
Abstract
A clean-up method was developed to enable the determination of tri-decabrominated diphenyl ethers, isomer-specific hexabromocyclododecanes (HBCDs), 1,2-bis(2,4,6-tribromophenoxy)ethane (BTBPE), decabromodiphenyl ethane (DBDPE), (2-ethylhexyl)tetrabromobenzoate (TBB), and bis(2-ethylhexyl)tetrabromophthalate (TBPH) in the same dust sample extract using reasonable amounts of solvents and without dividing the sample. After extraction, the sample was separated on a silica column into three fractions that were subsequently cleaned up individually. The polybrominated diphenyl ethers (PBDEs) and DBDPE were eluted in Fraction I, TBB, TBPH, and BTBPE in Fraction II, and HBCDs in Fraction III. Fractions I and II were analyzed using gas chromatography/mass spectrometry and Fraction III using liquid chromatography/mass spectrometry. The method gave good recoveries (60–120%), precise results using 13 C-labelled internal standards and was accurate when comparing results to certified values (PBDEs in NIST SRM 2585). The method was applied to dust samples from the Stockholm (Sweden) area. All the emerging brominated flame retardants (BFRs) studied, except BTBPE, were present in all the samples in quantifiable concentrations, often higher than the PBDEs. BTBPE was quantified in only one sample. It is evident that emerging BFRs are present in Swedish homes, and these compounds should be included in the BFR analyses of indoor environments.

Clean-up method for determination of BFRs in dust
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Brominated flame retardants (BFRs) are used in a wide variety of products to enhance fire resistance. Many BFRs are only physically mixed into the material [1], and due to the lack of covalent bonds between the flame retardant (FR) and the polymer, the release of these compounds into the environment is evident. Polybrominated diphenyl ethers (PBDEs), a group of BFRs, have been used as three different commercial mixtures, two of which (penta- and octaBDE mixtures) were banned by the European Union in 2004 [2]. The use of the decabrominated diphenyl ethers (decaBDE) mixture has been banned in electronic and electrical equipment in the EU in 2008 [3], and the production is being phased out in the US by 2013 [4]. Hexabromocyclododecane (HBCD) is also coming under scrutiny, and the European Commission recently announced a ban set for mid-2015 [5]. The restricted PBDEs are being replaced by other FR chemicals by the industry. 1,2-Bis(2,4,6-tribromophenoxy)ethane (BTBPE) and decabromodiphenyl ethane (DBDPE) have been detected in various biotic and abiotic matrices, indicating their distribution into different compartments of the environment [6]. Two emerging BFRs (2-ethylhexyl)tetrabromobenzoate (TBB) and bis(2-ethylhexyl)tetrabromophthalate (TBPH), are the major components in the FR mixture Firemaster® 550 (FM 550) and have recently been detected in house dust in the US [7] and in marine mammals from Hong Kong, China [8]. TBPH is also present in the mixture DP-45™ (Chemtura Inc., Philadelphia, Pennsylvania, USA), which is used as a flame retardant plasticizer.
PBDEs have been found to cause effects on neurobehavioral development, reproduction, and the thyroid system in laboratory animals, and associations have also been seen for similar effects in humans [9–16]. Toxicological data on the emerging BFRs are very scarce [17–21]. The similarities in physico-chemical properties suggest similar behavior to PBDEs. PBDEs have been monitored in the environment and humans for several years. Now, there is a need to include the emerging BFRs in monitoring programs. It would be favorable to be able to determine the established and emerging BFRs in the same samples without several sample preparations.
Previous methods to include TBB and TBPH in the analysis of BFRs in dust required splitting the dust sample into smaller aliquots, each of which underwent separate extraction and clean-up [7]. This was due to the fact that these compounds are sensitive to breakdown in acid, and thus, the effective and easy clean-up procedure using concentrated sulphuric acid cannot be applied. Another method used fractionation of the extract to separate some of the BFRs from each other but did not include HBCDs [22]. In a recently published study, a number of established and emerging FRs were separated on a Florisil column into two fractions that were first analyzed with gas chromatography/mass spectrometry (GC-MS). HBCDs were found in both fractions, and these had to be recombined before isomer-specific analysis on liquid chromatography/mass spectrometry (LC/MS) could be performed [23]. This method did not include the analysis of nonaBDEs or several of the octaBDEs (BDE-196, BDE-197, BDE-203).
The objective of the current study was therefore to develop an analytical method for determination of all the above-mentioned BFRs (PBDEs, HBCD, BTBPE, DBDPE, TBB, and TBPH) in the same dust sample extract. This required developing a fractionation method for the different analyte groups before clean-up and analysis. Clean-up methods such as gel permeation chromatography require relatively large amounts of solvents enhancing the risk of blank problems and thus higher detection limits. BTBPE co-eluted with a heptaBDE from the GC column and also produced the same ions in the MS and had to be separated from the PBDEs before the instrumental analysis. The HBCDs were isomer-specifically determined with LC/MS and were conveniently separated into one fraction from the other target compounds before the instrumental analysis. The clean-up method developed here was validated using a standard reference material (house dust) and was then applied to six dust samples from the Stockholm (Sweden) area.
Experimental
Chemicals and materials
Dichloromethane (DCM) and n-hexane (both LiChrosolv) were obtained from Merck (Darmstadt, Germany); diethyl ether (DEE) and iso-octane (both HPLC-grade) from LabScan (Gliwice, Poland); acetonitrile and methanol (B&J Brand) from Honeywell (Seelze, Germany); and sulphuric acid (AnalaR, BDH) from VWR International (Pennsylvania, USA). Water was obtained from a Milli-Q water purification unit (Millipore AB, Solna, Sweden).
Other materials used were silica gel 60 (0.063–0.200 mm) from Merck; anhydrous Na2SO4 (reagent grade) from Scharlau (Barcelona, Spain); ISOLUTE aminopropyl columns, empty reservoirs, and frits from Biotage (Uppsala, Sweden); and Bulk Isolute Sorbent, Isolute HM-N, from International Sorbent Technology Ltd, UK. The vacuum evaporator was a Syncore® Line from Büchi (Flawil, Switzerland). The standard reference material (SRM 2585, “Organic Contaminants in House Dust”) from the National Institute of Standards and Technology (NIST, Gaithersburg, MD, USA) was considered to be a homogenous dust matrix and was used in the validation of the method.
The surrogate/reference standards used were purchased from Wellington Laboratories Inc. (Guelph, Canada), Cambridge Isotope Laboratories Inc. (Andover, USA), and AccuStandard Inc. (New Haven, USA); see details in Tables S1 and S2 (Electronic supplementary material). 13 C-labelled α-, β-, and γ-HBCDs; BTBPE; BDE-183; BDE-197; BDE-207; and BDE-209 were used as surrogate standards, and d18-β-HBCD and 13 C-CB-180 as recovery standards. Reference standards were prepared in iso-octane from native TBB, TBPH, BTBPE, DBDPE, α-, β-, and γ-HBCDs, BDE-28, BDE-35, BDE-47, BDE-49, BDE-66, BDE-85, BDE-99, BDE-100, BDE-153, BDE-154, BDE-183, BDE-184, BDE-196, BDE-197, BDE-203, BDE-206, BDE-207, BDE-208, and BDE-209, at 4–12 levels covering the concentration ranges of interest (0.041–1000 pg/ul).
Samples
House dust samples from six apartments in Stockholm, Sweden, were analyzed with the validated method. The samples were collected in a previous project studying BFRs in indoor air and dust [24]. The dust was collected on surfaces 1 m above the floor, using cellulose filters in styrene-acrylonitrile holders (Krim. Teknisk Materiel AB, Bålsta, Sweden) installed in the nozzle of a vacuum cleaner. No field blanks were available for this study, but, in a previous study by our laboratory [25], the field blank levels were similar to laboratory solvent blank levels for PBDEs for these samples.
Extraction
Sample extraction was performed according to Thuresson et al. [24] with minor modifications. In short, 7–100 mg of dust was weighed into a 15-mL glass test tube, and a mixture of the following 13 C-labelled surrogate standards: BTBPE (287 pg), BDE-183 (290 pg), BDE-197 (239 pg), BDE-207 (261 pg), BDE-209 (912 pg), α-HBCD (2580 pg), β-HBCD (2540 pg), and γ-HBCD (2300 pg) in iso-octane was added. The samples were extracted twice with 15 mL DCM in an ultrasonic bath for 30 min, and the extracts were combined. The sample volume was reduced to 1 mL in a vacuum evaporator, and the solvent was changed by the addition of 5 mL n-hexane and further reduction to 1 mL.
Clean-up
The sample fractionation was performed on a column packed with 2 g silica (deactivated with 2.5% H2O) and 1 g dry Na2SO4. Before the sample extract was applied, the column was washed and conditioned with about 40 mL n-hexane. The analytes were eluted in three fractions: PBDEs and DBDPE with 30 mL n-hexane (I), TBB, TBPH, and BTBPE with 10 mL 5% DEE in n-hexane (II), and HBCDs with 10 mL 50% DEE in n-hexane (III); see Fig. 1. Fractions I and III were evaporated to 2 mL, and the solvent was changed to n-hexane in fraction III. Five milliliters concentrated H2SO4 was added to fractions I and III to remove less persistent, co-extracted compounds; the test tubes were rocked gently 20 times, centrifuged to separate the two phases, and the organic phase from each test tube transferred to new test tubes. Fraction II was evaporated to 0.5 mL, the solvent changed to n-hexane, and the sample applied to a pre-washed (12 mL n-hexane) aminopropyl functionalized silica column (0.5 g). TBB, TBPH, and BTBPE were eluted with 12 mL n-hexane. Fractions I and II were evaporated to ∼500 μL under a gentle stream of nitrogen, transferred to GC vials (conical bottom) containing 50 μl recovery standard (13 C-CB-180, 21 pg/μL), and the volume further reduced to 50 μL. The solvent in fraction III was changed to acetonitrile, evaporated to ∼900 μL under a gentle stream of nitrogen, transferred to liquid chromatography (LC) vials with 100 μL recovery standard (d18-β-HBCD, 4 pg/μL) added, and the volume further reduced to 100 μL. All final volume adjustments were done in marked vials.

The clean-up procedure for dust samples
Instrumental analysis
GC-MS
Fractions I and II were injected (1 μL) into a gas chromatograph (GC) (Trace GC Ultra) coupled to a mass spectrometer (MS) (DSQ II MS; both Thermo Scientific, Waltham, USA) to determine PBDEs, TBB, TBPH, BTBPE, and DBDPE. The GC was equipped with a programmable temperature vaporizer injector and DB-5MS fused silica column (J&W Scientific, Folsom, CA, USA, 0.25 mm inner diameter, 0.1 μm film thickness). Two column lengths were used, a shorter (12 m) column for the analysis of octa-decaBDEs, DBDPE, TBB, TBPH, and BTBPE, in order to minimize thermal degradation of the higher brominated compounds [26], and a longer one (30 m) to achieve chromatographic separation of the other PBDEs (chromatograms of a dust sample run on both columns are shown in Fig. S1, S3, Electronic supplementary material). The GC methods used are described in detail in the Electronic supplementary material (Table S3). Helium (purity 4.6, Aga, Lidingö, Sweden) was used as the carrier gas (1.5 mL/min). Electron capture negative ionization (ECNI) with ammonia (purity 5.0, Aga) as moderating gas (5.0 mL/min) was used, and the MS was operated in selected ion monitoring mode recording the bromide ions (m/z −79 and −81) and also heavier mass fragments for the different analytes (Table S3, Electronic supplementary material). The quantification was performed with XCalibur 2.0.7 (Thermo Finnigan, San Jose, CA, USA).
UPLC/MS
Fraction III was injected (2 μL) into an ultra performance LC (ACQUITYTM UPLC) coupled to a tandem-quadrupole MS (Xevo™ TQ-S) to determine the three major HBCD stereoisomers (α-, β-, and γ-HBCD). The UPLC/MS instrument and columns used were from Waters (Milford, USA). The separation was performed on a UPLC column (ACQUITY UPLC® HSS C18; 1.8 μm; 2.1 × 100 mm), with a pre-column (ACQUITY UPLC™ HSS C18; 1.8 μm VanGuard™; 2.1 × 5 mm) coupled. A mobile phase linear gradient from 78:22 to 93:7 methanol/H2O was applied. Electrospray ionization in negative mode was applied for the ionization of the analytes, and the MS was run in multiple reaction monitoring mode, measuring the quasi-molecular ions [M–H]− as parent ions and bromide ions as daughter ions. The instrumental parameters and method are described in Table S-4, and a chromatogram of a dust sample is shown in Fig. S4, Electronic supplementary material.
Recovery study
The standard reference material SRM 2585 is a house dust that is certified for a number of PBDE congeners [27]. This is a homogenous matrix and was considered suitable for the validation of the extraction/clean-up method. Dust aliquots (10 mg), unspiked and spiked with the emerging BFRs (TBB, TBPH, and DBDPE) at two levels (five of each, Table S5, Electronic supplementary material), were analyzed. Three aliquots of an inert material (Bulk Isolute Sorbent, Isolute HM-N (diatomaceous earth), prewashed with DCM, often used for filling up pressurized solvent extraction cells) were spiked at one level and analyzed in parallel to test for matrix effects on recoveries/responses. Two laboratory (solvent) blanks were processed together with the recovery study samples. All the samples were also spiked with the 13 C-labelled surrogate standards (α-, β-, and γ-HBCDs, BTBPE, BDE-183, BDE-197, BDE-207, and BDE-209).
Quality control
All glassware was heated to 470 °C and rinsed with acetone before use. UV-filters were mounted on windows and light fixtures in the laboratory to minimize degradation of the analytes. When possible, amber glassware was used; otherwise, the glassware was covered with aluminium foil. Two quality control (QC) samples (SRM 2585) and two laboratory (solvent) blanks were processed together with the real dust samples.
Degradation of BDE-209 to nona- and octaBDEs is unavoidable to some extent during sample preparation/analysis, which complicates the determination of the latter, especially if 13 C-labelled octa- and/or nonaBDEs are to be used as surrogate standards [28]. In order to monitor the degradation and be able to correct for it, the formation of 13 C-octa-BDEs and 13 C-nonaBDEs was measured. 13 C-BDE-207 was added as surrogate standard to all the samples and thus could not be monitored in terms of degradation. However, 13 C-BDE-206 and 13 C-BDE-208 were measured in every individual sample. The formation of 13 C-BDE-207 in relation to the other nonaBDEs was tested by adding only 13 C-BDE-209 to one of the QC samples, while all surrogate standards were added to the other. The 13 C-octa-BDEs and 13 C-nonaBDEs formed in the first QC sample were measured, and the ratio of 13 C-BDE-207/13 C-BDE-208 was used to derive the formation of 13 C-BDE-207 in the individual dust samples. With the assumption that 13 C- and native BDE-209 are degraded equally, the amounts of octa- and nonaBDEs formed from the native BDE-209 present in the samples could be quantified and corrected for.
Quantification was performed using surrogate standards and 4–12-point linear calibration curves (for details see Table S6, Electronic supplementary material). For the analytes lacking 13 C-labelled equivalents, the relative recovery to the surrogate standard was used to correct the results. The instrumental limits of detection and quantification were defined by signal-to-noise (S/N) ratio of 3 and 10, respectively. The method limits of quantification (mLOQ) were derived from low-level dust samples as minimum-amount analyte present in the sample giving a S/N of 10. mLOQ was calculated individually for each sample with respect to the different sample intakes (mLOQ per milligram sample, see Table 4). The method limit of detection (mLOD) was estimated as one third of mLOQ. For analytes present in the blanks, mLOD and mLOQ were set to the mean blank values plus 3 and 5 times the standard deviation of the blanks, respectively. The mLOD and mLOQ for the nonaBDEs were defined for each sample, depending on the concentration/degradation of BDE-209.
Results and discussion
Method validation
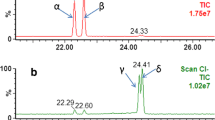
The method recoveries of the 13C-labelled surrogate standards and the emerging BFRs that were spiked to the SRM 2585 dust are shown in Table 1. The recoveries were generally high, close to 100%, with no differences between spike levels (TBB and BTBPE). For TBPH, the low spiking level was too low compared with what was present in the SRM dust, and the recovery is reported for the high spiking level only. The similar results for the two matrices (SRM dust and the inert sorbent material) seem to indicate that the method is robust for matrix differences (Table 1). TBPH and α-HBCD partially overlapped when eluted from the silica column (Fig. 1), resulting in a small proportion of TBPH being lost in Fraction III and a small proportion of α-HBCD being lost in Fraction II. This resulted in somewhat lower recoveries for these compounds (60% and 76%, respectively). There are no isotopic labelled standards available for TBB and TBPH; so, for these, the 13C-BTBPE was used as surrogate standard as it eluted in the same fraction. The differences in absolute recoveries between surrogate standard and analytes were taken into account when quantifying the samples. Although there is a 13 C-labelled DBDPE commercially available, this could not be used because the only fragment ions produced in ECNI from this compound are the bromide ions. The relative recovery of 13 C-BDE-209 and DBDPE was close to 100%, and therefore 13 C-BDE-209 was selected as a suitable surrogate standard for DBDPE. For all tri- to heptaBDEs, 13 C-BDE-183 was used as surrogate standard.
Only BDE-47, BDE-197, BDE-207, and BDE-209 were detected in the laboratory solvent blanks at much lower levels than in the samples (about 2% and less than 0.5% of the lowest sample concentration for BDE-47 and BDE-209, respectively, mean of blanks). No blank corrections were performed for BDE-47 and BDE-209. The levels of BDE-197 and BDE-207 in the blanks originated from the 13 C-internal standards added and were corrected for.
The mLOD and mLOQ ranged between 0.2–60 and 0.8–200 pg/sample for PBDEs, 20–100 and 60–300 pg/sample for the emerging BFRs, and 2–10 and 7–30 pg/sample for the HBCDs (Table S6, Electronic supplementary material). The mLOD and mLOQ for the nonaBDEs were defined individually for each sample considering the concentration and degradation of BDE-209. (The degradation of BDE-209 was estimated by measuring 13 C-nonaBDEs in each individual sample, as described in the section “Quality control” under “Experimental”). The peak area of the nonaBDE present in the sample itself (A original) was obtained by subtracting the nonaBDE peak area initiated from the degradation of BDE-209 during clean-up/analysis (A degradation) from the total (measured) nonaBDE peak area (A total), see Eq. 1 below. The original nonaBDE levels were considered quantifiable when exceeding 20% of A total (Eq. 2).
No 13 C-octaBDEs were detected as degradation products from 13 C-BDE-209. The method recoveries for 13 C-BDE-197 and 13 C-BDE-207 were equal enabling the use of the former as surrogate standard for both octa- and nonaBDEs in the analyses. The addition of 13 C-BDE-207 can therefore be left out in the future, and thus the monitoring of the 13 C-BDE-209 degradation in the sample preparation/analysis for all three 13 C-nonaBDEs can be achieved. This would result in more confidence in the quantification of BDE-207.
The method performance for the PBDEs for which we did not have 13 C-labelled surrogate standards was evaluated by comparison with their certified concentrations in the SRM 2585 dust. The PBDE concentrations found in this study were comparable to the certified values (Table 2). The higher standard deviations obtained in this study are possibly due to the much smaller sample intake used here compared with the NIST experiments (10 mg versus 500–1,000 mg) and a smaller number of samples. The lower concentration of BDE-206 found in this study compared with the certified NIST value can be explained by the correction made for the additional amount of the nonaBDE in the sample due to degradation of BDE-209. No degradation of BDE-209 to octaBDEs was observed in this study, which may be a possible explanation for our lower BDE-203 concentrations as compared with the NIST certified value. We have currently no explanation for the higher BDE-209 concentration found in this study compared with the certified value, but higher concentrations have been observed previously [24, 29].
There are no certified values for TBB, TBPH, BTBPE, or DBDPE in SRM 2585 to compare our results with, but a couple of studies have reported analytical values (Table 3). The concentrations of TBB and BTBPE in this study were similar to those reported by Ali et al. [22] and Van den Eede et al. [23] but very different from those reported by Stapleton et al. [7]. The TBPH levels here were much higher than those found in the other studies. DBDPE was not detected in any of the studies. The standard deviations in this study are fairly low and comparable to those of PBDEs (Table 2) in the same SRM material. This indicates that the SRM 2585 is homogenous also for these emerging compounds.
There are also no certified values for α-, β-, and γ-HBCD in the SRM 2585, and our results could only be compared with those from three other studies (Table 3). Our results were consistent with the preliminary values obtained from NIST (personal communication, J. Keller, NIST, USA). The α- and β-HBCD concentrations were similar in all the studies, but the concentrations of γ-HBCD found by Abdallah et al. [30] and Van den Eede et al. [23] were higher than the levels found by NIST and the current study (Table 3). In our first attempt to analyze HBCDs in SRM dust, we used a sample intake of only 10 mg. This resulted in very large differences in concentrations of the individual isomers between the replicate samples (n = 5) that could not be explained by the recoveries of the 13 C-HBCDs. The analysis was repeated with larger sample intakes (100 mg, n = 5), and the standard deviations obtained were more consistent with those for the other BFRs analyzed (Tables 2 and 3). This could be an indication that SRM 2585 is not homogenous with regard to the HBCDs.
Analysis of dust samples from homes in Stockholm
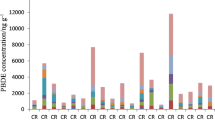
A wide range of PBDEs were detected in the house dust samples with BDE-209 contributing to ∼90% of the total PBDE concentrations. The levels were in the same range as previously published results from apartments/homes in Stockholm [24]. All of the emerging BFRs studied, except BTBPE, were found in quantifiable concentrations in all six dust samples analyzed (Table 4). BTBPE was detected in one sample only. DBDPE was found in high levels in all the samples, often higher than those of the BFR it has replaced, BDE-209. The DBDPE concentration in one of the samples was one order of magnitude higher than in the other samples. The levels of the emerging BFRs found in this study are in the same range as levels reported recently from homes in Belgium [31], the UK [32], and the US [7] but are higher than seen in a previous Swedish study [33]. In this study, TBB and TBPH were detected in all samples with TBPH generally found at higher concentrations than TBB, which is not consistent with the ratio present in the FM 550 technical product. This suggests that there are additional sources, such as DP-45TM, for TBPH in Sweden. Stapleton et al. [7] reported a wide variation (0.05–50) in the ratio of TBB/TBPH in dust samples in the US. However, the same study showed a significant correlation between TBB and TBPH in the dust samples. Another study conducted by Ali et al. [31] found no correlation between the concentrations of TBB and TBPH in indoor dust from Belgian houses and offices and from day-care centers and schools in the UK. No correlation between TBB and TBPH concentrations was found in the six samples analyzed in the current study. To our knowledge, this is the first time TBB and TBPH have been detected in house dust in Sweden.
α-, β-, and γ-HBCD were present in quantifiable amounts in all the six dust samples analyzed (Table 4), with the concentrations in one sample being one order of magnitude higher than in the other. The predominant stereoisomer in all samples was α-HBCD contributing 49–71% of the total HBCD concentrations. β- and γ-HBCD concentrations varied between 13–27% and 14–37% of the total HBCD, respectively. This is different from the composition of the technical HBCD mixture, which contains predominantly γ-HBCD (∼82%), with α- and β-HBCD contributing about 12% and 6%, respectively [34]. Higher proportions of α-HBCD than γ-HBCD have also been seen in dust from Belgium [35] and a general shift from γ-HBCD to α-HBCD has been seen in other dust studies [36, 37]. Harrad et al. [38] reported a photolytically mediated shift from γ-HBCD to α-HBCD in indoor dust when exposed to UV radiation from sunlight. Thus, photolytic conversion from exposure to sunlight could be a possible explanation for the higher proportion of α-HBCD seen in our dust samples.
References
Hutzinger O, Sundström G, Safe S (1976) Chemosphere 5:3–10
de Wit CA, Alaee M, Muir DCG (2006) Chemosphere 64:209–233
European Court of Justice (2008) Cases C-14/06 and C-295/06, Judgement of the Court, 1 April 2008, Directive 2002/95/EC and Commission Decision 2005/717/EC. Available at http://curia.europa.eu. Accessed March 2012
Hess G (2009) Industry to phase-out decaBDE. Chem. Eng. News. Available at http://pubs.acs.org/cen/news/87/i51/8751notw12.html. Accessed March 2012
Wilson A (2011) Flame retardant used in polystyrene to be banned by EU. Environ. Building News. Available at http://www.buildinggreen.com/live/index.cfm/2011/2/22/Flame-retardant-used-in-polystyrene-to-be-banned-by-EU. Accessed April 2012
de Wit CA, Herzke D, Vorkamp K (2010) Sci Total Environ 408:2885–2918
Stapleton HM, Allen JG, Kelly SM, Konstantinov A, Klosterhaus S, Watkins D, McClean MD, Webster TF (2008) Environ Part Sci Technol 42:6910–6916
Lam JCW, Lau RKF, Murphy MB, Lam PKS (2009) Environ Part Sci Technol 43:6944–6949
Birnbaum LS, Staskal DF (2004) Environ Health Perspect 112:9–17
Costa LG, Giordano G (2007) Neurotoxicology 28:1047–1067
Darnerud PO (2003) Environ Int 29:841–853
Darnerud PO (2008) Int J Androl 31:152–160
Akutsu K, Takatori S, Nozawa S, Yoshiike M, Nakazawa H, Hayakawa K, Makino T, Iwamoto T (2008) Bull Environ Contam Toxicol 80:345–350
Turyk ME, Persky VW, Imm P, Knobeloch L, Chatterton R, Anderson HA (2008) Environ Health Perspect 116:1635–1641
Herbstman JB, Sjödin A, Kurzon M, Lederman SA, Jones RS, Rauh V, Needham LL, Tang D, Niedzwiecki M, Wang RY, Perera F (2010) Environ Health Perspect 118:712–719
Harley KG, Marks AR, Chevrier J, Bradman A, Sjödin A, Eskenazi B (2010) Environ Health Perspect 118:699–704
Nakari T, Huhtala S (2010) Environ Toxicol 25:333–338
Tomy GT, Palace VP, Pleskach K, Ismail N, Oswald T, Danell R, Wautier K, Evans B (2007) Environ Part Sci Technol 41:4913–4918
Bearr JS, Stapleton HM, Mitchelmore CL (2010) Environ Toxicol Chem 29:722–729
Wang F, Wang J, Dai J, Hu G, Wang J, Luo X, Mai B (2010) Environ Sci Technol 44:5655–5660
Egloff C, Crump D, Chiu S, Manning G, McLaren KK, Cassone CG, Letcher RJ, Gauthier LT, Kennedy SW (2011) Toxicol Lett 207:25–33
Ali N, Harrad S, Muenhor D, Neels H, Covaci A (2011) Anal Bioanal Chem 400:3073–3083
Van den Eede N, Dirtu AC, Ali N, Neels H, Covaci A (2012) Talanta 89:292–300
Thuresson K, Björklund JA, de Wit CA (2012) Sci Total Environ 414:713–721
Björklund JA, Sellström U, de Wit CA, Aune M, Lignell S, and Darnerud PO (2011) Indoor air. doi:10.1111/j.1600-0668.2011.00765.x
Kierkegaard A, Sellström U, McLachlan MS (2009) J Chromatogr A 1216:364–375
National Institute of Standards & Technology, Gaithersburg, MD, USA. (2005) Certificate of analysis: Standard Reference Material 2585 "Organic contaminants in house dust”
Kierkegaard A, Asplund L, de Wit CA, McLachlan MS, Thomas GO, Sweetman AJ, Jones KC (2007) Environ Sci Technol 41:417–423
Vorkamp K, Thomsen M, Frederiksen M, Pedersen M, Knudsen LE (2011) Environ Int 37:1–10
Abdallah MAE, Ibarra C, Neels H, Harrad S, Covaci A (2008) J Chromatogr A 1190:333–341
Ali N, Harrad S, Goosey E, Neels H, Covaci A (2011) Chemosphere 83:1360–1365
Harrad S, Ibarra C, Abdallah ME, Boon R, Neels H, Covaci A (2008) Environ Int 34:1170–1175
Karlsson M, Julander A, van Bavel B, Hardell L (2007) Environ Int 33:62–69
Heeb NV, Schweizer WB, Kohler M, Gerecke AC (2005) Chemosphere 61:65–73
Roosens L, Abdallah MA-E, Harrad S, Neels H, Covaci A (2009) Environ Health Perspect 117:1707–1712
Abdallah MAE, Harrad S, Ibarra C, Diamond M, Melymuk L, Robson M, Covaci A (2008) Environ Part Sci Technol 42:459–464
Abdallah MAE, Harrad S, Covaci A (2008) Environ Sci Technol 42:6855–6861
Harrad S, Abdallah MAE, Covaci A (2009) Environ Int 35:573–579
Acknowledgments
We thank Tomas Alsberg for assistance with the UPLC/MS instrumentation; Ulla Eriksson for help with the GC/MS; and Karin Sjöstedt for laboratory work. Amelie Kierkegaard is acknowledged for valuable discussions. This study was funded by the Swedish Research Council for Environment, Agricultural Sciences, and Spatial Planning (FORMAS).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 411 kb)
Rights and permissions
About this article
Cite this article
Sahlström, L., Sellström, U. & de Wit, C.A. Clean-up method for determination of established and emerging brominated flame retardants in dust. Anal Bioanal Chem 404, 459–466 (2012). https://doi.org/10.1007/s00216-012-6160-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6160-y