
Abstract
A procedure for the determination of traces of mercury by liquid-phase microextraction based on solidification of a floating organic droplet for separation and electrothermal atomic absorption spectrometry for final measurement has been developed. For this purpose, 50 µL of pre-heated (50 °C) undecanoic acid (UA), are added to 25 mL of aqueous sample solution at pH 5. The mixture, maintained at 50 °C, is stirred for 10 min using a high stirring rate in order to fragment the UA drop into droplets, thus favoring the extraction process. Next, the vial is immersed in an ice bath, which results in the solidification of the UA drop that is easily separated. Injection into the atomizer is carried out after gentle heating. The pyrolytic atomizers are coated with electrolytically reduced palladium that acts as an effective chemical modifier for more than 500 firings. Under the optimized conditions, the detection limit was 70 ng L−1 mercury with an enrichment factor of 430. The relative standard deviation of the measurements was in the 2.1–3.5% range. Recovery studies applied to the determination of mercuric ions in bottled and tap water samples were in the 92–104% range.

LPME-SFO allows liquid phase and the solidified organic reagent to be easily separated
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Miniaturized preconcentration methods based on liquid–liquid or solid-phase extraction [1–4] have recently aroused a great interest due to the favorable characteristics they present. The use of organic reagents is minimized and may even be avoided, and high preconcentration factors are obtained. Today, there is a whole array of such miniaturized techniques available involving liquid-phase microextraction (LPME). In some cases, a drop of organic extractant is held in a capillary tip and immersed in the sample solution [5–8] or maintained in its head-space [9, 10]. In other approaches, the extractant is placed inside a hollow-fiber [11, 12] or on the surface of a stir bar [13].
Another group of LPME techniques does not use a physical support for the organic solvent, and it is directly added to the sample solution instead. When using this approach, the way to proceed depends on the density of the organic liquid. For solvents heavier than water, dispersive liquid–liquid microextraction (DLLME) [14, 15] in which a disperser agent is added together with the extractant, has proved useful for a number of analytes. In this case, after a brief shaking step, the organic extract is recovered by centrifugation. For organic solvents lighter than water, DLLME is less suitable and liquid-phase microextraction by solidification of a floating organic droplet (LPME-SFO) [16] can be used instead. In such cases, a few microliters of an organic solvent with low-melting point are used. The aqueous sample solution is stirred and maintained at such a temperature that the organic droplet remains melt for the extraction to occur. Once the process is finished and the liquid-cooled, the solidified extractant floats on the aqueous sample and can be easily separated by means of a spatula. This technique has been successfully applied using undecanol and dodecanol to concentrate substances such as trihalomethanes [17] organochlorine [18] and organophosphorus pesticides [19] polycyclic aromatic hydrocarbons [16] pyrazoline derivatives [20], and some metals [21–23]. Recently, a new technique which combines DLLME and LPME-SFO has been reported [24–26].
In this paper, the separation and preconcentration of low amounts of mercury ions by using undecanoic acid (UA) is reported. This weak acid, that acts both as the complexing and extracting agent, melts at a low temperature (28–31 °C), and has recently [27] proved to be suitable for LPME-SFO methodology. The mercury contained in the low-volume organic extract obtained can be measured by electrothermal atomic absorption spectrometry (ETAAS), provided that an effective chemical modifier is present. The procedure described could be of interest for fully exploiting the ETAAS instruments that are nowadays present in the vast majority of laboratories.
Materials and methods
Instrumentation
A model 939QZ atomic absorption spectrometer (ATI-Unicam, Cambridge, UK) equipped with a Zeeman-based correction device, a longitudinally heated graphite furnace atomizer and pyrolytic graphite platforms inserted into pyrolytically coated graphite tubes were used. Argon was the inert gas, the flow rate being 300 mL min−1 during all the stages, except for atomization when the flow was stopped. Measurements were carried out in the peak-area mode using a mercury hollow cathode lamp operated at 6 mA and 253.7 nm with a 0.5 nm spectral bandwidth. The instrumental parameters used are summarized in Table 1.
All glassware and plastic (polypropylene) vessels used for preparing and storing solutions were nitric acid-washed and rinsed with ultrapure water. Pipette tips were also of polypropylene.
Reagents
Ultrapure water, obtained using a Milli-Q system (Millipore, Bedford, MA, USA), was used exclusively. The Hg(II) stock standard solution (1,000 mg L−1) was obtained from Panreac (Barcelona, Spain). Working standard solutions were obtained by appropriate dilution of the stock standard solutions with 2% (v/v) nitric acid. The palladium modifier solution was obtained from a 10.0 ± 0.3 g L−1 Pd (as the nitrate) stock solution (Fluka, Buchs, Switzerland) with 15% (v/v) nitric acid. For checking other modifiers, 1,000 mg L−1 Au and Pt atomic absorption standard solutions (Fluka) were used. Undecanoic acid was obtained from Sigma-Aldrich (Sigma-Aldrich Chemie GmbH, Germany). The rest of the chemicals used were obtained from Fluka.
Treatment of the tubes and platforms with palladium
Both tube and platform were introduced into a vial containing a 10 g L−1 Pd(II) solution and left overnight (approximately 12 h). Next, the atomizer was placed in a small plastic tube containing 250 µL of the palladium solution and connected to a 3-V power source, the electrolysis being carried out during 3 min using a small platinum wire as the anode. Finally, the tube and platform were dried for 15 min at 120 °C and then preconditioned by running three times the heating program given in Table 1
Analytical procedure
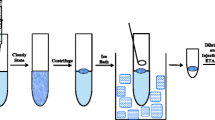
To 25 mL of sample placed in a 30-mL screw cap vessel vial with flat bottom, 0.25 mL of a 1 mol L−1 acetate buffer solution of pH 5 was added. The solution was heated at 50 °C using a thermostatic block placed on a magnetic stirrer, and next stirred using a high speed (at least 1,000 rpm) in order to obtain a vortex in the solution. Then 50 µL UA, pre-heated to 50 °C, were incorporated with a micropipette and the stirring maintained for 10 min. The stirring speed was next decreased (300 rpm approximately) for 2 min in order to group all the droplets and the vial was cooled in an ice bath. The solidified UA was removed with a spatula, placing it in a 2-mL Eppendorf vial. The vial was heated in a heat block at 50 °C, a 10-µL aliquot of the liquid was taken and injected into the atomizer and the program given in Table 1 was run. Calibration was carried out using aqueous standards submitted to the same sample treatment. For samples containing more than 50 mg L−1 calcium a back-extraction procedure is recommended (see below).
Results and discussion
Preliminary experiments showed that mercuric ions were extracted into a small volume of UA thus allowing the analyte to be efficiently preconcentrated. Taking into account the volatility of mercury, it is clear that a chemical modifier is mandatory to carry out the ETAAS measurement. A number of chemicals have been reported for the purpose [28], iridium as a permanent modifier [29, 30] and palladium salts being the most commonly recommended [31]. In the case here studied, two immiscible phases are involved, and so the addition of an aqueous palladium solution to the liquid UA would not produce an effective modification effect. Consequently, instead of using an aliquot of chemical modifier for each firing, the pre-treatment of the atomizer to obtain a long-term modifying action, effective for a large number of firings, was studied. To select the most suitable modifier, a large number of experiments were carried out by impregnating the pyrolytic material (both platforms and tubes) with solutions containing palladium, platinum, or gold salts overnight, and then adding a reducing agent (ascorbic acid) to the pretreated atomizers. The pyrolytic material thus preconditioned was used to obtain atomization profiles of mercury. In all cases, 25 mL of a 2-µg L−1 mercury solution were extracted into 50 µL of UA and 10 µL of each extract were injected. The characteristic masses obtained were 150, 125, and 103 pg mercury when using gold, platinum, and palladium, respectively. In addition, to provide the best sensitivity, reduced palladium proved effective for a large number of measurements. Since iridium has been recommended for stabilizing mercury [29, 30], additional experiments were carried out for comparing its performance with that shown by palladium. The results were similar when aqueous solutions were used, but in the presence of UA the sensitivity worsened (characteristic mass 120) for the case of iridium. As can be seen in Fig. 1, the use of palladium allowed a long-term mercury stabilization. However, after approximately 120 firings, the sensitivity worsened, indicating that a new impregnation is required. The literature reports that for chemical modification purposes, electrolytic coating is more effective than chemical coating by means of a reducing agent, and so this pre-treatment was checked. By using a very simple electrolytic procedure, as indicated in the experimental section, the coating with palladium proved so effective that no noticeable changes in sensitivity or premature loss of mercury was observed during almost 600 consecutive heating cycles in the conditions summarized in Table 1. To ensure complete reliability of the results, it is recommended to impregnate and re-coat the atomizer after 500 firings.

Analytical signals obtained of a 2 µg L−1 mercury solution using atomizers treated with gold, platinum and palladium salts, curves a–c, respectively. Each point is the average of ten measurements. Vertical bars indicate the standard deviation in each case
Ashing–atomizing curves were studied and the results obtained when using the atomizer coated electrolytically with palladium are shown in Fig. 2a. The recommended values are 450 and 1,400 °C for the pyrolysis and atomization stages, respectively. Using the optimized conditions, the characteristic mass was found to be 67 pg, which agrees with previous reports [28]. A hot injection (80 °C are recommended) of the UA extract is required to avoid problems caused by solidification of the organic reagent inside the micropipette tip. The final temperature program is given in Table 1, and a typical firing showing the low background signal obtained during the atomization stage is shown in Fig. 2b.

a Ashing (a) and atomizing (b) graphs obtained of a 2 µg L−1 mercury solution using palladium electrolytically reduced as the modifier. b Atomization profile of a 1 µg L−1 mercury solution (curve a). Curve b is the background signal
Optimization of the microextraction stage
The usual practice of LPME-SFO involves that a small volume of a low-melting point solvent floats on the top of the aqueous sample solution while it is being stirred. This means that the contact surface between the organic and aqueous phases is small and, consequently, a relatively long extraction time is required. To speed-up the microextraction process, it is more convenient to use a high stirring speed in such a way that a pronounced vortex originates in the solution. Thus, when the organic solvent is incorporated, the drop is fragmented into many droplets that are submitted to repeated displacements towards the bottom and the top of the sample solution. The large contact surface in this way obtained increases the rate of the extraction process. Once the microextraction process is considered to be finished, it is sufficient to decrease the stirring speed in order the droplets regroup and, after a suitable cooling, the low-melting point extractant solidifies and can easily be separated from the aqueous phase. For most of the experiments here reported, a 29-mm outer diameter vial containing 25 ml of aqueous solution was used. When the solution was stirred (1,000 rpm) by means of a 10 × 3 mm stir bar, a vortex that extends down to the base of the vial was obtained. In this way, a 50 μL UA drop is fragmented into small droplets that easily regroup when the stirring speed is reduced to 300 rpm. On the other hand, the extraction temperature has to be high enough to allow UA (melting point 28 °C approximately) to be in the liquid form. The recommended value is 50 °C. It is important to note that this behavior shown by UA permitted an easy-to-perform effective separation, while some other low-density, low-melting point solvents checked (1-undecanol, 1-decanol, and decanoic acid) proved inappropriate for the purpose since the droplets did not regroup easily.
On the contrary, an inconvenience of using UA lies in the fact that it is slightly soluble in water, the solubility increasing with the pH, hindering the extraction above pH 7. The effect of the acidity of the aqueous solution on the extraction of mercury by UA was studied and maximum extraction was found at pHs close to 5. Consequently, the rest of the experiments were carried out in the presence of a diluted (0.01 mol L−1) acetate/acetic acid buffer solution of pH 5.
The optimal volume of UA to be used was studied in the 5–50 µL range, while the volume of the aqueous phase was considered in the 10–50 mL range. It is clear that for a maximum preconcentration, the UA volume should be kept at a minimum while that of the aqueous phase should be as large as possible. Finally, taking into account that very low UA volumes are not advisable because a part of the reagent dissolves in water, and too large volumes for the aqueous phase would increase the extraction time, a solution of compromise was adopted by selecting 50 µL UA and 25 mL sample solution. When using the recommended conditions, the volume of UA recovered after the microextraction stage was approximately40 µL, which allowed 10-µL aliquots to be injected in duplicate.
The effect of the extraction time was studied and the results are shown in Fig. 3. As can be seen, the signal obtained (curve a) was practically constant and reached a maximum after 10 min. The graph also shows (curve b) the rate of the process. On the other hand, to verify the high percentage of extraction, each solution was extracted twice, the analytical signal being also obtained after the second extraction and a value of 85% was calculated. This value has to be considered only as approximate, due to the way in which it was obtained. In addition, the enhancement factor was also evaluated. For this purpose, the ratio between the slope of a calibration line obtained from aqueous solutions submitted to the microextraction process and the slope of a calibration line obtained for aqueous solutions that were not extracted (for obtaining this line, the heating program was slightly modified by using 130 °C as the drying temperature in order to avoid spattering) was calculated, and a value of 430 was found. This value has again to be considered as an experimental estimation, since it is affected by factors such as the different response of ETAAS measurements in UA and aqueous solutions, as well as by the slight solubilization of the organic reagent in water. In any case, taking into account the experimental errors, this estimation agrees with the already mentioned high percentage of extraction.

Effect of the stirring time (line a) on the analytical signal of a 1.5 µg L−1 mercury solution. The rate of change of the signal is given by line b
Analytical figures of merit
Using 25 mL sample solution and 50 µL for the organic reagent, a calibration graph was obtained in the 0.2–3 µg L−1 mercury range. The relative standard deviation (five measurements in each case) was in the 2.1–3.5% range. The detection limit calculated on the basis of three times the standard error from the calibrating regression (s/x) was 0.07 µg L−1 mercury. Due to the selectivity of ETAAS measurements no significant interferences caused from ions commonly present in waters was observed, with the exception of calcium. It was experimentally verified that for calcium concentrations exceeding 50 mg L−1, approximately, the UA molten drop was covered by a white solid attributed to the calcium salt. The atomization of an aliquot of this calcium-enriched extract led to a high background that rendered mercury measurement unreliable. To overcome this drawback, 25 µL of the UA extract were back-extracted with 25 µL of a 1 mol L−1 hydrochloric acid solution. The addition of 25 µL cyclohexane allowed a rapid separation of the phases without the need of centrifugation. Mercury was then measured by injecting 10 µL of the aqueous phase. In this case, since an aqueous solution was injected, the drying temperature of the heating program was lowered to 130 °C to avoid spattering. This treatment permitted well-shaped mercury atomization profiles to be obtained with a very low background.
The procedure was applied to seven water samples, namely, five bottled mineral water samples, tap water and tap water treated by means of a domestic purification system. No mercury was detected in any case. The samples were then spiked by incorporating 0.5 and 1 µg L−1 mercury and the recoveries (five extractions for each sample at each level, the final solutions being measured in duplicate) were in the 92–104% range. The tap water sample had a relatively high calcium content that made necessary to use the back-extraction procedure to overcome the drawback caused by a high background during atomization. In this particular case, the recovery of a 1 µg L−1 mercury spike was 97% (mean value for five experiments, a single measurement for each final aqueous solution obtained after back-extraction)
Table 2 summarizes the main characteristics of relevant procedures for mercury traces based on final ETAAS measurements. The preconcentration factor as well as the characteristic mass of mercury achieved in the procedure here studied compare favorably with these procedures, the detection limit being somewhat higher than that shown by the methodology based in cold vapor followed by retention into the atomizer.
References
Pena-Pereira F, Lavilla I, Bendicho I (2009) Spectrochim Acta 64B:1–15
Nerín C, Salafranca J, Aznar M, Batlle R (2009) Anal Bioanal Chem 393(3):809–833
Liu JF, Chi YG, Jiang GB, Tai C, Peng JF, Hu JT (2004) J Chromatogr A 1026:143–147
Dadfarnia S, Shabani AMH (2010) Anal Chim Acta 658:107–119
Liu HH, Dasgupta PK (1996) Anal Chem 68:1817–1821
He Y, Lee HK (1997) Anal Chem 69:4634–4640
Batlle R, Nerín C (2004) J Chromatogr A 1045:29–35
Yangcheng L, Quan L, Guangsheng L, Youyuan D (2006) Anal Chim Acta 566:259–264
Ye CL, Zhou OX, Wang XM, Xiao JP (2007) J Sep Sci 30(1):42–47
Romero J, López P, Rubio C, Batlle R, Nerín C (2007) J Chromatogr A 1166:24–29
Zhu LY, Zhu L, Lee HK (2001) J Chromatogr A 924:407–414
Pezo D, Salafranca J, Nerín C (2007) J Chromatogr A 1174:85–94
Jiang XM, Lee HK (2004) Anal Chem 76:5591–5596
Rivas RE, Lopez-Garcia I, Hernandez-Cordoba M (2009) Spectrochim Acta 64B(4):329–333
Liu Y, Zhao EC, Zhu WT, Gao HX, Zhou ZQ (2009) J Chromatogr A 1216(6):885–91
Khalili-Zanjani MR, Yamini Y, Shariati S, Jönsson JA (2007) Anal Chim Acta 585(2):286–293
Farahani H, Norouzi P, Dinarvand R, Ganjali MR (2009) J Sep Sci 32(2):314–320
Farahani H, Yamini Y, Shariati S, Khalili-Zanjani MR, Mansour-Baghahi S (2008) Anal Chim Acta 626(2):166–173
Khalili-Zanjani MR, Yamini Y, Yazdanfar N, Shariati S (2008) Anal Chim Acta 606(2):202–208
Sobhi HR, Yamini Y, Esrafili A, Adib M (2008) J Pharmaceut Biomed 48(4):1059–1063
Dadfarnia S, Salmanzadeh AM, Shabani AMH (2008) Anal Chim Acta 623(2):163–7
Bidabadi MS, Dadfarnia S, Shabani AMH (2009) J Hazard Mater 166(1):291–296
Dadfarnia S, Shabani AMH, Kamranzadeh E (2009) Talanta 79:1061–1065
Xu H, Ding ZQ, Lv LL, Song DD, Feng YQ (2009) Anal Chim Acta 636(1):28–33
Leong MI, Huang SD (2008) J Chromatogr A 1211(1–2):8–12
Baghdadi M, Shemirani F (2008) Anal Chim Acta 613:56–63
Rivas RE, Lopez-Garcia I, Hernandez-Cordoba M (2009) Anal Methods, in press doi: 10.1039/B9AY00237E
da Silva AF, Welz B, Curtius AJ (2002) Spectrochim Acta Part B 57:2031–2045
Bermejo-Barrera P, Moreda-Piñeiro J, Moreda-Piñeiro A, Bermejo-Barrera A (1997) J Anal At Spectrom 12:317–321
Vereda-Alonso E, Siles-Cordero MT, García de Torres A, Cañada-Rudner P, Cano-Pavón JM (2008) Talanta 77:53–59
Welz B, Schlemmer G, Mudakavi JR (1992) J Anal At Spectrom 7:499–503
Jiang HM, Hu B, Jiang ZC, Qin YC (2006) Talanta 70:7–13
Aranda PR, Gil RA, Moyano S, de Vito I, Martinez LD (2008) Talanta 75:307–311
Li Y, Jiang Y, Yan XP, Ni ZM (2002) Environ Sci Technol 36:4886–4891
dos Santos WNL, Dias FS, Reboucas MV, Pereira MG, Lemose VA, Teixeira LSG (2006) J Anal At Spectrom 21:1327–1330
Acknowledgments
The authors are grateful to the Spanish MICINN (Project CTQ2009-08267/BQU) and to Fundación Séneca (CARM Project 11796/PI/09) for financial support. Ricardo E. Rivas acknowledges a fellowship from Departamento de Formación del Personal Académico de la Universidad Centroccidental Lisandro Alvarado (Venezuela)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
López-García, I., Rivas, R.E. & Hernández-Córdoba, M. Liquid-phase microextraction with solidification of the organic floating drop for the preconcentration and determination of mercury traces by electrothermal atomic absorption spectrometry. Anal Bioanal Chem 396, 3097–3102 (2010). https://doi.org/10.1007/s00216-010-3500-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-3500-7