
Abstract
Raman microspectroscopic imaging provides molecular contrast in a label-free manner with subcellular spatial resolution. These properties might complement clinical tools for diagnosis of tissue and cells in the future. Eight Raman spectroscopic images were collected with 785 nm excitation from five non-dried brain specimens immersed in aqueous buffer. The specimens were assigned to molecular and granular layers of cerebellum, cerebrum with and without scattered tumor cells of astrocytoma WHO grade III, ependymoma WHO grade II, astrocytoma WHO grade III, and glioblastoma multiforme WHO grade IV with subnecrotic and necrotic regions. In contrast with dried tissue section, these samples were not affected by drying effects such as crystallization of lipids or denaturation of proteins and nucleic acids. The combined data sets were processed by use of the hyperspectral unmixing algorithms N-FINDR and VCA. Both unsupervised approaches calculated seven endmembers that reveal the abundance plots and spectral signatures of cholesterol, cholesterol ester, nucleic acids, carotene, proteins, lipids, and buffer. The endmembers were correlated with Raman spectra of reference materials. The focus of the single mode laser near 1 μm and the step size of 2 μm were sufficiently small to resolve morphological details, for example cholesterol ester islets and cell nuclei. The results are compared for both unmixing algorithms and with previously reported supervised spectral decomposition techniques.

Morphological details in tissue sections are resolved by Raman imaging and might contribute together with chemical information to improved diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Raman spectroscopy has been suggested to complement histopathology in disease diagnosis and to distinguish tissue types at exposed locations (e.g. skin), by minimally invasive endoscopy or during open surgery [1]. In histopathology, tissue sections are processed, stained by dyes for contrast enhancement, and subsequently inspected by light microscopy. Standard procedures for tissue processing encompass fixation by formalin, dehydration by organic solvents, embedding in paraffin as support medium, and removal of paraffin by organic solvent after sectioning. A consequence of this process is that the composition of the sample is altered and virtually all hydrophobic molecules, for example cholesterol and lipids, are lost. Changes in lipid composition and content have been identified as valuable diagnostic properties in pathology, e.g. for atherosclerosis [2] and brain tumors [3]. Raman spectroscopy enables examination of tissue sections in their unprocessed, non-dried condition without any staining, washing, or drying processes. Raman spectra probe inherent vibrations that constitute a specific fingerprint of molecules. In the context of tissue, the vibrations of all molecules can be probed simultaneously and the Raman spectrum of all overlapping contributions can be regarded as a hyperspectral signature of the biochemical composition that is correlated with the diagnosis and provides contrast in images. Because of the complexity of the hyperspectral signatures, multivariate data analysis is required for Raman-based tissue classification. The spectral variations that can be used to distinguish tissue types or identify pathological tissues are sometimes smaller than the spectral variations arising from sample preparation. Therefore, well-defined preparation procedures have to be performed before classification models give accurate results. A prominent example is that air drying of thin tissue sections is frequently used as a surprisingly effective fixation procedure. When the water content has been vaporized in a dry atmosphere, the proteins are precipitated to form an insoluble mass that is resistant to further degradation [4]. Further alterations include the denaturation of nucleic acids and crystallization of lipids. In the context of brain tumor specimens, microcrystals of cholesterol and cholesterol ester have been detected on dried tissue sections [5, 6]. These alterations make it difficult to transfer results from tissue morphology and chemistry from dried tissue sections to non-dried tissues that are relevant for in-vivo applications of Raman spectroscopy.
Here, tissue sections were prepared from brain tumors and immersed in aqueous buffer to prevent drying during acquisition of Raman microscopic images. The Raman images were processed by use of hyperspectral unmixing algorithms called vertex component analyses (VCA) and N-FINDR. The results of both methods correlated well with the chemical composition and morphology in all recorded regions.
Materials and methods
Tissue samples
Brain tumor specimens were obtained from five patients undergoing neurosurgical craniotomies. All procedures were approved by the local ethics commission and informed consent was obtained. The samples were snap frozen in liquid nitrogen, cut at −23 °C in a cryotome, transferred on to calcium fluoride slides, shipped on dry ice, and stored at −80 °C until use. Before Raman experiments the samples were thawed in a Petri dish containing PBS solution buffered at pH 7.4. Histopathologic analysis classified the eight tissue sections as follows: molecular (a) and granular (b) layer of cerebellum, the cerebrum without (c) and with (d) scattered tumor cells of an astrocytoma WHO grade III, an ependymoma WHO grade II (e), an astrocytoma WHO grade III (f), and a glioblastoma multiforme WHO grade IV with sub-necrotic (g) and necrotic (h) regions.
Reference material
DNA sodium salt from calf thymus, proteins (bovine serum albumin and concanavalin A), cholesterol, cholesterol ester, brain extract from bovine brain containing mainly lipids, beta-carotene and PBS buffer were all from Sigma–Aldrich, Germany. Raman spectra were collected from the materials without further purification.
Raman microspectroscopy
Eight regions of interest were imaged with the CRM300 confocal Raman microspectrometer coupled to a 785 nm single mode diode laser (Witec, Germany). A 60×/NA 1.0 water-immersion objective focused the laser on to the sample and collected the scattered light in the backreflection geometry. Each image was composed of 21 × 21 spectra at a step size of 2 μm covering areas of 40 by 40 μm2. Laser power was 70 mW and the exposure time was set to 5 s per spectrum. The spectral range was registered from 299 to 2037 cm−1 with a spectral resolution of approximately 2 cm−1.
Data processing
A quality test was performed using in-house Matlab routines (The Mathworks, USA) to identify and subsequently remove spectra from the dataset with cosmic spikes or contamination from dust particles or bacteria that were accidentally trapped by the excitation laser. All images were combined in one data set and subjected to the spectral unmixing algorithms VCA and N-FINDR [7]. The VCA and N-FINDR algorithms decompose data sets into endmembers corresponding to most dissimilar spectra and abundances corresponding to concentration of endmembers. VCA and N-FINDR differ in the way endmembers are determined. VCA calculates endmember signatures using the variations in the data set. Although these endmembers are claimed to correspond to pure spectra, they might contain overlapping contributions from different spectra. This frequently happens if the number of endmembers is smaller than the number of distinguishable chemical components. After the N-FINDR endmember signatures have been determined, the algorithm searches for the most similar original input spectrum and sets it as the endmember. Then, both unmixing algorithms describe each spectrum as a linear combination of endmember spectra using non-negativity constrained least-squares fitting.
Results and discussion
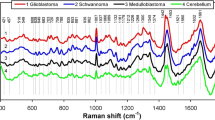
Figures 1 and 2 show seven out of ten endmember spectra obtained by using the N-FINDR (solid traces) and VCA (dotted traces) algorithms. Reference spectra of pure components are included for comparison (black traces). The remaining three spectra are dominated by noise or outliers (not shown). The first component in Fig. 1 is assigned to cholesterol (traces 1–3). The endmember spectra coincide well with typical cholesterol bands near 424, 491, 545, 606, 702, 845, 926, 1133, 1440, and 1670 cm−1. The Raman spectra of brain lipids have previously been reported in detail [8]. The coincidence of the N-FINDR endmember (trace 1) with the cholesterol spectrum (trace 3) is better than the coincidence of the VCA endmember (trace 2) with the cholesterol spectrum (trace 3). This is evident from the broader band near 1670 cm−1 probably due to amide I of proteins or OH deformation of water.

Endmember spectra using the N-FINDR (solid traces 1, 4, and 7) and VCA (dotted traces 2, 5, and 8) algorithms, and Raman spectra of the pure components (black traces 3, 6, and 9) as references for cholesterol, cholesterol ester, and lipid. Same numbering and color as in Fig. 3

Endmember spectra using the N-FINDR (solid traces 10, 13, 16 and 19) and VCA (dotted traces 11, 14, 17 and 20) algorithms, and Raman spectra of the pure components (black traces 12, 15, 18 and 21) as references for carotene, protein, DNA, and phosphate buffer. Numbering is continued from Fig. 1 and also used in Fig. 3. Same color code as in Fig. 3
The endmember spectra of cholesterol ester, CE (traces 4 and 5), contain more intense bands at 1302 and 1441 cm−1 due to fatty acids and 1731 cm−1 due to the ester group. In addition, bands of the steroid skeleton of cholesterol are shifted to 428 and 613 cm−1, and the band intensity at 547 cm−1 is reduced. These changes agree with the Raman spectrum of pure CE (trace 6). Differences between the endmembers and CE spectrum are evident in the less intense fatty acids bands at 1064, 1132, and 1302 cm−1 relative to the band at 701 cm−1 and the doublet near 1670 cm−1. These differences might be indicative of:
-
1.
co-localization of cholesterol and CE which prevents proper unmixing;
-
2.
averaging effects of cholesterol without fatty acids and CE with fatty acids;
-
3.
shorter fatty acids chains in CE compared with the reference CE; or
-
4.
the non-dried environment of fatty acids.
The endmember spectra of lipids labeled as 7 and 8 show bands of the choline group of phosphatidylcholine and sphingomyelin near 719 and 875 cm−1, the phosphate groups of phospholipids near 1087 cm−1, fatty acids side chain groups near 1063, 1130, 1298, and 1439 cm−1, and C = CH and C = C bands of unsaturated fatty acids near 1270 and 1660 cm−1, respectively. Most significant changes between the reference spectrum of lipids and endmembers are observed near 702 cm−1, because of co-localization with cholesterol and CE, and near 1002 cm−1, because of co-localization with proteins (see below). The lipid reference spectrum was collected from lipids that were extracted from brain tissue and contained virtually no cholesterol.
Figure 2 shows endmember spectra (traces 10 and 11) that contains spectral contributions of cholesterol, lipid, proteins, and carotene. Intense bands of carotene are confirmed near 1157 and 1526 cm−1 in the reference spectrum (trace 12). Similar to CE, the algorithms did not resolve a pure carotene spectrum because the component co-localized with other components and never occurred in its pure form. The detection of carotene in pathological tissue has been reported before in atherosclerosis [2] and breast cancer [9]. Because carotene has a higher cross section than all other tissue constituents it can be detected even in low quantities.
The endmember spectra labeled as 13 and 14 show all the characteristic bands of proteins (trace 15) such as aromatic amino acids (e.g. phenylalanine at 624, 1004, and 1033 cm−1, tyrosine at 645, 831, and 851 cm−1, tryptophan at 760, 1343, and 1550 cm−1), aliphatic amino acids near 1446 cm−1, C–C stretching vibrations at 1129 cm−1 and the peptide backbone (amide I near 1667 and amide III near 1250 cm−1). The CH2 deformation band is shifted from 1439 cm−1 in lipids to 1446 cm−1 in proteins as a consequence of the lower CH2 to CH3 ratio in proteins. Differences between the reference spectrum (trace 6) and the endmembers (traces 4 and 5) are found near 1250 and 1667 cm−1. These differences are assigned to different secondary and tertiary structures. The reference spectrum represents a mixture of the alpha helical protein bovine serum albumin and the beta sheet protein concanavaline A. The protein composition in brain tumor tissue is more complex and deviates from this mixture. Furthermore, the conformations differ for hydrated proteins in non-dried tissues and non-hydrated proteins in the lyophilized reference material.
The endmember spectra labeled as 16 and 17 are dominated by spectral contributions of nucleic acids. Bands near 498, 683, 727, 788, 1255, 1340, 1376, 1488, and 1580 cm−1 are assigned to nucleotides adenine, guanine, cytosine, and thymine. The band at 1094 cm−1 is assigned to the phosphate backbone in the B-DNA conformation. These bands coincide well with the reference spectrum of DNA (trace 18). Differences between the reference spectrum and endmembers near 1002, 1250–1300, and 1660 cm−1 are due to proteins. This can be explained by the biological fact that DNA in cell nuclei does not occur in an isolated form but in complexes with histone proteins that are involved in wrapping of DNA into superstructures.
The endmember spectra labeled as 19 and 20 are assigned to the buffer solution with typical bands of OH deformation vibrations of water near 1640 cm−1 and phosphate ions near 990 cm−1. Both endmembers coincide with a reference spectrum of the buffer solution (trace 21). Comparing the endmember spectra reveals that the signal-to-noise ratio (SNR) is better after VCA unmixing than after N-FINDR unmixing. This general feature can also be observed for the other endmembers. This effect, which is most pronounced for the buffer solution, can be explained in the following way. Whereas SNR of N-FINDR endmember is typical for single spectra, the SNR of VCA endmember is the result of a VCA model which uses all spectra of the data set and represents the total acquisition time. Consequently, VCA endmembers are expected to have better SNR than N-FINDR endmembers.
Figures 3 and 4 show the abundance plots for eight Raman images using VCA and N-FINDR, respectively. Each Raman image was unmixed into seven components whose endmembers are shown in Figs. 1 and 2 with the same colors and numbering as in Figs. 3 and 4. The letters refer to the histopathology analysis given in the “Materials and methods” section.

Abundance plots of eight Raman images using the VCA algorithm. Each image is composed of 21 × 21 spectra at step size 2 μm covering areas of 40 × 40 μm2. Pathology findings (column): molecular (a) and granular (b) layer of cerebellum, cerebrum without (c) and with (d) scattered tumor cells of an astrocytoma III, ependymoma II (e), astrocytoma III (f), and glioblastoma multiforme IV with sub-necrotic (g) and necrotic (h) regions. Endmember components (row): protein, lipid and cholesterol with carotene (11, cyan), DNA (17, yellow), cholesterol (2, green), cholesterol ester (5, green), buffer solution (20, blue), protein (14, red), and lipid (8, magenta). Last rows display composite images. Same numbering and color code are used as in Figs. 1 and 2

Abundance plots of eight Raman images using the N-FINDR (B) algorithm. Each image is composed of 21 × 21 spectra at step size 2 μm covering areas of 40 × 40 μm2. Pathology findings (column): molecular (a) and granular (b) layer of cerebellum, cerebrum without (c) and with (d) scattered tumor cells of an astrocytoma III, ependymoma II (e), astrocytoma III (f), and glioblastoma multiforme IV with sub-necrotic (g) and necrotic (h) regions. Endmember components (row): protein, lipid, and cholesterol with carotene (10, cyan), DNA (16, yellow), cholesterol (1, green), cholesterol ester (4, green), buffer solution (19, blue), protein (13, red), and lipid (7, magenta). Last rows display composite images. Same numbering and color code are used as in Figs. 1 and 2
Raman images (a)–(c) represent the typical morphology and chemical components of normal brain tissue sections that are characterized by cell nuclei with diameters between 5 and 8 μm and regular cell density (rows 16, 17). This is shown for molecular layer of cerebellum (a) or cerebrum (c). The subcellular resolution of cell nuclei agrees with previous reports on liver tissue [10] and cartilage [11]. The highest abundance of DNA is found in the granular layer of the cerebellum (b). Because of their high density, individual cell nuclei were not completely resolved. Furthermore, CE (rows 4, 5), protein (rows 13, 14), and lipid (rows 7, 8) are quite homogenously distributed over the regions of interest. Cerebellum (a) and (b) was found to contain more protein and fewer lipids than cerebrum (c). Whereas these observations agree for N-FINDR and VCA, the CE distributions differ (rows 4, 5). On the one hand, the VCA algorithm revealed more CE in the cerebellum than in the cerebrum. On the other hand, the N-FINDR algorithm revealed fewer CE in the cerebellum than the cerebrum. A possible explanation is that the abundance values depend on the intensities of the endmembers. Although the normalized signatures of CE in Fig. 1 (traces 4 and 5) are similar, their relative intensities might be different.
Raman images (d)–(h) represent different malignancy grades of primary brain tumors. Individual cell nuclei were resolved in images (d)–(g). The diameter and number of cell nuclei increase compared with normal tissue (rows 16, 17). In particular, the astrocytoma III sample (f) contains several large cell nuclei with maximum DNA abundance that are consistent with the high malignancy of the brain tumor. In glioblastoma multiforme IV (g), the most malignant primary brain tumor, the diameter, number, and abundance values of cell nuclei were lower than astrocytoma III (f). This observation is consistent with subnecrotic regions of the tumor. No cell nuclei and abundance values near zero for the DNA component were found in the necrotic portion of the tumor (h). The necrosis is also characterized by maximum abundance of proteins (rows 13, 14), the presence of carotene (rows 10, 11), and different cholesterol composition (rows 1, 2). Spectral contributions of carotene were also detected in the cerebrum with scattered astrocytoma III cells (d). The expression of carotene or its accumulation might be because of the dedifferentiation of malignant tumor cells.
The highest abundance of CE (rows 4, 5) is found for the astrocytoma III specimen (d) by VCA and N-FINDR. Here, an islet of elevated CE content is formed. Such morphology has not previously been observed by Raman imaging of dried tissue sections, because CE forms microcrystals after dehydration. Detection of high CE content is consistent with malignant brain tumors. Up to 100-fold increase has been reported in brain tumors compared with normal brain tissue [12].
The lipid (rows 7, 8) to protein (rows 13 14) ratio in IR and Raman spectra of brain tissue has been suggested as a diagnostic marker [13, 14]. Here, the protein abundances are high in the cerebellum control sample (a) and (b) and necrosis (h). The protein abundances are lower in the cerebrum (c), brain tumor specimens of astrocytoma III, and ependymoma II (d)–(f). Lipid abundances are similar for all samples (a)–(g) except for necrosis (h). Local minima of lipid abundances within each image are because of non-lipid features, for example cell nuclei (b), (e), and (f), CE (d), and buffer solution (e). The abundance of the buffer solution (rows 19, 20) has maximum intensity for ependymoma II (e) which points to lower sample density than for the other regions.
More and larger regions of interest will be studied to improve correlations between the chemical composition and the morphology that are evident from spectral unmixing by VCA and N-FINDR. The chemical composition is an important diagnostic property and can be probed using Raman-based techniques. Previous Raman studies used pure components, for example cholesterol, lipids, proteins, and buffer, to approximate the chemical composition of breast [9] or brain tissue[14]. In the context of Raman spectroscopy from arterial tissue, lipids were extracted to obtain remnants of, primarily, proteins [2]. This component was added to the set of pure references. In particular for the nucleic acid, protein, and lipid component, it is challenging to prepare an appropriate reference compound, because the exact composition is unknown. Spectral unmixing algorithms offer a novel way of obtaining Raman spectra of pure components to analyze the tissue composition and its disease-related changes.
Raman imaging has already been applied for real-time assessment of brain tissue under in-vivo [15] and ex-vivo [14] conditions after coupling to fiber optic probes. However, individual features such as cell nuclei were not resolved in these Raman images because of the 100 μm step size. A related vibrational spectroscopic technique is infrared spectroscopy. However, the spatial resolution obtained in infrared spectroscopic images is inferior because of the longer wavelengths in the mid-infrared range, and the penetration of infrared radiation into non-dried tissue is limited to few micrometers because of the strong absorbance of water. Another promising technique for assessing extended regions of interest at subcellular resolution with exposure times of seconds is coherent anti-Stokes Raman scattering (CARS). The application of CARS in the context of brain tumors has recently been reported [16, 17]. In future developments the objective is to transfer the current spontaneous Raman results to CARS microscopy, and, finally, bring CARS methodology into the operating theatre.
Conclusions
The Raman spectroscopic images reported here for non-dried brain tissue at a step size of 2 μm constitute significant progress, because such data can be correlated with histopathologic findings at the microscopic level and the results can be transferred to in-vivo collected spectra better than results from dried tissue sections. Raman microscopic imaging combined with hyperspectral unmixing has been shown to be an innovative method for visualizing biochemical and morphological features of unprocessed brain tissue. The advantages of unsupervised unmixing algorithms include that reference spectra are generated directly from the Raman data sets and external reference spectra are not required. This Raman-based approach can complement standard histopathologic procedures because molecular contrast is obtained on the subcellular level without preparation.
References
Krafft C, Steiner G, Beleites C, Salzer R (2008) Disease recognition by infrared and Raman spectroscopy. J Biophoton 2:13–28
Römer TJ, Brennan JF, Schut TC, Wolthuis R, van den Hoogen RC, Emeis JJ, Van der Laarse A, Bruschke AV, Puppels GJ (1998) Raman spectroscopy for quantifying cholesterol in intact coronary artery wall. Atherosclerosis 141:117–124
Campanella R (1992) Membrane lipids modifications in human gliomas of different degree of malignancy. J Neurosurg Sci 36:11–25
Romeo M, Mohlenhoff B, Diem M (2006) Infrared micro-spectroscopy of human cells: Causes for the spectral variance of oral mucosa (buccal) cells. Vib Spectrosc 42:9–14
Koljenovic S, Choo-Smith LP, Bakker Schut TC, Kros JM, van den Bergh H, Puppels GJ (2002) Discriminating vital tumor from necrotic tissue in human glioblastoma tissue samples by Raman spectroscopy. Lab Invest 82:1265–1277
Krafft C, Sobottka SB, Schackert G, Salzer R (2006) Raman and infrared spectroscopic mapping of human primary intracranial tumors: a comparative study. J Raman Spectrosc 37:367–375
Hedegaard M, Matthäus C, Hassing S, Krafft C, Diem M, Popp J (2011) Spectral unmixing and clustering algorithms for assessment of single cells by Raman microscopic imaging. Theor Chem Acc 130:1249–1260
Krafft C, Neudert L, Simat T, Salzer R (2005) Near infrared Raman spectra of human brain lipids. Spectrochim Acta A 61:1529–1535
Shafer-Peltier KE, Haka AS, Fitzmaurice M, Crowe J, Myles J, Dasari RR, Feld MS (2002) Raman microspectroscopic model of human breast tissue: implications for breast cancer diagnosis in vivo. J Raman Spectrosc 33:552–563
Krafft C, Alipour Didehroshan M, Recknagel P, Miljkovic M, Bauer M, Popp J (2011) Crisp and soft algorithms visualizes cell nuclei in Raman images of liver tissue sections. Vib Spectrosc 55:90–100
Bonifacio A, Beleites C, Vittur F, Marsich E, Semeraro S, Paoletti S, Sergo V (2010) Chemical imaging of articular cartilage sections with Raman mapping, employing uni- and multi-variate methods for data analysis. Analyst 135:3193–3204
Nygren C, vonHolst H, Mansson JE, Fredman P (1997) Increased levels of cholesterol esters in glioma tissue and surrounding areas of human brain. Br J Neurosurg 11:216–220
Krafft C, Sobottka SB, Schackert G, Salzer R (2004) Analysis of human brain tissue, brain tumors and tumor cells by infrared spectroscopic mapping. Analyst 129:921–925
Krafft C, Sobottka SB, Schackert G, Salzer R (2005) Near infrared Raman spectroscopic mapping of native brain tissue and intracranial tumors. Analyst 130:1070–1077
Kirsch M, Schackert G, Salzer R, Krafft C (2010) Raman spectroscopic imaging for in vivo detection of cerebral brain metastases. Anal Bioanal Chem 398:1707–1713
Evans CL, Xu X, Kesari S, Xie XS, Wong STC, Young GS (2007) Chemically-selective imaging of brain structures with CARS microscopy. Opt Express 15:12076–12087
Meyer T, Bergner N, Bielecki C, Krafft C, Akimov D, Romeike BF, Reichart R, Kalff R, Dietzek B, Popp J (2011) Nonlinear microscopy, infrared, and Raman microspectroscopy for brain tumor analysis. J Biomed Opt 16:021113
Acknowledgements
This work was funded by the European Union via the “Europäischer Fonds für Regionale Entwicklung” (EFRE), the “Thüringer Ministerium für Bildung, Wissenschaft und Kultur” (project B714-07037), and the “Bundesministerium für Bildung und Forschung” (BMBF) within the MediCARS project (FKZ: 13N10774).
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is dedicated to Professor Reiner Salzer on the occasion of his 70th birthday to honor his substantial achievements in analytical chemistry and vibrational spectroscopy.
Rights and permissions
About this article
Cite this article
Bergner, N., Krafft, C., Geiger, K.D. et al. Unsupervised unmixing of Raman microspectroscopic images for morphochemical analysis of non-dried brain tumor specimens. Anal Bioanal Chem 403, 719–725 (2012). https://doi.org/10.1007/s00216-012-5858-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-5858-1