
Abstract
Farnesyl- and geranylgeranylpyrophosphate (FPP and GGPP) are isoprenoid intermediates in the mevalonate pathway. They play a crucial role in cell survival, growth and differentiation due to their attachment (isoprenylation) to small GTPases (Ras, Rho, etc.). Isoprenoid formation seems to be tightly regulated within the mevalonate pathway and its perturbation has been linked to certain diseases (e.g., cancer, Alzheimer’s disease), but tissue levels are unknown. It is therefore of the utmost importance to quantify these isoprenoids in diseased tissue or in tissue after drug administration. The current work describes an isolation procedure utilizing a combination of Extrelut® liquid/liquid and reversed-phase solid-phase extraction (SPE) for homogenized human frontal cortex tissue. In addition, after a careful validation of an HPLC–fluorescence method, this assay allowed the determination of nanomolar concentrations of endogenous FPP and GGPP levels (4.5 and 10.6 ng/mg protein, respectively) in human brain tissue. The method is selective, precise (<15% RSD), accurate (<15% relative error) and sensitive over a linear range of 10–400 ng/mL for FPP and 50–1000 ng/mL for GGPP according to the current FDA criteria for bioanalytical method validation. Overall, this new method introduces the ability to simultaneously quantify FPP and GGPP in human brain tissue, and is potentially applicable to several other tissues and species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The two isoprenoids farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP) are intermediates in the mevalonate pathway, which provides the only source of these compounds in mammals [1]. FPP and GGPP play decisive roles in cell signaling, and their presence is crucial to cell survival [2]. Their synthesis seems to be tightly regulated within the mevalonate pathway and especially through the rate-limiting activity of hydroxymethylglutaryl-coenzyme A reductase (HMG-CoA reductase). FPP is the branch point of this pathway, serving as a precursor of cholesterol and its products and as an immediate precursor of GGPP [3]. These two isoprenoids provide the basis for all longer-chain isoprenoids such as heme A, dolichol and ubiquinone. Furthermore, FPP and GGPP are required substrates for the posttranslational modification (isoprenylation) of certain proteins [1]. They serve as their lipophilic anchors and therefore affect protein activity, function and localization in membranes. Approximately 300 proteins in the human proteome are prenylated by GGPP and FPP, particularly members of the Ras superfamily of small GTPases (e.g., Ras, Rab, Rho, etc.). Involvement of prenylated proteins in functions such as cell growth and survival, metabolism, differentiation and cytoskeletal organization are of significant importance in cellular function [4], and perturbation of the mevalonate pathway appears to be associated with certain diseases such as Alzheimer’s disease (AD), cardiovascular disease and different cancers [5–7]. For example, clinical trials are being undertaken to test anticancer agents inhibiting the two prenylating enzymes FTase and GGTase that belong to this pathway [8, 9]. An estimated 10–15% of all human cancer types are due to a mutation in the Ras protein [10, 11] (proto-oncogene), underscoring the importance of research on isoprenoids. Against this background, there is keen interest in determining tissue concentrations of FPP and GGPP under conditions which may affect signaling pathways. Such effects might be due to modified cholesterol synthesis or may be a result of drugs targeting this pathway. The most popular drugs that act on cholesterol synthesis are statins, which represent competitive inhibitors of the HMG-CoA reductase. In addition, these drugs are known for their cholesterol-independent effects, such as improvement of endothelial function, stabilization of atherosclerotic plaques, anti-inflammatory and immunomodulating effects, etc., which are thought to be due to alterations in isoprenoid levels [12]. It has been discussed that the potentially beneficial effect of statins in neurodegenerative disorders [13] is at least partly due to the reduction of isoprenoids in the central nervous system [5].
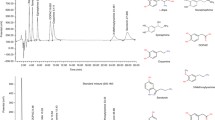
In spite of the great interest in isoprenoids and cell signaling, there is a lack of data on FPP and GGPP levels in different tissues under both normal and pathological conditions. Therefore, it is of the utmost importance to quantify FPP and GGPP levels. To the best of our knowledge, no validated method for the nonradioactive quantitative determination of FPP and GGPP in mammalian tissue has been published. A radioactive method for FPP has been described by Bruenger et al. [14] for the investigation of endogenous levels in the liver of mice and rats. Another approach was published by Saisho et al. [15], who quantified FPP in human and dog plasma after cleavage of the diphosphate group and successive coupling to 9-anthroyl cyanide. The most relevant studies in regard to the current work were undertaken by Tong et al., who measured FPP and GGPP concentrations in statin-treated NIH 3T3 mouse cells [16] and in different mouse tissues [17]. The authors developed a sensitive and selective derivatization procedure, coupling the two isoprenoids to their corresponding dansyl-labeled pentapeptides, resulting in the fluorescent derivatives shown in Fig. 1 (designated as FPP* and GGPP* throughout this manuscript). The method described in the current work expands on the work by Tong et al. [16, 17] by taking the fast degradation of diphosphates by endogenous phosphatases into account [18], resulting in an assay with much improved sample stability during storage, handling and analysis. Until now, there has not been a validated method for the simultaneous determination of FPP and GGPP in any mammalian tissue. Furthermore, the method was developed under the Food and Drug Administration (FDA)’s guidelines for full method validation of bioanalytical methods [19], including the novel incorporation of 5-(dimethylamino)naphthalene-1-(4-nonylphenol)-sulfonic acid ester (DNP) as an internal standard (IS). Advantages of this new validated method are the selectivity, accuracy and precision of the assay and its results.

Structures of the two isoprenoids after enzymatic coupling to their appropriate dansyl-labeled pentapeptides: FPP* (= D*GCVLS-F; n = 2; R= –OH) and GGPP* (= D*GCVLL-GG; n = 3; R= isopropyl)
In the current work we describe a robust validated HPLC-fluorescence detection (HPLC-FLD) method, for the quantitative determination of FPP and GGPP in human brain tissue. Due to the universal nature of the extensive clean-up procedure along with the high purity of the extracts of the analytes that result from it, this application provides the potential to analyze FPP and GGPP in several other tissues via HPLC-FLD or via other sensitive methods, including mass spectrometry. This method allows access to crucial information concerning isoprenoid levels (FPP and GGPP) in the diseased state and/or after pharmacological intervention, as well as the regulation of the mevalonate pathway and other basic regulatory mechanisms in cell metabolism.
Materials and methods
Chemicals and reagents
FTase and GGTase were obtained from Jena Bioscience (Jena, Germany) and D*-GCVLS (dansyl Gly-Cys-Val-Leu-Ser) and D*-GCVLL (dansyl Gly-Cys-Val-Leu-Leu) from Calbiochem (Darmstadt, Germany). Ammonium hydroxide solution 28–30% was purchased from Alfa Aesar (Karlsruhe, Germany), and the phosphatase inhibitors Halt® and PhosStop® from Thermo-Fisher/Pierce (Bonn, Germany) and Roche Diagnostics GmbH (Mannheim, Germany), respectively. All solvents were of analytical grade or higher quality. Acetonitrile was obtained from Carl Roth GmbH (Karlsruhe, Germany), while 1-butanol, n-hexane, 2-propanol, methanol and acetone as well as ammonium acetate, Tris-HCl, MgCl2, ZnCl2 and Na2CO3 from Merck (Darmstadt, Germany). FPP, GGPP, octyl-β-D-glucopyranoside and dithiothreitol were from Sigma-Aldrich (Schnelldorf, Germany). Radioactive [3H]FPP (26.2 Ci/mmol) and [3H]GGPP (23.0 Ci/mmol) were from PerkinElmer (Waltham, MA, USA). Millipore water was used for all solutions (Schwalbach, Germany).
Internal standard
The synthesis of DNP was conducted according to Naasnser et al. [20] After 16 hours of reaction time, the mixture was evaporated to dryness under reduced pressure. The residue was dissolved in acetonitrile for repeated preparative clean-up using an analytical C18 endcapped Nucleodur 100–5 column (250 × 4 mm, 5 μm, Macherey–Nagel, Dueren, Germany). A 1H-NMR (300 MHz) measurement was conducted for product confirmation and the compound was checked for the absence of fluorescent impurities by HPLC-FLD analysis. Stock solution (2.8 μM) aliquots were evaporated to dryness and kept under nitrogen at 4°C.
Sample preparation
Frozen human brain tissue was homogenized with a rotor-stator homogenizer at 1100 rpm in 100 mM Tris buffer (pH 8.5) with 5 μL Halt® and 10 μL PhosStop® phosphatase inhibitor. The homogenate was vigorously mixed with 1 mL 100 mM Tris buffer (pH 8.5). A 10 μL aliquot from the homogenate was retained for protein determination and thereafter spiked with 15 μL 2.8 μM solution of the internal standard. The homogenate was loaded onto Merck Extrelut® NT1-columns (Darmstadt, Germany) and after 15 min was washed off with three separate 2 mL steps of a 1-butanol/ammonium hydroxide/ water mixture (10:1:2, v/v/v). The filtrate was centrifuged for 10 min at 29,000×g to remove precipitated proteins. The supernatant was evaporated under reduced pressure and dissolved again in 5 mL 5% methanol. After sonication the solution was brought onto Oasis® HLB (3 cc; 60 mg) solid-phase extraction cartridges from Waters (Eschborn, Germany) that had previously been conditioned with n-hexane, 2-propanol and methanol. The extract was washed with a 2% methanol solution and finally eluted with an ammonium hydroxide/2-propanol/n-hexane mixture (1:7:12, v/v/v). The filtrate was vacuum-dried in order to be redissolved in assay buffer for the enzymatic reaction.
Prenylation assay
The enzymatic reaction was carried out adhering to the work of Tong et al. [15], with the following changes. The dried residue was dissolved in 44 μL Tris–HCl assay buffer and spiked with 2 μL of a 50 μM solution of D*-GCVLS and D*-GCVLL (dansyl-labeled peptides) and 250 ng FTase and GGTase, respectively. The mixture was incubated at 37 °C in an Eppendorf thermomixer comfort (Wesseling-Berzdorf, Germany) programmed for 90 min (per minute: 5 s; 500 rpm). After stopping the reaction, the mixture was centrifuged (4 °C; 15000×g; 5 min) prior to HPLC-FLD analysis.
Chromatographic conditions
The chromatographic separation was carried out on a Jasco HPLC-system (LG-980–02, PU-980, AS-950; Gross-Umstadt, Germany) with gradient elution on an Ascentis® Express C18 reversed-phase analytical column from Supelco (150 × 2.1 mm, 2.7 μm; Munich, Germany) protected by a Phenomenex Security guard column (C18, 4 × 2.0 mm; Aschaffenburg, Germany). Two solvents were used for gradient elution: solvent A, 20 mM ammonium acetate in 40% acetonitrile, and solvent B, 20 mM ammonium acetate in 90% acetonitrile. The gradient was initiated at 35% B for 1.5 min, and subsequently a linear gradient led to 100% in 6.5 min, which was maintained for 6 min and brought back to 35% B within 2 min. Total run time was 20 min, with a constant flow rate of 0.5 mL/min at 20 °C. The labeled analytes were monitored by a fluorescence detector (Gilson, Middleton, WI, USA) set at an excitation wavelength of 340 nm and 525 nm for emission. The retention times for FPP* and GGPP* were 4.1 min and 11.0 min, respectively and 12.1 min for the IS.
FTICR measurements for the characterization of FPP* and GGPP*
Accurate mass measurements were performed on a QFT 9.4 T triple-quadrupole Fourier transform ion cyclotron resonance (FTICR) mass spectrometer (IonSpec, Lake Forest, CA, USA) working in the negative ion mode. FPP* and GGPP* fractions were manually collected from spiked brain samples after HPLC separation and infused into the ESI source after mixing with internal mass calibration solution at a flow rate of 7 μL/min. The needle potential of the ESI source was set to –3.8 kV, the cone voltage to –45 V, and the extractor to –10 V. Ion accumulation times between 500 and 1000 ms in Q3 were used. Chirp excitation was performed at 155 V for 16 ms (for m/z 75– 2500). Mass accuracies were calculated using the MIDAS molecular formula calculator software (NHMFL, Tallahassee, FL, USA) with atom constraints for C, H, N, O and S set to the respective number of atoms in the precursor ions.
Validation samples
Thawed human cortex samples were pooled and homogenized in 100 mM Tris–HCl buffer (pH 8.5), resulting in a stock solution with 200 mg wet weight tissue per 1 mL. Aliquots were stored at −20 °C throughout the validation. Two separate batches from different brain samples were prepared, one for the calibration curves and one for the quality control samples (QCs). Stock solutions (1 mg/mL) of FPP and GGPP in methanol/aqueous ammonium hydroxide (7:3, v/v) were kept at −20 °C. Different working solutions containing FPP and GGPP were prepared by diluting the stock solution with methanol/aqueous ammonium hydroxide (7:3, v/v). The IS was prepared by dissolving the dried residue in an appropriate volume of acetonitrile to give a 2.8 μM solution. Calibration standards were prepared daily by adding 1 mL of brain homogenate to 1 mL 100 mM Tris–HCl spiked with 15 μL of IS solution and 10 μL of the appropriate FPP-GGPP working solutions, resulting in concentrations of 10, 20, 40, 60, 80, 100, 200 and 400 ng/mL for FPP and 50, 75, 100, 125, 150, 250, 500 and 1000 ng/mL for GGPP. Three different concentrations of QCs (FPP/GGPP: QC-L 30/80, QC-M 150/400 and QC-H 300/800 ng/mL; low, medium and high) were prepared analogously to the calibration standards.
Method validation
To prove the suitability of the developed preparation procedure and the analytical method, validation was performed according to the current FDA bioanalytical method validation guidelines [20]. In order to exclude interference with endogenous FPP or GGPP, sealed brain samples were thawed and kept at room temperature for two days prior to homogenization and storage at −20 °C. To ascertain the complete degradation of FPP and GGPP and to assure the consistency of the matrix, zero samples (matrix sample spiked with IS) and spiked samples were compared, respectively. The selectivity of the method was shown by the comparison of blank and zero sample chromatograms from six brain homogenate samples with different origins. Linearity in the brain homogenate was verified using five calibration curves measured on five separate days. Each one consisted of eight non-zero calibration standards with concentrations ranging from 0.03 to 1.06 μmol for FPP and 0.11to 2.15 μmol for GGPP. The calibration curves were calculated by linear regression using weighting for both analytes. The back-calculated values are required to be within 15% of the relative standard deviation (RSD) and 20% of RSD at the lower limit of quantification (LLOQ) of the nominal concentration. The acceptance criterion for the correlation coefficients (r 2) of the calculated regression curves was 0.99 or greater.
To determine the LLOQ, six different brain homogenate samples were spiked with FPP and GGPP at the lowest level of the calibration curve (10 ng/mL and 50 ng/mL, respectively) and recalculated with a freshly prepared calibration curve. The intraday accuracy and precision were determined by analyzing four replicates of each QC sample (in the lower, middle and upper ranges) together with a complete set of calibration standards in one analytical run within one day. The interday accuracy and precision were established by measuring four replicates of each QC sample on four consecutive days together with all calibration standards freshly prepared for each day. The mean, the standard deviation (SD), the RSD and bias were calculated. The recoveries of FPP and GGPP from brain homogenate were determined by analyzing triplicates of each QC sample. Responses were compared to extracted blank brain homogenate (n = 3 for each concentration) spiked with the same nominal concentrations prior to the prenylation assay. For further proof of reproducibility, both recoveries were additionally determined by analyzing each QC sample after isolation using [3H]FPP and [3H]GGPP. Counts were measured on a Canberra Packard Tricarb 1900 scintillation counter (Rüsselsheim, Germany) and referred to those obtained from samples spiked with the same nominal concentrations.
The stabilities of FPP and GGPP were tested in the presence of both phosphatase inhibitors in brain homogenate (n = 4) for two concentration levels (low and high) after storage at room temperature for 2 h, after three freeze and thaw cycles, and after storage for one and three months at −80 °C. Furthermore, the autosampler stability was tested using QC-M samples (n = 4) and measured at the following time points: 0, 5, 10 and 20 h storage at 4 °C in the autosampler.
Human brain samples
Human frontal cortex samples were obtained from the Human Brain and Spinal Fluid Resource Center (Los Angeles, CA, USA) and stored at −80 °C. Patients were between 71 and 80 years old; the post mortem interval (PMI) for the removal of the donor brain after exitus varied between 11.5 and 18.3 h, and neuropathological diagnoses of the brain were attested to be “essentially normal.” PMI did not correlate to levels of either isoprenoid. It appears that the endogenous phosphatases are less active in the dead but intact tissues in comparison to the removed and homogenized brain slices. However, since a complete preservation of all phosphorylated compounds could not be guranteed in the intact cells after cutting off the nutrient supply, we are using similar mean post mortem intervals for the comparison groups. For sample preparation, five slices were taken from the freezer and immediately dissected into small pieces of white and gray matter (approx. 200 mg w.w. each). The frozen samples were homogenized as described above.
Protein determination
3Protein concentrations were measured using the BCA Protein Assay Kit from Thermo-Fisher/Pierce. To facilitate a valid comparison between the results, we conducted the BCA assays on the same day and, if possible, on the same plate for all samples. In addition, all experiments were measured in triplicate. Accuracy and precision measurements were all conducted in the exact same matrix. The results for isoprenoid measurements were not related to protein content during the method validation; that is, possible experimental errors from the protein assay did not influence the validation results.
Results and discussion
The present study describes the development of a validated assay for the endogenous isoprenoids FPP and GGPP. Importantly, the procedure described here meets all of the criteria outlined by the Food and Drug Administration (FDA) for full method validation of bioanalytical methods [19]. These criteria include selectivity, linearity, stability, accuracy, precision and repeated measurements at the lower limit of quantitation (LLOQ). The technique shown here also makes use of a weighing factor for the calibration curves in order to improve accuracy. The major challenges encountered in the current work originated from endogenous phosphatases, resulting in fast analyte degradation from dephosphorylation reactions in the homogenate. Of course, the enzymatic reaction used to derivatize the analytes prior to analysis is specific for the diphosphate moiety, thus reducing the substrate concentration in the samples. Preliminary tests showed that despite handling the samples at 4 °C, distinct analyte loss occurred, and only the introduction of two different phosphatase inhibitors suppressed phosphatase activity sufficiently. Furthermore, a significant current shortcoming is the lack of an appropriate internal standard for FPP and GGPP that has similar chemical properties and behavior during isolation, separation and detection without being sensitive to the enzymatic derivatization reaction. Several compounds were tested, including dansyl- and nitrobenzoxadiazole- (NBD) labeled compounds, e.g., phospholipids. However, these choices proved to be either unstable or unreliable. After various further experiments, DNP was identified as a suitable IS. Due to the lack of a commercial source, we synthesized DNP according to Naasner et al. [20]. A stock solution of 2.8 μM was obtained after repeated cleanup by HPLC. The structure was unequivocally confirmed via 1H-NMR and sufficient purity was demonstrated via HPLC analysis of the synthesized and purified IS compound (data not shown).
For analyte confirmation, FPP and GGPP peak fractions were collected from spiked tissue samples after HPLC separation and analyzed using FTICR-MS. The measured accurate masses for FPP* and GGPP* corresponded well to the calculated exact masses (<5 ppm measurement uncertainties for all experiments), thus confirming the identities of the derivatized isoprenoids.
Method validation
Selectivity
To determine the degradation of endogenous FPP and GGPP levels in the two brain homogenate batches, zero samples were isolated and subjected to the enzymatic reaction. The resulting samples showed no detectable isoprenoid concentrations due to the complete degradation of the diphosphate moiety [18]. To further ensure the consistency of this brain matrix, samples were spiked before and after storage at room temperature and analyzed, and again no significant differences in their chromatograms were observed.
Furthermore, the selectivity of the assay was successfully demonstrated. There were no significant interfering signals observed in six blank brain homogenate samples with different origins at the retention times for FPP* (t = 4.1 min) and GGPP* (t = 11.0 min), as well as in the related zero samples spiked with the IS.
Linearity
Good linearity was shown over the investigated calibration range of 10–400 ng/mL for FPP and 50–1000 ng/mL for GGPP in brain homogenate on five separate days. The best fit for the calibration curves was achieved using a weighting factor of 1/x 3 for FPP and 1/x 2 for GGPP, evaluated by comparing the r 2 values and the sums of the absolute values of the relative errors for the calibration curves using different weighting factors (data not shown). The correlation coefficients (r 2) were consistently greater than 0.992 and 0.989, resulting in mean values of 0.994 ± 0.0012 and 0.991 ± 0.0014 for FPP and GGPP, respectively. The relative deviations of the calculated standard concentrations from their nominal values were always less than 15% for all values, including the LLOQ, thus meeting the FDA requirements [19].
LLOQ
Determination of the LLOQ at 10 ng/mL for FPP and at 50 ng/mL for GGPP was carried out in six spiked brain homogenate samples, yielding an average of 9.8 ng/mL ± 4.3% (bias −2.1%) for FPP and 45.6 ng/mL ± 6.2% (bias −8.9%) for GGPP. The general criteria for accuracy and precision for the lowest quantification level were thus fulfilled. An overlay in Fig. 2 illustrates a chromatogram at the LLOQ compared to its respective blank sample.

Representative chromatograms. Upper trace, blank brain homogenate; lower trace, FPP and GGPP at the LLOQ
Accuracy and precision
The values for the intra- and interday accuracies and precisions are summarized in Table 1, fulfilling the international acceptance criteria for bioanalytical method validation, which require a maximum of 15% RSD for the value of each QC [19].
Recovery
We evaluated a wide range of solid-phase extraction (SPE) materials and liquid/liquid (l/l) extraction procedures during the method development phase of this study. The best results were obtained using the combined approach of matrix-assisted l/l extraction and SPE with a mixed mode material (see “Experimental” for more details about the procedure). The recoveries for FPP and GGPP were determined in the low, medium and high ranges of the calibration curves. Mean values were 64.6 (± 1.1), 63.1 (± 4.1) and 52.9% (± 5.7%) for FPP and 15.0 (± 6.3), 16.2 (± 2.7) and 26.5% (± 4.9%) for GGPP, respectively. Despite the relatively poor recoveries, especially for GGPP, the results were reproducible throughout the validation. This was further confirmed by the recovery tests using [3H]FPP and [3H]GGPP, yielding a FPP retrieval of 54% for all three concentrations and 18.5, 12.7 and 23.1% for GGPP in the low, medium and high concentration ranges.
Stability
The stability tests showed no significant degradation of FPP or GGPP in brain homogenate after storage at room temperature in the presence of two phosphatase inhibitors, at −80 °C for one and three months, and after three freeze and thaw cycles. Storage in the autosampler gave unsatisfying results for 20 h at 4 °C due to degradation of the analytes and IS, whereas results measured at the other time points showed acceptable results, as noted in Table 2. This was also demonstrated by comparing the mean peak areas for each compound at 0, 5 and 10 h (data not shown). Consequently, the samples were kept for a maximum of 10 h in the autosampler.
Analysis of human brain samples
Here, for the first time, we report endogenous FPP and GGPP levels in human brain samples. White and gray matter from five different human frontal cortex specimens were prepared and analyzed. The results of these experiments are shown in Table 3. The data clearly demonstrate a significantly higher concentration of GGPP in all samples and a much higher abundance in the white matter. A potential explanation for these higher GGPP levels is that elevated cholesterol abundance in the white matter results in a negative feedback mechanism, reducing the turnover of HMG-CoA activity directed at isoprenoid synthesis [1]. FPP shows a less distinct distribution between the two areas, and lower concentrations than GGPP in general. A plausible explanation for this observation is that FPP is at a branch point and that two separate pathways could potentially diminish this precursor pool [5].
Conclusions
The current work describes the development, validation and application of a novel method of quantifying endogenous FPP and GGPP levels in the human brain. Knowledge of these levels is very important, as there is great interest in the role of prenylated proteins in neurodegenerative diseases, particularly Alzheimer’s disease [5]. There is currently no data on the levels of free isoprenoids in the brain, severely impeding our understanding of the role and regulation of isoprenoids in the human brains of both normal and diseased individuals. Researchers have investigated isoprenoid/protein complexes using mass spectrometry [9, 21] as well as semiquantitative immunoassay techniques [22, 23]. Complementary data from our studies on the free isoprenoids will be extremely important for fully describing the regulatory mechanisms governing isoprenylation. Future research will focus on other peripheral tissues and the effects of statin administration on endogenous isoprenoid levels. Furthermore, we are currently in the process of transferring the assay to a more sensitive and rapid LC-MS/MS technique.
In this study, the sample matrix for calibration samples was different from that for the QC samples, and both extracts were produced independently of each other. The use of matrix-matched calibrators generally minimizes (or even completely removes) ambiguities arising from possible coeluting coextractants. We exploited the instability of the analytes to obtain a true blank control matrix. There is a small risk of additional error, however. The control matrix may now contain higher levels of metabolites of the analytes than the sample matrix, and these metabolites could interfere directly or indirectly. This situation is analogous to pharmacokinetic/ADME studies, where the presence of analyte metabolites in the sample matrix (“incurred samples”) but not in the control matrix necessitates reanalysis of at least some incurred samples [24, 25]. A strategy using isotope-labeled internal standards, as described recently [26, 27], could be used in future LC/MS/MS studies.
References
McTaggart SJ (2006) Cell Mol Life Sci 63:255–267
Calleros L, Lasa M, Rodriguez-Alvarez FJ, Toro MJ, Chiloeches A (2006) Apoptosis 11:1161–1173
Liao JK (2002) J Clin Invest 110:285–288
Buhaescu I, Izzedine H (2007) Clin Biochem 40:575–584
Cole SL, Vassar R (2006) Neurobiol Dis 22:209–222
Motoshima H, Goldstein BJ, Igata M, Araki EJ (2006) Physiol 574:63–71
Duncan RE, El-Sohemy A, Archer MC (2005) Mol Nutr Food Res 49:93–100
Brunner TB, Hahn SM, Gupta AK, Muschel RJ, McKenna WG, Bernhard EJ (2003) Cancer Res 63:5656–5668
Barnidge DR, Dratz EA, Martin T, Bonilla LE, Moran LB, Lindall A (2003) Anal Chem 75:445–451
Downard J (2003) Nat Rev Cancer 3:11–22
Sahai E, Marshall CJ (2003) Nat Rev Cancer 2:133–142
Farooqui AA, Ong WY, Horrocks LA, Chen P, Farooqui T (2007) Brain Res Rev 56:443–471
Eckert GP, Müller WE, Wood WG (2007) Future Lipidol 2:423–432
Bruenger E, Rilling HC (1988) Anal Biochem 173:321–327
Saisho Y, Morimoto A, Umeda T (1997) Anal Biochem 252:89–95
Tong H, Holstein SA, Hohl RJ (2005) Anal Biochem 336:51–59
Tong H, Wiemer AJ, Neighbors JD, Hohl RJ (2008) Anal Biochem 378(2):138–143
Adair WL, Cafmeyer N, Keller RK (1984) J Biol Chem 7:4441–4446
CDER (2001) Guidance for industry: bioanalytical method validation. Center for Drug Evaluation and Research (CDER), US Food and Drug Administration, Rockville, MD (see http://www.fda.gov/cder/guidance/4252fnl.pdf, last accessed 25 July 2008)
Naasner M, Mergler M, Wolf K, Schuphan I (2001) J Chromatogr A 945:133–138
Appels NM, Rosing H, Stephens TC, Schellens JH, Beijnen JH (2006) Anal Chem 78:2617–2622
Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth GJ (2005) Biol Chem 280:34202–32409
Dursina B, Reents R, Delon C, Wu Y, Kulharia M, Thutewohl M, Veligodsky A, Kalinin A, Evstifeev V, Ciobanu D, Szedlacsek SE, Waldmann H, Goody RS, Alexandrov K (2006) J Am Chem Soc 128:2822–2835
Jemal M, Ouyang Z, Powell ML (2002) Rapid Commun Mass Spectrom 16:1538–1547
Viswanathan CT, Bansal S, Booth B, DeStefano AJ, Rose MJ, Sailstad J, Shah VP, Skelly JP, Swann PG, Viswanathan RW (2007) Pharm Res 24:1962–1973
Jemal M, Schuster A, Whigan DB (2003) Rapid Commun Mass Spectrom 17:1723–1734
Li W, Cohen LH (2003) Anal Chem 75:5854–5859
Acknowledgments
We would like to thank the Hanna Bragard-Apfel Foundation, the L.A. Brain Bank, and the Medical Research Council, as well as to acknowledge the provision of NIH grants AG23524, AG18357 and the support of the Department of Veterans Affairs. We also appreciate the technical support of Rambod Daneshfar.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hooff, G.P., Volmer, D.A., Wood, W.G. et al. Isoprenoid quantitation in human brain tissue: a validated HPLC–fluorescence detection method for endogenous farnesyl- (FPP) and geranylgeranylpyrophosphate (GGPP). Anal Bioanal Chem 392, 673–680 (2008). https://doi.org/10.1007/s00216-008-2306-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-2306-3