN group with microalbuminuria (micro-DN, 30 < UACR < 300 mg/g, n = 32), and overt DN group with macroalbuminuria (macro-DN, UACR > 300 mg/g, n = 26). All serum samples were stored at −80 °C.
To extract metabolites, the frozen sera samples were thawed and 100 μl aliquots were put into 900 μl of methanol that contained internal standards (20 μmol/l each of methionine sulfone and camphor 10-sulfonic acid). The internal standards were used to normalize the extraction efficiency of metabolites during sample preparation for both cationic (methionine sulfone) and anionic (camphor 10-sulfonic acid) metabolite analysis. The solutions were mixed well and then 400 μl of Milli-Q water and 1 ml of chloroform were added, followed by centrifugation at 4,600×g for 5 min at 4 °C. The aqueous layer was transferred to a 5-kDa cutoff centrifugal filter tube (Millipore, Billerica, MA, USA) to remove large molecules. The filtrate was centrifugally concentrated at 35 °C and reconstituted with 50 μl of Milli-Q water that contained reference compounds (200 μmol/l each of 3-aminopyrrolidine and trimesic acid) immediately before CE-TOFMS analysis. These reference compounds were added to eliminate the variation in migration time of individual peaks in electropherogram among multiple datasets.
Reagents
Methionine sulfone (internal standard) was purchased from Alfa Aesar (Ward Hill, MA), and hexakis-(2,2-difluoroethoxy)-phosphazene (Hexakis) from SynQuest Laboratories (Alachua, FL). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO) or Wako Pure Chemicals Industries Ltd. (Osaka, Japan). All chemicals used were of analytical or reagent grade. Water was purified with a Milli-Q purification system (Millipore, Billerica, MA).
Instruments
All CE-electrospray ionization (ESI)-TOFMS experiments were performed using an Agilent CE capillary electrophoresis system (Agilent Technologies, Waldbronn, Germany), an Agilent G3250AA LC/MSD TOF system (Agilent Technologies, Palo Alto, CA, USA), an Agilent 1100 series isocratic HPLC pump, a G1603A Agilent CE-MS adapter kit, and a G1607A Agilent CE-ESI-MS sprayer kit. The CE-MS adapter kit included a capillary cassette, which facilitated thermostating of the capillary, and the CE-ESI-MS sprayer kit, which simplified coupling of the CE system with the MS system, was equipped with an electrospray source. For system control and data acquisition, G2201AA Agilent Chemstation software was used for CE, and Agilent TOF (Analyst QS) software was used for TOFMS. The original Agilent SST316Ti stainless steel (Fe/Cr/Ni/Mo/Ti; 68:18:11:2:1) ESI needle was replaced with a platinum needle for anion analysis [17]. The resolution of the TOFMS instrument used in this study is higher than 3,000 at m/z 100 with high mass accuracy (<3 ppm).
CE-TOFMS analysis of cationic metabolites
CE-TOFMS analysis of cationic metabolites was performed as described previously [10]. Cationic metabolites were separated in a fused-silica capillary (50 μm i.d. × 100 cm total length) filled with 1 mol/l formic acid as the reference electrolyte. The sample solution was injected at 5 kPa for 3 s (approximately 3 nl), and a positive voltage of 30 kV was applied. The capillary and sample trays were maintained at 20 °C and <5 °C, respectively. The sheath liquid was methanol/water (50 % v/v) containing 0.1 μmol/l Hexakis and was delivered at 10 μl/min. ESI-TOFMS was operated in positive ion mode. The capillary voltage was set at 4 kV, and the nitrogen gas (heater temperature 300 °C) flow rate was set at 10 l/min. In TOFMS, the fragmenter voltage, skimmer voltage, and octapole radio frequency voltage were set at 75, 50, and 125 V, respectively. An automatic recalibration function was performed using the following masses of two reference standards: [13C isotopic ion of the protonated methanol dimer (2MeOH + H)]+, m/z 66.06306; and [protonated Hexakis (M + H)]+, m/z 622.02896. Mass spectra were acquired at a rate of 1.5 cycles per second from m/z 50 to 1000.
CE-TOFMS analysis of anionic metabolites
The CE-TOFMS analysis of anionic metabolites was performed as described previously [17]. Anionic metabolites were separated in a commercially available COSMO(+) capillary, which was chemically coated with a cationic polymer. Ammonium acetate solution (50 mmol/l, pH 8.5) was used as the electrolyte for CE separation. The sample solution was injected at 5 kPa for 30 s (approximately 30 nl) and a voltage of −30 kV was applied. Ammonium acetate (5 mmol/l) in methanol/water (50 % v/v) containing 0.1 μmol/l Hexakis was delivered as the sheath liquid at 10 μl/min. ESI-TOFMS was operated in negative ion mode. The capillary voltage was set at 3.5 kV. In TOFMS, the fragmenter voltage, skimmer voltage, and octapole radio frequency voltage were set at 100, 50, and 200 V, respectively. An automatic recalibration function was performed using the following masses of two reference standards: [13C isotopic ion of deprotonated acetic acid dimer (2CH3COOH-H)]–, m/z 120.03834; and [Hexakis + deprotonated acetic acid (M + CH3COOH-H)]–, m/z 680.03554. Mass spectra were acquired at a rate of 1.5 cycles per second from m/z 50 to 1,000.
Data processing
The raw data were processed using our proprietary software (MasterHands) [10, 12]. The overall data processing flow consisted of noise filtering, baseline correction, peak detection, and integration of the peak areas from 0.02 m/z-wide sections of the electropherograms. Subsequently, the accurate m/z of each peak was calculated by Gaussian curve fitting in the m/z domain, and the migration times were normalized to match the detected peaks among the multiple datasets. The peaks were identified by matching m/z values and normalized migration times of corresponding authentic standard compounds. Processed peak lists were exported for further statistical analysis.
Statistical analysis
The relative ratio of the detected peak area to that of the internal standard was used to eliminate systematic bias derived from injection volume variance and MS sensitivity. Data were analyzed with GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA) for statistical tests. The Kruskal–Wallis test and Dunn’s post test were used to assess the statistical significance of differences among non-DN, micro-DN and macro-DN samples. The Spearman’s rank correlation test was used to calculate correlations among UACR, eGFR, and the relative ratios of peak areas of the metabolites. Multiple logistic regression (MLR) models were developed to discriminate non-DN and DN cohorts. Biomarker metabolites for these models were selected in two procedures. First, normalized data were subjected to orthogonal partial least-squares discriminant analysis (OPLS-DA) using SIMCA-P + software (Version 12.0, Umetrics, Umeå, Sweden), and a model was built and used to identify marker metabolites that accounted for differentiation of non-DN and macro-DN cohorts. Next, a stepwise variable selection method (forward and backward selection) was conducted with a threshold of p < 0.25 for adding and eliminating features using JMP 8.0 (SAS Institute Inc., Cary, NC, USA). The generalization ability of the developed MLR model was evaluated using cross-validation methods. Tenfold cross-validation was conducted 20 times with different random seeds using WEKA (ver. 3.6.1, The University of Waikato, Hamilton, New Zealand) to split the datasets into training and validation data [18]. Bootstrap analysis was also conducted to estimate the optimistic bias in the given datasets. Two hundred replicates, including the same number of patients, were computationally generated with a random selection of individuals, this permitted redundant selection, and MLR models were developed and cross-validation tests were conducted on each generated dataset.
Results
Metabolome analysis of serum samples obtained from non-DN and DN patients
Serum metabolome profiles of 78 patients in three successive stages of DN were collected using a single standard protocol [non-DN (n = 20), micro-DN (n = 32) and macro-DN (n = 26)] and analyzed. Age distribution, gender and other clinical characteristics are listed in Table 1. The ages in the micro-DN and macro-DN groups were slightly higher than in the non-DN group (p = 0.0226). The macro-DN group had significantly higher creatinine contents and lower estimated glomerular filtration rates (eGFR) than the other groups (p < 0.0001), while no significant difference was seen between the non-DN and micro-DN groups. The macro-DN group also showed significantly higher triglycerides and systolic blood pressure (SBP) compared with the non-DN group (p = 0.0172 and 0.0083, respectively). The other clinical parameters showed no significant difference among all groups (p > 0.05).
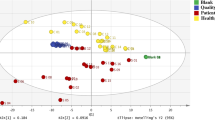
On average, 4400 peaks were detected from each serum sample with CE-TOFMS. After eliminating redundant peaks, such as noise, fragments and adduct ions, 289 metabolites remained. Using this dataset, we firstly performed principal component analysis (PCA), but the resultant score plots of the PCA showed no unequivocal stage-specific clusters (data not shown). Next, OPLS-DA was performed to discriminate between DN patients (micro-DN and macro-DN) and non-DN patients based on the profiled metabolites. The OPLS-DA model demonstrated satisfactory separation between non-DN and micro-DN patients (Fig. 1a) using one predictive component and one orthogonal component (R 2 X cum = 0.21, R 2 Y cum = 0.676, Q 2cum = 0.179), and clear separation between non-DN and macro-DN patients (Fig. 1b) using one predictive component and three orthogonal components (R 2 X cum = 0.353, R 2 Y cum = 0.946, Q 2cum = 0.599). These results indicate that serum metabolome profile can be used to distinguish DN patients from non-DN patients.

OPLS-DA based on comprehensive metabolites data from (A) non-DN (blue dots) and micro-DN (pink triangles) samples and (B) non-DN (blue dots) and macro-DN (red triangles) samples. The ellipse in each figure indicates the Hotelling T2 (0.95) range for this model
The resultant S plot of the developed OPLS-DA model between non-DN and macro-DN patients identified 19 metabolites (Table 2) that were highly correlated in the separation of the groups (|p(corr)| > 0.5). Of these, we were able to assign metabolite identities to eight metabolites by matching their m/z values and migration times with those of standard reagents. These metabolites were creatinine, aspartic acid, γ-butyrobetaine, citrulline, symmetric dimethylarginine (SDMA), kynurenine, azelaic acid, and galactaric acid. The composition formulae of the other metabolites were calculated based on their isotope distribution patterns as follows: C5H8N2O2 [metabolite ID (MID) 17], C9H17NO (MID 51), C9H19NO (MID 52), C2H4N2O3 (MID 158), and C6H6N4O (MID 202). Only the m/z values of the other metabolites are listed in Table 2 because of insufficient isotope peak size. The AUC values of MID 202 (0.765, 95 % CI); 0.649–0.880, p = 4.47 × 10−4) gave the best discriminating ability among these markers (Table 3).
Correlation between biomarker candidates and clinical parameters
Correlation analysis between these 19 serum biomarker candidates and currently available clinical parameters showed all candidate metabolites were significantly correlated with UACR (p < 0.009) (Table 4). The correlation coefficients for creatinine (r = 0.5701), aspartic acid (r = 0.4993), γ-butyrobetaine (r = 0.4942), citrulline (r = 0.4300), SDMA (r = 0.4820), kynurenine (r = 0.5351), MID 17 (r = 0.4968), MID 97 (r = 0.5223), MID 152 (r = 0.5336), MID 158 (r = 0.4980), and MID 202 (r = 0.6352) were positively correlated with UACR. Those of azelaic acid (r = –0.5210), galactaric acid (r = –0.4596), MID 51 (r = –0.4728), MID 52 (r = –0.4871), MID 96 (r = –0.3085), MID 114 (r = –0.3638), MID 127 (r = –0.2961), and MID 134 (r = –0.3669) were negatively correlated. Furthermore, 15 of 19 metabolites were significantly correlated with eGFR (p < 0.035). The correlation coefficients of creatinine (r = –0.8832), aspartic acid (r = –0.3912), γ-butyrobetaine (r = –0.6492), citrulline (r = –0.6531), SDMA (r = –0.7111), kynurenine (r = –0.5627), MID 17 (r = –0.5808), MID 97 (r = –0.7651), MID 152 (r = –0.7687), MID 158 (r = –0.6302), and MID 202 (r = –0.7455) showed negative correlation with eGFR. This indicates that they were positively associated with renal dysfunction. By contrast, those of azelaic acid (r = 0.3739), galactaric acid (r = 0.4152), MID 51 (r = 0.2204), and MID 134 (r = 0.2397) showed positive correlation with eGFR.
MLR model development
For the discrimination of DN (micro-DN and macro-DN) from non-DN patients, we developed a MLR model. Of the 19 biomarker candidates, γ-butyrobetaine, SDMA, azelaic acid, MID 114, and MID 127 were selected by stepwise feature selection as MLR variables. The developed model yielded high AUC values (0.927, 95 % CI, 0.870–0.983, p < 0.0001, Fig. 2a). The model also yielded high AUC values (±SD; 0.880 ± 8.62 × 10−3) in the cross-validation test. In a bootstrap test, the AUC values were 0.946 ± 0.0262 and 0.895 ± 0.0364 for training and cross-validation, respectively. To evaluate only the eight identified metabolites, we independently developed a MLR model. Stepwise feature selection selected aspartic acid, SDMA, azelaic acid, and galactaric acid as MLR variables. This MLR model also yielded high AUC values (0.844, 95 % CI, 0.754–0.934, p < 0.0001, Fig. 2b), although it performed slightly worse than the model with all metabolites, including unknown peaks. This model also yielded high AUC values (±SD; 0.792 ± 1.21 × 10−2) in the cross-validation test. In a bootstrap test, the AUCs were 0.875 ± 0.0419 and 0.820 ± 0.0543 for training and cross-validation, respectively. These results indicate that the developed model is sufficiently accurate, specific, and general.

ROC curve analyses in combination with (A) γ-butyrobetaine, SDMA, azelaic acid, MID 114, and MID 127, and (B) aspartic acid, SDMA, azelaic acid, and galactaric acid to discriminate non-DN and DN patients
Discussion
The aim of this study was to obtain metabolic markers for early detection of DN from patient serum samples. We used CE-MS-based metabolome analysis to find differences in the serum metabolites from non-DN, micro-DN, and macro-DN samples. OPLS-DA with 289 metabolites clearly separated non-DN from macro-DN. Adequate separation of micro-DN from non-DN was also achieved. These results show that OPLS-DA is useful in this type of analysis. The resultant S-plot of the developed OPLS-DA model identified 19 metabolites that were major contributors to the separation of macro-DN from non-DN (|p(corr)| > 0.5). These metabolites showed a gradual increase or decrease with progressive development of nephropathy. Among them, eight metabolites were identified, and these markers are discussed in comparison with other published reports below.
The concentration of serum creatinine was significantly increased in the micro-DN and macro-DN groups compared with the non-DN group (p < 0.0001), and positively correlated with UACR (r = 0.5701, p < 0.0001) and negatively correlated with eGFR (r = –0.8832, p < 0.0001). Accumulation of serum creatinine was also observed in DN patients by metabolic analysis [19]. In clinical practice, creatinine is widely used as a marker of DN that reflects the renal function. Although serum creatinine had high specificity for detecting decreased GFR, the sensitivity is not sufficient because its levels do not significantly increase until the GFR is reduced to less than 50 % of normal levels [20].
The levels of amino acids, including aspartic acid (p < 0.0001), citrulline (p = 0.0005), SDMA (p = 0.0004), and kynurenine (p = 0.0002), were significantly elevated in the DN groups compared with the non-DN group. These metabolites showed high positive correlations with UACR (aspartic acid, r = 0.4993, p < 0.0001; citrulline, r = 0.4300, p < 0.0001; SDMA, r = 0.4820, p < 0.0001; kynurenine, r = 0.5351, p < 0.0001) and negative correlations with eGFR (aspartic acid, r = –0.3912, p = 0.0004; citrulline, r = –0.6531, p < 0.0001; SDMA, r = –0.7111, p < 0.0001; kynurenine, r = –0.5627, p < 0.0001). Aspartic acid and citrulline are involved in the urea cycle. Urea, is a major end product of nitrogen metabolism, and is produced by free ammonia and aspartic acid. Citrulline is normally taken up by the kidneys and converted to urea via arginine. Chuang et al. found significant accumulation of urea cycle intermediates in the patients with end-stage renal disease [21]. Because the kidneys are important in conversion of citrulline to arginine, the increase in the serum level of citrulline in DN patients could be attributed to degradation of this function.
SDMA and asymmetric dimethylarginine (ADMA), which is a structural isomer of SDMA, are formed by the enzymatic methylation of arginine residues within proteins. These metabolites have been identified as biomarkers for chronic kidney disease [22]. ADMA is metabolized by dimethylarginine dimethylaminohydrolase (EC 3.5.3.18) into citrulline and dimethylamine in the kidneys, whereas SDMA is excreted directly into the urine without further modification [23]. In this study, ADMA was under the detection limit, but SDMA was positively correlated with a decrease in function of kidney. Therefore, SDMA is a more sensitive marker than ADMA of various renal diseases, including DN.
Tryptophan is metabolized to kynurenine and further metabolized to acetyl-CoA and NAD in the tryptophan-kynurenine pathway. The rate limiting enzymes of this pathway are indoleamine 2,3-dioxygenase (EC 1.13.11.52) in the kidney and tryptophan 2,3-dioxygenase (EC 1.13.11.11) in the liver. Both these enzymes metabolize tryptophan to N-formylkynurenine, and N-formylkynurenine is subsequently catabolized to kynurenine. Saito et al. showed the peripheral kynurenine pathway accelerates in renal insufficient rats, and the reaction rate was positively correlated with the severity of the case [24]. They also found increased serum kynurenine concentrations reflected increased tryptophan 2,3-dioxygenase and decreased kynureninase (EC 3.7.1.3) activity in the liver [24]. Integration of profiling of these enzyme activities and metabolites will increase understanding of these mechanisms.
We detected a significant increase in γ-butyrobetaine in DN patients (p < 0.0001). Toyohara et al. showed a negative correlation between γ-butyrobetaine and eGFR in plasma from the patients with chronic kidney disease [25]. Because γ-butyrobetaine is converted to l-carnitine by γ-butyrobetaine dioxygenase (EC 1.14.11.1), it is assumed the increased γ-butyrobetaine arises from inhibition of this enzyme in the kidney.
The levels of azelaic acid (p < 0.0001) and galactaric acid (p < 0.0001) were significantly lower in the DN groups than the non-DN group. These metabolites also showed high negative correlations with UACR (azelaic acid, r = –0.5210, p < 0.0001; galactaric acid, r = –0.4596, p < 0.0001) and positive correlations with eGFR (azelaic acid, r = 0.3739, p = 0.0007; galactaric acid, r = 0.4152, p = 0.0002). Azelaic acid is a saturated C9 dicarboxylic acid derived from oxidation of fatty acids and inhibits the generation of reactive oxygen species on neutrophils [26]. Galactaric acid, is a natural product found in various fruits, and acts as a growth substrate for many organisms, including Escherichia coli [27]. However, biological mechanisms of decreased serum azelaic acid and galactaric acid after onset DN need to be clarified.
In this study, the obtained 19 metabolites showed relatively high separation abilities (AUC values of receiver operating characteristic curves 0.643–0.765, Table 3). To increase the separation ability, we then applied a MLR model to this dataset. The developed MLR model included five metabolites, γ-butyrobetaine, SDMA, azelaic acid, MID 114, and MID 127. This model had a higher AUC value for diagnosis of DN (0.927, p < 0.0001) than single markers, and shows the use of multiple markers is advantageous (Fig. 2a). However, this model contained two unidentified metabolites. The model using only identified metabolites was even simpler and more versatile for actual diagnosis because it could be used with quantification by another technique, such as LC, LC-MS, or an enzymatic method. Thus, we developed another MLR model using only the identified metabolites, aspartic acid, SDMA, azelaic acid and galactaric acid (Fig. 2b). This model showed high separation ability (AUC value 0.844, p < 0.0001), and could also be used to diagnose DN. However, there are several limitations to be acknowledged for this study. For example, the developed model should be further validated using larger and independent new datasets. In addition, although we evaluated the generalization ability of the developed model using cross-validation, the specificity of the model was not assessed. Especially, the specificity for DN using data obtained from study of other kidney diseases (e.g., kidney cancer) should be addressed in future study.
In conclusion, we applied CE-MS-based metabolome profiling to serum samples from diabetic patients with or without existing DN. Biomarker candidates for the early diagnosis of DN were obtained. Although a further validation study is needed, this technique has potential as a tool for biomarker discovery studies.
References
Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T (2005) Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care 28(1):164–176
Kim HJ, Cho EH, Yoo JH, Kim PK, Shin JS, Kim MR, Kim CW (2007) Proteome analysis of serum from type 2 diabetics with nephropathy. J Proteome Res 6(2):735–743
Molitch ME, DeFronzo RA, Franz MJ, Keane WF, Mogensen CE, Parving HH, Steffes MW (2004) Nephropathy in diabetes. Diabetes Care 27(Suppl 1):S79–S83
Tabaei BP, Al-Kassab AS, Ilag LL, Zawacki CM, Herman WH (2001) Does microalbuminuria predict diabetic nephropathy? Diabetes Care 24(9):1560–1566
Dihazi H, Muller GA, Lindner S, Meyer M, Asif AR, Oellerich M, Strutz F (2007) Characterization of diabetic nephropathy by urinary proteomic analysis: identification of a processed ubiquitin form as a differentially excreted protein in diabetic nephropathy patients. Clin Chem 53(9):1636–1645
Mischak H, Kaiser T, Walden M, Hillmann M, Wittke S, Herrmann A, Knueppel S, Haller H, Fliser D (2004) Proteomic analysis for the assessment of diabetic renal damage in humans. Clin Sci (Lond) 107(5):485–495
Tiziani S, Lopes V, Gunther UL (2009) Early stage diagnosis of oral cancer using 1H NMR-based metabolomics. Neoplasia 11(3):269–276
Kind T, Tolstikov V, Fiehn O, Weiss RH (2007) A comprehensive urinary metabolomic approach for identifying kidney cancer. Anal Biochem 363(2):185–195
Sieber M, Wagner S, Rached E, Amberg A, Mally A, Dekant W (2009) Metabonomic study of ochratoxin a toxicity in rats after repeated administration: phenotypic anchoring enhances the ability for biomarker discovery. Chem Res Toxicol 22(7):1221–1231
Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, Esumi H, Soga T (2009) Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res 69(11):4918–4925
Soga T, Baran R, Suematsu M, Ueno Y, Ikeda S, Sakurakawa T, Kakazu Y, Ishikawa T, Robert M, Nishioka T, Tomita M (2006) Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J Biol Chem 281(24):16768–16776
Sugimoto M, Wong DT, Hirayama A, Soga T, Tomita M (2010) Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 6(1):78–95
Xia JF, Liang QL, Hu P, Wang YM, Li P, Luo GA (2009) Correlations of six related purine metabolites and diabetic nephropathy in Chinese type 2 diabetic patients. Clin Biochem 42(3):215–220
Jiang Z, Liang Q, Luo G, Hu P, Li P, Wang Y (2009) HPLC-electrospray tandem mass spectrometry for simultaneous quantitation of eight plasma aminothiols: application to studies of diabetic nephropathy. Talanta 77(4):1279–1284
Zhang J, Yan L, Chen W, Lin L, Song X, Yan X, Hang W, Huang B (2009) Metabonomics research of diabetic nephropathy and type 2 diabetes mellitus based on UPLC-oaTOF-MS system. Anal Chim Acta 650(1):16–22
Soga T, Ohashi Y, Ueno Y, Naraoka H, Tomita M, Nishioka T (2003) Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J Proteome Res 2(5):488–494
Soga T, Igarashi K, Ito C, Mizobuchi K, Zimmermann HP, Tomita M (2009) Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal Chem 81(15):6165–6174
Witten I, Frank E (2000) Data mining: practical machine learning algorithms with Java implementations. Morgan Kaufmann Publishers, CA
Xia JF, Liang QL, Liang XP, Wang YM, Hu P, Li P, Luo GA (2009) Ultraviolet and tandem mass spectrometry for simultaneous quantification of 21 pivotal metabolites in plasma from patients with diabetic nephropathy. J Chromatogr B Analyt Technol Biomed Life Sci 877(20–21):1930–1936
Perrone RD, Madias NE, Levey AS (1992) Serum creatinine as an index of renal function: new insights into old concepts. Clin Chem 38(10):1933–1953
Chuang CK, Lin SP, Chen HH, Chen YC, Wang TJ, Shieh WH, Wu CJ (2006) Plasma free amino acids and their metabolites in Taiwanese patients on hemodialysis and continuous ambulatory peritoneal dialysis. Clin Chim Acta 364(1–2):209–216
Fleck C, Janz A, Schweitzer F, Karge E, Schwertfeger M, Stein G (2001) Serum concentrations of asymmetric (ADMA) and symmetric (SDMA) dimethylarginine in renal failure patients. Kidney Int 59:S14–S18
Pahlich S, Zakaryan RP, Gehring H (2006) Protein arginine methylation: cellular functions and methods of analysis. Biochim Biophys Acta 1764(12):1890–1903
Saito K, Fujigaki S, Heyes MP, Shibata K, Takemura M, Fujii H, Wada H, Noma A, Seishima M (2000) Mechanism of increases in L-kynurenine and quinolinic acid in renal insufficiency. American Journal of Physiology-Renal Physiology 279(3):F565
Toyohara T, Akiyama Y, Suzuki T, Takeuchi Y, Mishima E, Tanemoto M, Momose A, Toki N, Sato H, Nakayama M, Hozawa A, Tsuji I, Ito S, Soga T, Abe T (2010) Metabolomic profiling of uremic solutes in CKD patients. Hypertens Res 33(9):944–952
Akamatsu H, Komura J, Asada Y, Miyachi Y, Niwa Y (1991) Inhibitory effect of azelaic acid on neutrophil functions: a possible cause for its efficacy in treating pathogenetically unrelated diseases. Arch Dermatol Res 283(3):162–166
Watanabe S, Yamada M, Ohtsu I, Makino K (2007) α-Ketoglutaric semialdehyde dehydrogenase isozymes involved in metabolic pathways of D-glucarate, D-galactarate, and hydroxy-L-proline. J Biol Chem 282(9):6685
Acknowledgments
We thank Dr. Astuko Watarai, Japan Labour Health and Welfare Organization Chubu Rosai Hospital, for assistance with sample collection. We also thank Maki Sugawara and Hiroko Ueda, Institute for Advanced Biosciences, Keio University, and Jiro Nakamura, Department of Endocrinology and Diabetes, Nagoya University Graduate School of Medicine, for technical support and fruitful discussions. This work was supported by a Health and Labour Sciences Research Grant “Research on Biological Markers for New Drug Development”, Grants from the Ministry of Health, Labour and Welfare of Japan “Research on Rare and Intractable Disease”, KAKENHI Grants-in-Aid for Scientific Research on Priority Areas “Systems Genomes” and “Lifesurveyor” from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and research funds from the Yamagata prefectural government and the City of Tsuruoka.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hirayama, A., Nakashima, E., Sugimoto, M. et al. Metabolic profiling reveals new serum biomarkers for differentiating diabetic nephropathy. Anal Bioanal Chem 404, 3101–3109 (2012). https://doi.org/10.1007/s00216-012-6412-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6412-x