
Abstract
This review discusses the characterisation of natural organic dyestuffs of historical interest by liquid chromatography–mass spectrometry. The structures of the most important natural organic dyestuffs traditionally used are presented and discussed from the perspective of their analytical chemical determination. The practical aspects of the determination of this inhomogeneous range of compounds with different structures, such as anthraquinones, flavonoids, indigoids or tannins, are discussed with their implications for sample preparation, liquid chromatographic separation and mass spectrometric detection. The particular focus of this review is the discussion of the mass spectral fragmentation patterns of the different classes of natural organic dyestuffs, which in the ideal case allow the identification of the dyestuff actually used, and thereby provide a key to the better characterisation and understanding of historical objects dyed with natural organic dyestuffs.

LC-MS allows characterisation of natural dyestuff constituents: the MS spectrum of alizarin is superimposed over a photo of a textile coloured using this red dye
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dyeing of textiles has been performed for more than 4,000 years, and natural organic materials have for the longest part of this time been the principal source of the dyestuffs, being replaced only during the last one and a half centuries or so by synthetic pigments and dyes which are much cheaper and more convenient to produce [1–3]. The role of textile dyeing was mainly a decorative one, to please the spectator and the person that wore dyed textiles, but at the same time a representation of power and wealth: Considering that during former times a good-quality dyestuff was as precious as gold or silver, it is clear why wearing dyed clothes was a sign of high social status, and during the early Roman period, only the emperor and high priests wore purple-dyed textiles. Later, in the Middle Ages, scarlet-dyed fabrics were exclusively reserved for important members of the clergy.
The dyestuffs used in that times were all of natural origin: all kinds of plants were used with their various parts for dyeing; furthermore certain animals, notably, insects and shellfish, as well as lichens were also used. The often limited availability of particularly the animal sources of dyestuffs and the difficulty of extracting and producing the dyestuffs from these sources explains why they have been so highly valued during certain periods.
There are at least three important reasons to investigate the identity of historically used dyestuffs: First, restorers and conservators may require this information for choosing appropriate means of conservative or restorative treatment. Second, the knowledge of the materials used can provide art historians with important information on the dyeing technique of an artist or craftsman and the technological understanding of that period. And third, the identification of individual dyes which came into use at different times may help in identifying the historic period of the art object, or its provenance.
Various methods have been developed for the characterisation of organic dyes. The choice of technique depends mainly on two factors: first, whether a sample can be removed from the object or not, that is, whether the measurement has to take place in situ or in the laboratory; second, whether the object may be sampled or not, that is, whether a destructive technique—at least on the scale of the sample size—can be used. Where in situ analysis is required, spectroscopic techniques such as UV/Vis spectrometry [4], fluorescence [5], infrared [6] or Raman spectroscopy [7], or XRF spectroscopy [8] (to determine the presence of metal ions used as mordants) can be applied. If the object of investigation can be brought to the laboratory, then spectroscopic analysis, chemical testing and chromatographic analysis can be performed, the last two requiring a sample to be extracted from the object.
The first step of a typical analytical identification scheme is—at least for those dyestuffs that can be solubilised—an appropriate extraction step. For blue dyestuffs there is a special microchemical test that takes account of the poor solubility of indigoid dyestuffs. As most natural dyestuffs contain more than one colouring matter, separation methods such as thin-layer chromatography (TLC) or high-performance liquid chromatography (HPLC) are typically employed. Owing to the simplicity and low cost of TLC, this technique is still widely used in conservation science laboratories, despite the superior separation and detection capabilities of HPLC. The organic extract of the coloured sample can also be analysed by spectroscopic techniques such as Fourier transform infrared (FTIR) spectroscopy. A scheme of a typical sequence for the identification of natural dyestuffs is given in Fig. 1.

Identification scheme for natural organic dyestuffs (redrawn from [1])
As colour is imparted to a substance by specific absorption in the visible region of the electromagnetic spectrum, it is obvious that the primary detection technique for HPLC analysis of organic dyestuffs is UV/Vis absorbance [9, 10]. Despite the widespread use of this technique, it has some limitations with respect to its sensitivity, selectivity and above all its ability to identify unknown compounds. UV/Vis spectra may thus be considered an excellent tool for quantitation, and for the confirmation of the identity of an analyte which either is available as reference substance or whose absorbance spectrum is available in a previously established spectral library. It cannot, however, be thought of as a tool for the positive identification of an unknown substance.
This is where mass spectrometric (MS) detection for HPLC becomes particularly useful: HPLC-MS is nowadays becoming a routine technique [11–14], with ease of use comparable to UV/Vis or fluorescence detection (FLD). Depending on the type of mass spectrometer used, information is provided on the molecular mass of a detected compound, and, moreover, structural information is also gained from the interpretation of the specific fragmentation pattern observable under certain instrumental conditions, or with particular types of mass spectrometers [15]. Although the initial costs of such instrumentation were for a long time prohibitive for conservation science laboratories, HPLC-MS is now becoming popular as a tool for the investigation of natural organic (historical) dyestuffs [16]. It is thus the aim of this review to present analytical strategies for the identification of natural organic dyes by HPLC-MS, to discuss the methodological and instrument-related aspects and to demonstrate what particularly rich information can be drawn from the interpretation of LC-MS spectra. To start with, some general terms shall be explained, and also the main classes of natural organic dyes will be presented.
Classification of organic dyestuffs
There are different ways of classifying the vast number of natural organic dyestuffs. One common way is to distinguish them according to colour, which is the approach of an artist; another is to classify them according to their chemical structure, which would be the approach of an analytical chemist; and third they can be classified according to their source of origin, which is perhaps how a biologist would look at this task.
A practical way of classifying dyestuffs is according to their method of use, which allows us to distinguish mordant dyes, vat dyes and direct dyes [1, 3, 17].
Classification according to method of application
Mordant dyes form the largest class of natural organic dyestuffs. A mordant dye is a colouring matter which is bound to a textile material for which otherwise it would have only little or no affinity by the addition of a mordant, a chemical, in most cases a metal salt, whose cation forms through complexation a “bridge” to the fibre or fabric. Most mordant dyes yield different shades or colours with different mordants. Mordant dyes can be used with silk and wool, but are hardly ever used with cellulose fibres (e.g. cotton) since these have to be chemically modified before they can be dyed with a mordant dye.
For the identification of the colouring matter, this metal complex needs to be decomposed to allow extraction of the colouring matter into an aqueous/organic solvent for subsequent chromatographic analysis, while the metal used as a mordant is typically identified by spectroscopic techniques such as scanning electron microscopy with energy-dispersive detection (SEM-EDS) [18–20] or x-ray fluorescence (XRF) spectroscopy [21]. The former technique is preferred due to its higher spatial resolution which allows one to investigate minute sample amounts. The decomposition of the metal–dyestuff complex requires relatively rigid conditions, such as the treatment with 1:1 diluted hydrochloric acid. Many dyestuffs can resist this relatively harsh treatment. More sensitive dyestuffs such as saffron, orchill or turmeric will, however, be decomposed and require a milder method of hydrolysis.
Among the natural organic dyestuffs, there are only three—albeit very important—dyestuffs that belong to the class of vat dyes: woad, indigo and Tyrian purple. A vat dye is insoluble in common solvents such as water, methanol or ethanol in its coloured form. It can, however, be transformed by reduction into a soluble but colourless (leuco-)form in which the fibre or textile is impregnated. After re-oxidation, the dyestuff becomes insoluble again and also resumes its original colour. The limited solubility of the vat dyes makes them more difficult to analyse by chromatographic techniques and for this reason they are often identified by microchemical tests preceding extraction and chromatographic analysis.
Direct dyes are water-soluble organic molecules which can be applied as such to the fibres or textiles since they have some affinity and are taken up directly. Direct dyes are easily applied and yield bright colours. However, due to the nature of the (weak) chemical interaction, their fastness to washing is poor, although this can be improved by special after-treatment. The yellow dye saffron is one example for a direct dye. Examples of the three classes of dyes are given in Table 1.
In order to have a consistent usage of the common terms, we should define the following expressions [1, 17]: A dyestuff is a water-soluble substance, mostly of organic origin, with which a fibre or textile can be dyed directly or with the use of additional chemicals. The colouring component (also called the colouring principle) penetrates into the fibres and dyes the textile throughout. In contrast to this, a pigment is an insoluble coloured compound. Most important pigments are inorganic, such as the metal oxides and sulfides, but there are some important examples of organic pigments as well. They are produced by complex formation between the organic dyestuff and a mordant, such as alum (KAl(SO4)2) in solution, and further addition of a metal salt, such as chalk (CaCO3), which leads to precipitation and the formation of an insoluble Ca–Al–dyestuff complex, also called a lake. When pigments are used to paint or draw, they are typically bound to the surface of textile or paper fibres by means of a suitable binding medium (e.g. drying oils, proteins and gums).
The following section will discuss a different way of classification of the dyestuffs, and relate the colour of a dyestuff to its chemical structure. This not only appears a logical approach from the chemist’s point of view, but also is the base for the identification of dyestuffs from their mass spectra.
Classification according to colour and chemical structure
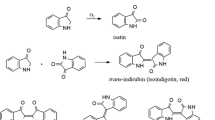
Organic dyes are traditionally classified into groups according to their colour, i.e. red, orange, yellow, brown, blue and violet. Note that few natural organic dyestuffs are truly green: in most cases, artists use a mixture of blue and yellow. Since most of the yellow dyestuffs are less stable than the blue dyestuffs or pigments, green colours eventually change into blue. This is why many of the famous Flemish tapestries now have blue shades where originally green areas have been [22]. The most important blue pigment of natural origin is indigo, which—despite its sophisticated procedure of use—is one of the longest-known blue dyestuffs [23]. The dyeing species, indigo(tin) is isolated from the leaves of a number of Indigofera species, of which Indigofera tinctoria (mainly occurring in Asia) is the commercially most important. This source had, by the mid-seventeenth century, almost completely replaced the European source of indigo, the woad (Isatis tinctoria), and both natural sources have towards the end of the nineteenth century lost much of their original importance after the introduction of synthetic indigo by Baeyer and Drewson in 1882 [24]. Indigo is often used in combination with red, yellow and brown dyes and produces then purple, green and black colours. The main structures and reactions leading to the formation of indigotin (1) are given in Fig. 2. In tropical and subtropical climate, Indigofera species contain indican (2a), whereas in the more temperate European climate, indigo-containing plants contain indican (2a) and isatan (2b). The hydrolysis of the glycosides leads to the indoxyl (3) which either is oxidised to leuco-indigo (6), or may be overoxidised to form isatin (4). Isatin (4) may react with further indoxyl (3) to form indirubin (5). Since indirubin (5) is a red pigment, it gives the mixture a purple hue. Unfortunately, relatively little information about the origin of a textile can be extracted from the blue pigments, since there is hardly a difference in the detectable components from different sources.

Precursors and reactions leading to indigo (reprinted from Ferreira et al. [2] with permission from the Royal Society of Chemistry)
No other blue organic dyestuff has the same historical, artistic and economic importance as indigo. Other blue dyes have received only limited attention, such as the Aztec dyestuffs texotil and matlali whose active colouring matters are still today not elucidated [25].
The purple dyes obtained from shellfish can also be chemically classified as derivatives of indigotin (1). Owing to their historical significance, purple dyes have been thoroughly investigated [26]. As in the case of vegetable sources for indigo, the dye is not present as such in the live mollusc. It is produced by enzymatic hydrolysis of precursors found in the animals’ hypobranchial glands (thereby forming derivatives of indoxyl (3)), followed by photochemical conversion to the purple pigment [26]. Since only very small amounts of dyes can be obtained from each mollusc (only about or less than 1 mg which suffices to dye about 1 g of wool), these dyes consequently are very expensive. Three species of molluscs were mainly used in the Mediterranean region to produce purple: the spiny dye-murex (Bolinus brandaris L. or Murex brandaris), rock-shell (Thais haemastoma L. or Purpura haemastoma) and banded dye-murex (Hexaplus trunculus L. or Murex trunculus) [27].
The active compounds of shellfish purples are brominated indigotin derivatives: Starting from a brominated precursor 7 , 6,6′-dibromoindigotin (8) and, to a smaller extent, also 6,6′-dibromoindirubin (10) are formed. Mixtures of these two and also of monobrominated derivatives, as well as of the unsubstituted indigoids are responsible for the different shades and hues of purple. Thus, it is known that the presence of (mono)bromoindigotin (9) and indigotin (1) gives a bluish hue in the final product when compared with purple derived from other sources. It is also worthwhile mentioning that many of the dye precursors have sulfur-containing substituents in the 2-position of the indoxyl derivative (e.g. 11) which release mercaptans in the final oxidation stage. This leads to a repelling odour during the dyeing process which is well documented [26].

Different shades of purple can be obtained from different species of molluscs, and they are also used in combination [28]. In addition, the dyeing technology may also influence the hue. It has, for example, been claimed that exposure of the vat dyebath to sunlight will give the final result a stronger blue hue. This was attributed to the fact that the reduced form of 6,6′-dibromoindigotin (8) can suffer debromination upon exposure to light which would result in the formation of some 9 and 1 [29]. More recent studies, however, propose that this colour change is a thermochroic phenomenon which appears when monobromoindigotin is heated to temperatures above 60 °C, leading to an irreversible colour change from purple to blue [30]. The historically most important form of purple was the Tyrian purple, a purple with a red hue [31]. Indigoid dyes are remarkable stable. Wool textiles dating back from the third century BC were found where Tyrian purple could still be identified (based on UV and x-ray fluorescence spectroscopy) [32].
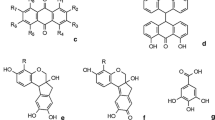
The most important natural red dyes are anthraquinone ( 12 ) derivatives which can be of plant or animal origin. Of great historical importance are the red dyes obtained from scale insects which mostly are plant parasites from the Coccidea family. Important species are extracted from American cochineal (Dactylopius coccus Costa), Polish cochineal (Porphyrophora polonica L.), Armenian cochineal (Porphyrophora hemelii Brandt), kermes (Kermes vermilio Planchon) and lac (Kerria lacca Kerr). All anthraquinone-derived red dyes were mainly used as mordant dyes in which the carbonyl group and the adjacent phenolic group(s) establish the fibre–mordant–dye complex. Kermes was known and has been used in Europe from Roman times, or even before. It is obtained from the dried egg-filled female insects which grow on the Mediterranean oak Quercus coccifera L. Due to the drastic decrease of the number of these trees in the Mediterranean, the parasite is eventually losing its habitat and is threatened by extinction. Kermes dye is a mixture whose main components are kermesic acid (13) (>75%) and flavokermesic acid (14) (also called laccaic acid D). For many centuries, kermes dye was the most important red natural dye in Europe and Asia. Kermes dye was used in the garments worn by high clergymen after shellfish purple dyeing declined. In the sixteenth century, kermes was widely replaced by the American cochineal.

The main constituent of all cochineal dyes is carminic acid ( 15 ). The various species have, however, characteristic fingerprints of other minor anthraquinone components (including kermesic acid (13) and flavokermesic acid (14)), as well as of some still unidentified anthraquinoid compounds commonly named dcII, dcIV and dcVI which allows one to distinguish them in historical samples (Table 2) [33]. Lac differs in its composition from the cochineals in that its main constituents are the two laccaic acids A and B (16 and 17, respectively).

Armenian cochineal is obtained from the cysts of the females of Porphyrophora haemli Brandt. Armenian cochineal occurred in Armenia, Turkey and Iran and was reportedly used from the eight century BC [27]. The largest component of Armenian cochineal is carminic acid (15) (>95%). Polish cochineal is derived from the cysts of the females of Porphyrophora polonica Linnaeus. It contains up to 30% of kermesic acid, as well as the abundant carminic acid. Its use is documented from the sixth century AD, and this dyestuff was mainly used in central and northern Europe, especially at times when kermes was difficult to obtain, such as in times of war [17]. The introduction of the American cochineal led to a rapid decline of the use of Polish cochineal. Although American cochineal was used in South and central America long before, it was introduced to Europe by the Spaniards only in the late sixteenth century [1]. It is obtained from the egg-filled female insects of Dactylopius coccus Costa, a parasite of the Opuntia cactus family after drying in the sun or in an oven. The main constituent of American cochineal again is carminic acid (15), with minor amounts of flavokermesic acid (14) and kermesic acid (13). In the century from the mid-1700s to the mid-1800s, it is estimated that 27,000 tons of cochineal were exported from Mexico to Europe, making this the second most important trade item next to silver. The high demand for American cochineal in Europe was due to the fact that the content of active colouring matter was higher than in the scale insect dyes [17]. Also, it was discovered that American cochineal was capable of producing a brighter red when using a tin (instead of aluminium) mordant. Prior to this discovery, dyes from plants and scale insects produced colours of similar quality and shade. After several unsuccessful attempts, farmers finally succeeded in the 1830 s to breed the cochineal in Europe, particularly on the Canary Islands, which from then on became one of the main suppliers for this dyestuff in Europe.
Lac is obtained from the female insects of the species Kerria Lacca Kerr. Lac was used in India and the Far East long before its introduction in Europe in the late eighteenth century. The major constituents of lac are laccaic acid A (16) (>70%) and laccaic acid B (17) (<20%).
Note that the data presented in Table 2 and discussed before are based on the original work by Wouters and Verhecken [33] who reported relative fractions of the individual constituents based on the UV absorption at 275 nm (which was also summarised by Hofenk-de Graaff in her book [1]). Although this method has found common acceptance owing to its practicality, results can only be considered semiquantitative, since the molar absorption coefficients of the investigated compounds can be considered similar for carminic and kermesic acid, but are likely to be significantly different for flavokermesic acid and dcII at this wavelength.
Unlike the purple dyes, there are very important plant sources for red dyestuffs, many of them based on the anthraquinone ring system 12. These dyes were obtained from the roots of different species of the Rubiaceae family, most importantly madder (Rubia tinctorium L.), then wild madder (Rubia peregrina L.), munjeeet (Rubia cordifolia L.), ladies’ bedstraw (Galium verum L.) and some different species of Relbunium found in South Africa. From a chemical point of view it is remarkable that the anthraquinone structures derived from plants are only substituted on one ring which distinguishes them from the insect-derived dyestuffs.
The most important red plant dye was madder which originally came from India [34]. During the period of high demand of red dyestuffs, however, its cultivation was also started in Europe and the Middle East. Although madder has less dyeing power than the scale insects, it is advantageous in that it allows one to obtain various shades ranging from pink, red, purple to black with different mordants. One of the most valued colours of former times, Turkish red, a very bright red dye, was obtained from madder by a very complex method (involving about twenty individual steps) and was particularly suited for cotton dyeing [35]. Once the secret of the famous Turkish red was elucidated, this dyeing technology was employed in many countries on an industrial scale. Investigations on the chemical nature of the colouring principles in madder date back to the first half of the eighteenth century, leading to the identification of alizarin (18) and its derivatives as important chromophores. It should be emphasised that alizarin is not always detected when extracting samples from historical artworks, whereas purpurin and/or other anthraquinoides typically are. This has prompted the interpretation that the absence of alizarin is an indication that the textile may have been dyed with Rubia peregrina (wild madder) [36]. It is therefore suggested that detailed measurements of the relative peak areas of the anthraquinoids be undertaken for the identification of the madder source, because alizarin may, even if being initially absent in yarn dyed with Rubia peregrina, occur due to the treatment of the sample with acidic alcoholic solution [37]. Under such circumstances, the concentration of alizarin related to purpurin (the dominant chromophore in wild madder) would still be relatively low. This situation may also apply for other Rubia species.
Graebe and Lieberman in Germany and Perkin in the UK independently found synthetic routes for alizarin (in 1868 and 1869) which opened the way for its production on an industrial scale [38]. Synthetic alizarin thus became available at a fraction of the price of natural madder. Although alizarin is, owing to its lightfastness, the most desired constituent in various (but probably not all) madder species, it is but one representative of a large family of anthraquinone derivatives that are formed in madder roots and that are both characteristic of the actual species, as well as of the vegetative status of the plant: While from young (i.e. about 1-year-old) plants, only a few tenths of a percent of a relatively simple mixture of anthraquinoid dyes can be isolated (relative to the weight of the dried root), mainly consisting of alizarin (18), purpurin (19), pseudopurpurin (20) and rubiadin (21), both the number and the concentration of anthraquinoid compounds increases in mature plants, yielding up to 2% by weight of the root, where 19 anthraquinones have already been isolated and identified [39].
Other plants are also known to produce red dyes from the anthraquinone family. Among these are munjeet (Rubia cordifolia L.), wild madder (Rubia peregrina L.) and also the roots of different Galium species (which also belong to the Rubiaceae family). Particularly the roots of ladies’ bedstraw (Galium verum L.) have been shown to contain alizarin (18) (ca. 70% of the extractable anthraquinones) and purpurin (19) (ca. 15%) as the main colouring principles. It is still an open question whether the anthraquinoid substance profile of ladies’ bedstraw changes as strongly as that of root madder with the age of the plant, or whether it is the decomposition of less-stable compounds which alters the profile of anthraquinoid substances found in historical textiles [10].
The roots of the Relbunium species (a plant originating from South America that also belongs to the Rubiaceae family) were also used as red dyes. The main anthraquinone species in this plant is pseudopurpurin (20), but munjistin (22), purpurin (19), xanthopurpurin (23), and also alizarin methyl esters were detected.
A further important class of natural red dyes are the redwoods, known for about seven centuries in Europe, and classified into the soluble redwoods (with neoflavonoids as the main colouring principle) and the insoluble redwoods (in which the principal colouring agents are condensed biflavonoids). The soluble redwood dyes are extracted from various tree species such as Brazilwood, Peachwood, Sappanwood, or Limawood, which often are collectively termed as Brazilwood. The colouring principles are readily soluble in water, and they are normally used as mordant dyes. In all species, brazilin ( 24 ) has been isolated which through oxidation yields brazilein (26), the main chromophore in brazilwoods. The identification of soluble redwoods was described by Nowik and is based on a brazilein derivative (named Bra’) together with another yellow compound (denoted “compound C”) [40]. Brazilwoods were valued less than madder or cochineal because of their poorer fastness and they were therefore usually applied in combination with other dyes [41].

Structurally related to brazilin is haematoxylin (25), which results from the hydrolysis of the glycosidic precursor contained in the fresh logwood or campeachy wood. Again, through oxidation the strong chromophore haematein (27) is formed which itself is red, but can assume different (darker) colours, depending on the mordant used. These colours were widely used owing to their good fastness.
A number of sources are known for the insoluble redwoods: sandalwood (or saunderswood, from South India), narrawood (Burma, Philippines), barwood and camwood (West Africa). As their name suggests, they are hardly soluble in water, which makes their use more difficult, but provides colours of good fastness. They are typically used in combination with other colours to obtain dark red and brown colours.
Among the red dyes of plant origin, safflower (Carthamus tinctorius L.) still deserves particular mention: Despite its limited lightfastness, it has often been used as a direct dye because of its ability to produce beautiful pinks, particularly on silk. Safflower derives from the petals of certain plant species indigenous to Turkey, but was later also cultivated in Egypt, South East Asia and Europe. Its application requires the removal of the more abundant yellow dyestuff also present in the plant petals. Since the red pigment is insoluble in water at neutral pH, but can be solubilised at alkaline pH, the fibres are introduced into the dyebath which then is acidified to precipitate the pigments inside the fibres. The structure of the red pigment carthamin ( 28 ), was only elucidated in the 1980s [42, 43], more than 70 years after its isolation.

The yellow colour of natural dyes is very often due to the presence of flavone ( 29 ) and flavonol (3-hydroxyflavone) (30) in the parent plants, in which they mostly occur as sugar derivatives (glycosides); however, during dyeing (and/or during analysis) they are hydrolysed to the parent aglycon flavonoid. They are used as mordant dyes where the metal ion is complexed by the carbonyl group and the adjacent phenolic group. Owing to the abundance of plants containing flavonoids, no individual dye source has achieved paramount importance. This can advantageously be used to assign the provenance of textiles, since sources of yellow dyestuffs can be expected to be of local origin. Flavones are more resistant to oxidation than flavonols; plants containing a higher proportion of the former are thus preferred for dyeing.

In Europe, weld (Reseda luteola L.), young fustic (Cotinus coggygria Scop.), dyer’s greenweed (Genista tinctori L.), sawwort (Serratula tinctoria L. Gaud.) and dyer’s camomile (Anthemis tinctoria L.) have often been used in traditional recipes for yellow colours [44].
Weld is a plant which grows wild in most of Europe, but has also been cultivated to satisfy the higher demand for dyestuff production. Weld produces bright and fast yellow colours with alum as a mordant [45]. Weld’s main colour-active substances are luteolin ( 31 ) and apigenin (32), mostly occurring as their sugar derivatives, which are, however, mostly hydrolysed to their parent flavonoids in the dyebath. The flavonoid content in weld is up to 2% by weight [46]. Frequently used in combination with indigo or woad, weld yields fast green dyes.

In dyer’s greenweed (Genista tinctoria L.), a variety found in large parts of Europe, the flavones luteolin (31) and apigenin (32) and the isoflavone genistein (33) are the main colour-active substances, along with the genistein 4′-methyl ether (see, e.g. [3, 47]). The same colouring principles are also found in the leaves of sawwort (Serratula tinctoria L.). Dyer’s chamomile (Anthemis tinctoria L.) also contains the glycosides of luteolin (31) and apigenin (32) as main colouring principles, together with other minor components such as quercategin (6-hydroxyquercetin) and patuletin (6-methoxyquercetin-7-O-glucoside).
The main colourants contained in young fustic (used as a mordant dye to produce orange-yellow to strong red-brown colours) are two flavonols, fisetin (34) and myricetin (35), together with sulfuretin ( 43 ). The lightfastness of this dyeing material is limited.
In old fustic (a large tree found mainly in the Caribbean) the main colorants are the flavonol morin (42) with small amounts of kaempferol (39), and the benzophenone maclurin (44).

Persian berries contain a wide range of flavonoids. In addition to the common flavonoids quercetin (36) and kaempferol (39), there are some derivatives more characteristic for this species, including rhamnetin (37), rhamnazin (38), rhamnocitrin (40), together with xanthorhamnin (a 3-O-glycoside of rhamnazin). Nonflavonoid colouring components, such as the anthraquinone derivative emodin (45), are also present.
Quercitron bark, the inner bark of the North American oak (Quercus tinctoria L.), contains quercetin (36), quercitrin (quercitin 3-O-rhamnoside) and kaempferol (39) as its main colour-active constituents.
Other flavone-type yellow dye sources are only of minor or local importance. There is, however, another important group of natural yellow dyes, the so-called polymethin dyes. The main representatives of this class are saffron, turmeric and annatto or orellana. Saffron has been used as a direct dye for more than two thousand years and is obtained from the stigmatas of the flowers of Crocus sativus L. Evidence of its use dates back to Egyptian times. Later it was replaced by cheaper dyes, like weld, that also had better fastness. Saffron produces a bright orange-yellow colour when used as a direct dye, but can also be used with alum as a mordant. The main colour-imparting constituent is crocin ( 46 ), a glucoside of crocetin (47). Similar in its characteristics is the dye annatto or orellana, which is extracted from the fruit of the shrub Bixa orellana L. Its main chromophore is bixin (48) which also is a polyunsaturated diacid useful as a direct dye for cotton, silk and wool [45]. Being used mainly in South America, this dyestuff never became important in Europe.

Turmeric or curcuma (known also as Indian saffron) is derived from the ground roots of Curcuma domestica Valet (also Curcuma longa L.), a widespread plant in the East Indies and China. It was used as a direct dye for cotton, wool and silk, mainly in combination with other dyes. Its main colour-active constituents are the three curcumins I, II and III (49–51). There is also one pigment of animal origin, Indian yellow [48], which is the magnesium salt of euxanthic acid (52), an anthraquinoid dyestuff and a pathological metabolite found in urine of cows fed with mango leaves.
Brown and black dyes are strong absorbers over almost the entire visible spectrum. Their chemical structure is not always easy to determine, as they tend to form oligomers and polymers. Oak galls and sumac are the best-known mordant dyes for brown and black. In both cases, the colour-active substances are formed from hydrolysable tannins which finally produce gallic acid ( 53 ) and sugars. The galls are formed as a reaction of the buds of Quercus infectoria L. to the infection by the eggs of a certain insect (Cynis spp.). Before the insects come out, the buds are collected, dried and ground to produce a lightfast black dye when used with an iron mordant, a procedure widely used in the production of black inks. High levels of tannins are also found in the leaves of various sumac species (Rhus spp.). This dye has also been used from ancient times for dyeing and leather tanning. Other hydrolysable tannins are found in the bark of oak and chestnut trees. Typical structures are, for example, those of ellagic acid (54), and of catechin and epicatechin (55) (from the bark of the oak species Quercus robur L.).

Measurement techniques
Sample preparation
In addition to the complexity of dyestuffs derived from natural sources and of their mixtures, there are two major difficulties in the analysis of organic dyestuffs used for textile dyeing: the relatively low concentrations in which dyes are used relative to the mass of fibre or fabric (typically ≤1 mg g−1 fibre or fabric), and the very low amounts of sample that are typically available from authentic art objects (often significantly less than 1 mg). Both preclude the direct analysis of the dyestuff on the fibre with spectroscopic techniques in most instances. Instead, an extraction step is required to solubilise the dyestuff for subsequent HPLC analysis. A widely adopted approach for extraction is the use of (concentrated) hydrochloric acid/methanol/water 2:1:1 (v/v) for 10 min at reflux temperature (e.g. [49, 50]) by which particularly anthraquinone dyes are well solubilized. Methanolic hydrochloric acid is also effective in dissolving mordant dyes, since the metal ions typically used (Al3+, Fe3+, Sn4+) form chloro complexes, and at the same time the dissociation of phenolic groups (which often establish the bond to the metal mordant) is strongly suppressed. The metal used as mordant is determined separately by x-ray fluorescence [51] or other spectroscopic techniques [52]. Many natural organic dyes (especially those used as direct and mordant dyes) can be efficiently extracted this way. Vat dyes, particularly from the indigo family, however, remain largely insoluble. The relatively harsh approach of extraction with methanolic HCl will additionally hydrolyse glyosidic bonds of flavonoid dyestuffs. On the one hand, this simplifies the analysis, since the flavonoid aglycones can more easily be identified. On the other hand, this simplification is offset by a loss of information, since particularly for the yellow flavonoid dyes the presence of the glycosidic moiety may provide valuable information to trace the actual plant source from which the dyestuff was derived [53–55]. An improved extraction procedure that combines the routine acid hydrolysis method with one using dimethylformamide (DMF) was proposed by Surowiecz et al. [56]. The additional MeOH/DMF (1:1 v/v) extraction step provided an increased recovery of major flavonoid and anthraquinoid compounds, and a very good recovery of indigotin even in textiles with no visible colour, thereby complementing the MeOH/HCl method already in use.
Working with minute amounts of sample and solvent is common in the analysis of samples from historical art objects. Two interesting modifications have, however, been recently introduced to improve sample preparation and make it more expeditious. Dabiri et al. [57] reported the use of ultrasound-assisted extraction (UAE) and microwave-assisted extraction (MAE) for alizarin and purpurin in Rubiaceae plants. Compared with the conventional extraction method (extraction under reflux), UAE showed comparable recoveries, whereas MAE showed significantly higher recoveries, on average at about 140%. In addition, a drastic reduction of the extraction time (from 6 h for reflux extraction, over 2 h with ultrasonication to 20 min with MAE) and solvent consumption (20 mL for UAE and MAE versus 100 mL for reflux extraction) was achieved with a reproducibility comparable with that provided by the reflux extraction as a reference method (RSDs < 10%).
While speeding up sample preparation may be an issue when many samples have to be processed, the development of milder extraction schemes is a topic of general importance: As was discussed before, the vigorous conditions of extraction often hydrolyse the glycosides, particularly of the yellow flavonoid dyes, and therefore reduce the information content of the analysis. Milder extraction schemes aim to preserve the integrity of glycosidic bonds, while at the same time providing adequate recoveries for mordant dyes. Different approaches have therefore been proposed to account for this situation.
Sanyova and Reisse proposed the use of the relatively weak (but strongly complexing) acid HF as an extractant for anthraquinone dyes mordanted with Al or Fe [58]. Hydrofluoric acid allowed the recovery of components such as pseudopurpurin (20), munjistin (22) and their glycoside precursors, all of which were partially or totally hydrolysed when the HCl/MeOH/water extraction method was used. Furthermore, the amount of alizarin recovered was often higher with HF. These improvements are mainly due to the complexation of aluminum by fluoride ions, which allows the extraction of the colorants from the lakes (conveniently accompanied by a visible colour change), at pH > 1 and at room temperature. Thus, many glycosidic precursors of colorants can be found with HF extraction, which otherwise would have remained undetectable when using extraction with strong acid. A different approach to preserve the integrity of glycosidic dyestuffs or parent compounds was proposed by Zhang and Laursen [59] who studied the extraction using either formic acid/methanol 5:95 (v/v) at 40 °C for 30 min, or 0.001 M aqueous H2EDTA/acetonitrile/ methanol, 2:10:88 (v/v/v) at 60 °C for 30 min. After extraction, the solutions are evaporated and the residue dissolved in water/methanol 50:50 (v/v). The results obtained with either of these two procedures not only are equivalent or superior to the common HCl/MeOH/water extraction scheme, but also allow the detection of some unhydrolysed glyocosides of yellow flavonoidic compounds. This may support the identification of the plant source of the yellow dyestuffs. Although the performance of the two new extraction methods is comparable, the formic acid extraction seems to be advantageous for dyed wool, whereas the extraction with EDTA performs better for dyed silk.
The introduction of such milder extraction techniques has greatest impact on previous analyses and interpretation of the colouring agents’ pattern, since hydrolysis of the glycosidic bond can lead to very different effects such as the deepening of a colour (e.g. if ruberythric acid, the primeverose-O-glycoside of alizarin, is hydrolysed to form alizarin (18)) or it can accelerate fading when the less-stable flavonol is formed from the glycoside (e.g. when rutin (41) is hydrolysed to quercetin (36)) [60].
For the analysis of insoluble organic dyes, other approaches have to be taken for their solubilisation. Indigo and the indigoids can be dissolved in DMSO or DMF, often only upon heating [61, 62], while hot pyridine has proven suitable for the extraction of Tyrian purple [10]. Particular care has to be taken when trying to maximise extraction yields of indigoid substances since the individual representatives of this compound class will undergo oxidation during the extraction at higher temperatures in the presence of oxygen. Some colour-forming compounds of the so-called insoluble red dyewoods can be at least partially dissolved in solvents such as ethyl acetate, chloroform or carbon tetrachloride, or in alkaline aqueous solution [63]. Care must however be taken that, due to the changed conditions during chromatographic separation, the compounds do not become insoluble again and precipitate in the HPLC system.
Chromatographic separation
The chromatographic separation of colouring matter is almost exclusively performed with reversed-phase HPLC columns [1, 10, 29, 33]. Column dimensions typically are 4.6 mm × 250 mm with 5-μm particle diameter. These dimensions tend to be very ‘conservative’, and in view of the limited sample amount available and the need to achieve highest sensitivity, it is surprising that only few laboratories have adopted the use of columns of smaller diameter (2 or 3 mm), shorter length (100 or 150 mm) and smaller particle diameter (3 μm) [59, 63–65]. Apparently, the available instrumentation prevents the widespread use of narrow-bore and capillary columns, as does the quest for robust analytical procedures. Monolithic HPLC columns which have had a major impact on liquid chromatography in almost all other fields of application [66] have not been used so far in the analysis of natural organic dyestuffs to the best knowledge of the author. This may be due to the fact that speed of analysis is not such an important issue in art analysis as it is in the pharmaceutical industry, for example. Moreover, full advantage of the use of monolithic columns is only made at comparatively high flow rates which normally are not desirable, particularly not in combination with MS detection.
The stationary phases typically used for the analysis of dyestuffs are octadecylsilane (ODS, C18) modified and endcapped. Generic gradients are used, such as starting at H2O (0.1% TFA)/acetonitrile (0.1%TFA) 95:5, then changing to 100% acetonitrile (0.1% TFA) within 60 min, and were probably introduced by Halpine as early as 1996 [67] and adopted later on by several groups (e.g. [68]). The use of trifluoroacetic acid or acetic acid is in principle compatible with MS detection, whereas phosphoric acid is not and must be replaced by one of the volatile acids. Particularly when using TFA, however, concentrations should be kept at the minimum that still provides adequate chromatography (e.g. 0.001% instead of 0.1%) in order to avoid signal suppression due to the ion-pairing effect and to minimize corrosion effects for the LC/MS equipment [61, 69]. The use of acetic acid is therefore clearly preferable, if sufficient chromatographic resolution can be achieved.
Unfortunately, there is no general agreement on the gradient program used in the analysis of natural organic dyestuffs, not even on the fact that a linear gradient be used. Otherwise, conversion of the retention time from a separation carried out under one set of conditions to another would be possible and would greatly simplify the assignment of unknown peaks, establishing a system similar to the Kovacs retention index which is widely used in flavour analysis by gas chromatography [70]. Retention of analytes can easily be modelled in liquid chromatography [71], and the retention times obtained for a known set of analytes from two runs under different chromatographic conditions are typically sufficient to extrapolate retention behaviour to deliberate other separation conditions.
The analysis of the intact glycosides of the dyestuffs is often performed on more polar columns owing to the better water solubility of these compounds. Typical columns used for this purpose are C4-modified [65] or C8-modified [64] silica columns. The analysis of indigoid dyes is sometimes performed with a different mobile phase due to the limited solubility in water/methanol or water/acetonitrile eluents. A water/acetonitrile/THF gradient has been proposed for this purpose [68].
Mass spectrometric detection
In relation to the relatively large number of papers dealing with liquid chromatographic analysis of historical dyestuffs with diode array detection, it is surprising that the use of HPLC with MS detection has remained relatively limited so far [72, 73]. Apart from the lack of general availability of this more costly detector in the conservation science laboratory, two further reasons for this situation may exist: first, the limited sensitivity of mass spectrometric detection for these classes of substances, resulting in a sensitivity of MS detection in the scan mode that is comparable with the well-established UV/Vis diode array detection, and even in the SIM mode it is only in the most favourable cases one order of magnitude more sensitive [64]; second, MS detection in the liquid phase with a simple quadrupole or ion trap instrument and ionisation by one of the soft atmospheric pressure ionisation techniques can typically not provide the same richness of information and positive identification capability as GC/MS with electron impact ionisation does. In contrast to UV/Vis detection, the primary information that can be deduced is thus not the colour of the separated compound, but rather its molecular weight.
Early applications of LC-MS for the analysis of natural organic dyes made use of LC–thermospray MS [74–76]. With the commercial availability of the more robust and easy-to-use atmospheric pressure interfacing techniques, more recent work was almost exclusively performed with either atmospheric pressure chemical ionisation (APCI) [69, 77] or electrospray ionisation (ESI) [50, 59, 62, 65, 77–83]. These ionisation techniques are commonly known as soft ionisation techniques, implying that very little fragmentation of the analyte molecule typically occurs, thus also limiting structural information [84]. This lack of information can be overcome by different approaches: (a) collision-induced dissociation, (b) multidimensional mass spectrometers, and (c) high resolution mass spectrometers. In collision-induced dissociation (CID), instrumental conditions (pressures, voltages) are chosen in such a way that the analyte ions (typically molecular or quasimolecular ions) undergo fragmentation upon collision with residual gas molecules (N2) in an intermediate pressure zone after the atmospheric pressure ionisation region, but before entering the high-vacuum region of ion separation. The effect is a significant increase in fragmentation, along with a drastic decrease in sensitivity. Depending on the mode of ion detection (positive or negative) the fragments formed can be more or less informative (e.g. loss of small neutral molecules such as water, Δm = 18 amu, from OH groups in positive ion mode, or loss of more characteristic groups in negative ion mode). CID is often performed with single-quadrupole instruments as a simple option to increase information content of the signal. The price to be paid, however, is high in view of the significant reduction in sensitivity, and considering that selectivity is still poor due to the fact that the precursor ion has not been isolated, and thus fragment ions appear together with all other m/z signals detected in the sample. This makes this approach feasible only for the characterisation of reference substances or at higher analyte concentrations in real samples [84, 85].
A more suitable approach—if instrumentation is available—is the use of multidimensional mass spectrometers. These allow one to isolate one particular ion from the first MS dimension (often the molecular or quasimolecular ion), then to induce fragmentation by low-energy collision with a collision gas or the application of a radiofrequency pulse, and then to record the fragment ion spectrum. The two stages of ion selection and detection can either be separated in space (as is done in triple-quadrupole or MS/MS instruments) or in time (as for ion trap instruments). The latter type of instrument allows experiments of even higher order (MSn) to be performed with n ranging up to 5 or 6 where precursor ions are isolated and the fragment ion spectrum detected subsequently in each step.
The third approach to increase information content is the use of high resolution MS. If the exact mass of the molecular ion is determined with sufficient precision and accuracy, the molecular formula can be deduced from this information. The smaller the mass window, the smaller is the number of possible molecular formulae for a given nominal mass. Within a window of the exact mass ±10 ppm, about 200 possible candidates exist, within ±5 ppm about ten possible candidates, and for a window of ±2 ppm typically only about one or two candidates remain. Several types of instruments capable of high mass resolution are available including sector field (SF), time-of-flight (TOF) and ion cyclotron resonance–Fourier transform (ICR-FT) instruments. The use of SF instruments is nowadays decreasing in popularity due to the relatively unfavourable price/performance ratio of this technique. In many applications SF instruments have been supplanted by TOF instruments which currently are the commercially most successful type of high resolution mass spectrometers. Their relative simplicity of use and speed of data acquisition makes them highly attractive. ICR-FT instruments provide ultimate mass resolution and accuracy; however, the substantially higher price of this type of instrument related to the other high resolution mass spectrometers has precluded its widespread use.
Hybrid mass spectrometers are becoming more popular for various analytical tasks. They combine one or two different types of mass analysers, such as quadrupole–time-of-flight (q-TOF), two-dimensional time-of-flight (TOF/TOF) or ion trap–time-of-flight instruments (IT-TOF), to provide at the same time two-dimensional or multidimensional mass spectrometry with high resolution. The use of such hybrid LC-MS instruments for natural organic dyestuff analysis has not been reported yet, to the best of the author’s knowledge.
Modes of measurement
Both theoretical considerations and practical experiments show that the ranges of application of atmospheric pressure chemical ionisation and electrospray ionisation are complementary [85, 86]: APCI is reported to work well for low to medium molecular mass analytes of moderate to medium polarity, whereas ESI is more successful in the ionisation of medium to high polarity and even ionic analytes with hardly any restriction to the molecular mass (large biomolecules are multiply charged). Since very apolar compounds can hardly be ionised by either of the two techniques, a third atmospheric pressure ionisation technique has been introduced: atmospheric pressure photoionisation (APPI), which is reported to work very well for polycyclic aromatic hydrocarbons [87]. The potential of this ionisation technique has not yet been assessed for the analysis of natural organic dyestuffs. Due to the rather polar nature of most natural organic dyestuffs, ESI is the preferred mode of ionisation, although APCI has occasionally also been used. The disadvantage of electrospray compared with atmospheric pressure chemical ionisation is that the signal depends to a larger degree on the matrix, making measurements in different matrices more variable and also more susceptible to signal suppression. Table 3 and Fig. 3 compile the ionisation modes under which the various natural organic dyestuffs have been detected. It is important to emphasise that instrument design has a critical influence on the signals observed. In the extreme case, one mode of ionisation may produce good results with the interface from one manufacturer, while with an instrument of a different make signals may become detectable with inferior sensitivity.

Plot of the most sensitive modes of ionisation in LC-MS analysis of natural organic dyestuffs. Compound class names in brackets indicate that ions are detected albeit at lower sensitivity than in the other polarity
The polarity of ions formed in the atmospheric pressure ionisation mode is a function of the analyte’s structure and the mobile phase composition. Compounds with a high proton affinity such as nitrogen bases or the anthraquinoid structures can easily be protonated, whereas molecules with one or more (aromatic) hydroxyl groups are more easily deprotonated, yielding intensive negative molecular ion signals. Dye molecules very often are polyfunctional molecules, possessing both types of (proton acceptor and donor) functional groups. They can thus often be detected in either positive or negative ion mode. For those dyestuffs that can be detected in both modes—mainly flavones and anthraquinoids—the positive ion detection mode often produces larger signals. This is of course also a consequence of the use of acidic mobile phases which suppress the dissociation of the weakly acidic phenol groups of the analytes. However, as was noted by certain authors [50], the negative ion mode may still provide the better sensitivity owing to a better signal-to-noise ratio, and is thus preferred in most cases.
Structural elucidation
Although it was discussed above that the so-called soft ionisation techniques (ESI, APCI, APPI) provide relatively little structural information in the mass spectra, instrument settings can be chosen in a way to enhance in-source fragmentation which provides an increased degree of structural information even with single-quadrupole instruments. This versatile approach has also been applied in selected cases to elucidate the structure of the natural organic dyestuffs [62, 80].
In the following, we will thus discuss typical fragmentation patterns class by class to provide a means of identifying unknown colouring matter from natural sources.
Anthraquinoids
The mass spectra of anthraquinoids have been studied by various groups, both with direct reference to dyestuff analysis [77–81, 88] and with other focus [89]. Detection was reported to be possible in positive and negative ion modes, although the negative ion mode was the more sensitive one, and in the case of anthraquinone aglycans sometimes the only applicable one [81]. Laccaic acids were found to have similar sensitivity in both ionisation modes [77]. The main fragmentation reactions that the anthraquinoids exhibit are decarbonylation of one of the keto groups (Δm = 28 amu) or from the phenolic groups. Successive decarbonylation may lead to fragment ions [M−H−28]−, [M−H−56]− and [M−H−84]− (Fig. 4). For laccaic acids, decarboxylation of the carboxylic acid group [M−H−44]− is an important fragmentation pathway, followed by the loss of water [M−H−62]−. Further intensive fragments are [M−H−88]− and [M−H−106]− which correspond to the loss of two moles of CO2 and to the subsequent loss of water from the fragment ion, [M−H−2CO2−H2O]−, respectively.

Proposed fragmentation pattern for A alizarin, B xanthopurin and C monochloroalizarin (reprinted from Szostek et al. [77] with permission from Elsevier Ltd.)
Indigoids
Indigoids are difficult candidates for ESI [62, 77, 80] and APCI [69, 90] detection. They produce signals of relatively low intensity and are detectable only in positive ion mode. The mass spectra essentially feature only the protonated molecular ion [M+H]+. Fragment ions are detectable at [M+H−28]+, [M+H−44]+ and [M+H−57]+ which correspond to the subsequent loss of CO and NH2, and the loss of CO+HCO, respectively (Fig. 5). In the case of indigotin, the doubly charged cation [M+2H]2+ is also observed. It is worth noticing that, in contrast to other reports, Sztosek et al. were not able to detect indirubin at normal concentrations in ESI(+) mode [77]. The authors attributed this to the structural differences between indigotin and indirubin: The latter molecule can form intramolecular hydrogen bonds between the oxygen and the nitrogen atom which prevent the molecule from being efficiently protonated. This probably also prevents the molecule from being detected in APCI(+) mode, as reported by Karapanagiots et al. [69]. A similar observation was made for 6-monobromoindigotin and 6,6′-dibromoindigotin. In APCI(+) mode, at least the last two compounds could be detected and easily identified by the characteristic patterns of the bromine isotopes. Intensive fragment ions are seen for [M+H−Br]+ at [M+H−79]+. The indigo precursor isatan B was also detectable in ESI(−) in an extract of woad leaves [91].

Fragmentation pattern of indigoid dyes
Flavonoids
Fragmentation patterns of flavonoids have been widely studied, as these compounds not only are used as colouring matter in historical organic dyes, but as they are important natural constituents of fruit and vegetables [92]. Interest in the elucidation of structures of the flavonoids and their glycosides by ESI-MS therefore mainly comes from this area [93–95].
As the glycosidic bond is weaker than the bonds within the flavonoid molecule, the fragmentation pathway of O-glycosylated flavonoids typically starts with the cleavage of the glycosidic bond, leading to the elimination of the sugar moiety with the charge being retained on the aglycone fragment. In contrast, fragmentation of the sugar molecule is typically observed with C-glycosides. The mass difference between the presumptive molecular ion and the fragment ion is indicative for the type of sugar, i.e. if it is a pentose, a hexose, or a diglycan or triglycan. This implies, of course, that a mild extraction technique has been used that does not hydrolyse the glycosidic bonds. A mass difference of Δm = 162 amu is characteristic for the loss of a hexose, and a Δm = 146 amu suggests the loss of a deoxyhexose, while a mass difference of Δm = 132 amu is characteristic for the loss of a pentose [96]. An excellent example of this situation is the mass spectral detection of rutin (quercetin-3-O-(6′-O-rhamnosyl)-galactoside) [94]. With ESI(+), characteristic ions are detected at m/z 611, 465, 449 and 303 (and also a Na+ adduct quasimolecular ion at m/z 633, [97]). The signals correspond to the quasimolecular ion, the loss of the rhamnosyl moiety excluding and including the oxygen atom that establishes the bond to the galactose unit, and the loss of the galactose moiety (Fig. 6).

ESI mass spectra obtained from rutin: a ESI(+) in direct injection mode (MeOH/water 1:1) (reprinted from Stobiecki et al. [97] with permission from John Wiley & Sons Ltd.). b LSIMS negative ion spectrum, obtained with a magnetic sector instrument (reprinted from Cuyckens and Claeys [92] with permission from John Wiley & Sons Ltd.)
With respect to the analysis of flavonoid glycosides, LC-MS provides information on (a) the molecular mass, (b) the structure of aglycone (pattern of hydroxylation of the aglycone, and attachment points of rings B and C), (c) acylation of sugar hydroxyl groups and possible methylation or sulfatation of the aglycone hydroxyl groups, and (d) the number of sugar units, and in a few cases also on their configuration and the placement of the glycosidic bonds.
API-MS spectra (both with APCI and ESI) were shown to be relatively similar to FAB-MS spectra, although their activation energy is only in the range of a few electron-volts, instead of 2–20 keV [92]. This allows us to adopt large parts of the interpretation previously given for the FAB mass spectra of flavone glycosides [92, 98–100].
To describe the fragmentation pattern of flavonoid glycosides in a clear and comprehensive way, a special notation has been introduced by Ma et al. [98], based on preceding work by Domon and Castello [101]. This notation is illustrated in Fig. 7. The most informative fragments derive from the cleavage of C–C bonds of the C ring. These result in i,jA+ and i,jB+ fragments. The superscripts i and j denote the number assigned to the bond of the C ring of the flavonoid. These fragments can be seen as the products of a retro-Diels–Alder (RDA) reaction. They are the diagnostically most relevant, since they provide information on the number and type of substituents in the A and B rings. For the free aglycones, the i,jA+/i,jB+ notation refers to fragments that contain the intact A and B rings of the flavonoid. For conjugated aglycones, a subscript 0 is used to the right of the letter, to avoid confusion with the \({\text{A}}_{\text{i}}^{\text{ + }} \) and \({\text{B}}_{\text{i}}^{\text{ + }} \) (i ≥ 1) fragments of the carbohydrate units containing a terminal (nonreducing) sugar unit [101]. Under typical conditions, fragmentations are observed with C–C bond cleavage at positions 1/3, 0/2, 0/3, 0/4 or 2/4 of the C ring. Which of these fragmentations is favoured depends strongly on the substitution pattern of the particular flavonoid. In the case where there is a hydroxyl group in position 3 (that is, the compound is a flavonol), there are additional possibilities of fragmentation, such as 0,2A+, [0,2A−CO]+, [0,2A+2H]+ and [1,3B−2H]+. For flavones, signals 1,3B+, 0,4B+ and [0,4B−H2O]+ are typically observed. To illustrate this, the mass spectra of luteolin and kaempferol shall be discussed (Fig. 8). Although both compounds have the same molecular mass and quasimolecular ion (m/z 287), the flavone, luteolin, can readily be distinguished from the flavonol, kaempferol, on the basis of their i,jA+ and i,jB+ ions. Most characteristic for flavonols is the presence of an intense 0,2A+ ion (stabilised by the OH group in the 3-position) which is absent in the case of flavones. The 1,3A+ ion is typically very intense in the mass spectra of all flavonoids (although, in contrast to FAB-MS, this depends strongly on the ionisation conditions with atmospheric pressure ionisation techniques). Due to its biochemical pathway of formation (from acetate and malonate) the A ring often has hydroxyl groups in positions 5 and 7, so that the 1,3A+ ion is found at m/z 153. In the absence of the 4-keto group, that is, for flavanes and flavanaols, the signal of this fragment is found at m/z 139, and a signal at m/z 135 indicates that the A ring is substituted by only one OH group.

Nomenclature of ions formed in mass spectrometric analysis of flavonoid glycosides (illustrated for apigenin 7-O-rutinosid) (reprinted from Cuyckens and Claeys [92] with permission from John Wiley & Sons Ltd.)

Low-energy CID mass spectra of a luteolin and b kaempferol, obtained with a magnetic sector-quadrupole instrument (reprinted from Ma et al. [98] with permission from John Wiley & Sons Ltd.)
Further to these i,jA+ and i,jB+ ions, the loss of small neutral molecules or radicals from [M+H]+ is observed, such as loss of CO [M+H−28]+ or H2O [M+H−18]+. A loss of 15 mass units implies the presence of a methoxy-substituted flavonoid which is characterized by a very intense [M+H−CH3]+ peak. Although the above discussion relates to the positive ion mode, much of what was said also holds true for negative ion detection. Negative ion mode detection is typically significantly more sensitive for the flavonoid aglycans [62]; however, fragmentation is more difficult to achieve, and the mass spectra are thus less informative.
The product ions from conjugated flavonoids are identified according to the nomenclature proposed by Domon and Costello [101]. The ions that contain the aglycone are labelled k,lX j , Y j and Z j , where j denotes the number of the glycosidic bond that is broken, starting from the aglycone. The superscripts k and l denote cleavages within the carbohydrate ring. The glycosidic bond that links the flavonoid and the glycan is numbered 0. When the charge stays on the glycan moiety, the fragments that can further be formed are denoted k,lA i , B i and C i , where i (≥ 1) is the number of the glycosidic bond cleaved, counting from the nonreducing end. It is now possible to distinguish O-glycosides, C-glycosides or O,C-diglycosides on the basis of low-energy CID spectra [102]. The protonated O-glycosides typically produce \({\text{Y}}_{\text{1}}^{\text{ + }} \) and \({\text{Y}}_{\text{0}}^{\text{ + }} \) ions which stem from the rearrangement of the interglycosidic bond (Fig. 9). It appears that the hydrogen atoms from the hydroxyl groups of the sugars are required for the formation of \({\text{Y}}_{\text{n}}^{\text{ + }} \) ions. In the case of O,C-diglycosides only those \({\text{Y}}_{\text{1}}^{\text{ + }} \) ions are detected which are formed by the cleavage of the interglycosidic bond. In the case of C-glycosides, only the [M+H]+ signal is detected, with further signals from cross-ring cleavages and the loss of water. In the negative ion mode, deprotonated O-glycosides and di-O-glycosides produce both \({\text{Y}}_{\text{1}}^ - \) and \({\text{Y}}_{\text{0}}^ - \) ions.

Formation of Y0 and Y1 ions (illustrated with the example of protonated apigenin-7-O-neopheseridose). Any hydroxyl group from the sugar moiety can participate in the rearrangement reaction (reprinted from Li and Claeys [102] with permission from John Wiley & Sons Ltd.)
Mass spectrometric measurements allow one to deduce information on the carbohydrate sequence and the aglycone moiety. As an example, the case of rutin (quercetin-3-O-rutinoside) can be taken: the loss of 146 amu points to a terminal deoxyhexose rest (in this case, rhamnose) which yields a \({\text{Y}}_{\text{1}}^{\text{ + }} \) signal at m/z 465. The further loss of 162 amu (leading to a signal at m/z 303) indicates that the second sugar is a hexose. Alternatively, the direct loss of the rutinose moiety also results in the aglycone ion \({\text{Y}}_{\text{0}}^{\text{ + }} \) (Fig. 6).
In flavonoid-C-glycosides, there is a direct link between the flavonoid ring and the sugar via a nonhydrolysable C–C bond. For this reason, the mass spectra provide very little information other than molecular mass. Under harsher CID conditions, cross-ring cleavage of the sugar moiety is induced, and the loss of water is frequently encountered. As far as is known today, C-glycosides are only linked through a bond in the C-6-position or C-8-position of the flavonoid system. The challenge for mass spectrometric detection is thus to find out in which of these two positions substitution has occurred. This can in some instances be achieved by evaluating relative peak intensities: The loss of water from the protonated flavonoid-C-glycoside is a prominent reaction for both C-6 and C-8 glycosides. For the C-6 glycosides (particularly of flavonoids that are OH-substituted in position 5 and/or 7), the loss of one water molecule is even more pronounced due to the formation of a five-membered ring structure (Fig. 10) [103].

Loss of water observed for 6-C-glycosylflavonoids involving the hydroxyl group at the 2″-position of the sugar residue and the hydroxyl group at the 5-position or 7-position of the aglycone. This process is facilitated by hydrogen bonding between the ether oxygen of the sugar residue and a hydroxyl group at the 5-position or 7-position, respectively (reprinted from Li et al. [102] with permission from John Wiley & Sons Ltd.)
Neoflavones
Hulme and coworkers studied the CID fragmentation pattern of brazilein and haematein obtained with ESI(−) [82]. The authors proposed that for these two analogous compounds, which differ only by one OH group, a complex electron rearrangement mechanism of the deprotonated molecule leads to cleavage of the four-ringed system into a diphenolic compound and an o-naphthoquinoic structure. As the substitutent of ring A of the initial compound remains with the diphenolic moiety, the same characteristic naphthoquinonic fragment (at m/z 173) is detected for both compounds (Fig. 11).

Fragmentation pattern of brazilein and hematein (redrawn from Hulme et al. [82] with permission from Elsevier Ltd.)
The fragmentation pattern of the two reduced analogues, brazilin and haematoxylin, follows a different scheme and results in a substituted benzopyran (chromene) moiety and a benzosemiquinone. The three main signals observed are the deprotonated quasimolecular ion [M−H]−, a signal with Δm = 18 amu, corresponding to the loss of a water molecule (most likely from the aliphatic hydroxyl group), and signals at m/z 163 and 179, respectively (Fig. 12).

Fragmentation pattern of brazilin and hematoxylin (redrawn from Hulme et al. [82] with permission from Elsevier Ltd.)
Tannins
Tannins are a very heterogenous and complex class of compounds. Their structures are not yet elucidated completely [104], and it must be assumed that any type of sample preparation, such as extraction and/or change of the pH value of their native environment, changes the original tannin structure [105]. Extraction by methanol and acidification of the extract (which is required for successful chromatography) is thus expected to hydrolyse the labile bonds of the more complex tannins to simpler structures [106]. This explains why the mass spectra observed for tannins exhibit a ‘quasimolecular ion’ which can be attributed to the pentaester of a hexose, in this particular case, the pentagalloyl-glucose structure. The signal of this ‘quasi-molecular ion’ is indeed seen in negative ion detection at m/z 939 (corresponding to the structure in Table 4), accompanied by a further signal with a mass difference of 152 amu (leading to m/z 787) which can be attributed to the loss of one galloyl unit, [M−(GA−H2O)−H]−, with GA = gallic acid. Chromatograms of tannins extracted from natural samples do not show just one signal, but a multiplet of chromatographically incompletely resolved peaks, where the above-mentioned signal derives from the latest eluting peak. Under reversed-phase chromatographic conditions, seven individual peaks can be identified, resulting from the various degrees of substitution of the central glucose molecule by gallic acid residues (Fig. 13). The mass spectra of these peaks thus differ by a further 152 mass units: [M−(GA−H2O)2−H]− at m/z 635, [M−(GA−H2O)3−H]− at m/z 483, and also the dimer and monomer of gallic acid is detected: [GA2−H2O−H]− at m/z 321, and [GA−H]− at m/z 169. Negative ion detection is again more sensitive then detection in positive ion mode by a factor of 10–20, but also the ESI(+) spectra provide diagnostically highly useful spectra. While the signal of the protonated ‘quasi molecular ion’ is absent, a strong signal is seen for the ion that is produced from this structure by the loss of water [M−H2O+H]+ at m/z 923. Although ESI(+) produces a series of signals which can be attributed to the subsequent loss of gallic acid units, leading to [M−GA+H]+ at m/z 771, [M−GA−(GA−H2O)+H]+ at m/z 619, [M−GA−(GA−H2O)2+H]+ at m/z 467, [M−(GA−H2O)3+H]+ at m/z 449, and also the monomer of gallic acid is detected prior and after the loss of water: [GA+H]+ at m/z 171, and [GA−H2O+H]+ at m/z 153.

Excerpts from the chromatographic separation and mass spectra of tannic acid, recorded in a negative ion mode and b positive ion mode (reprinted from Pawlak et al. [62] with permission from John Wiley & Sons Ltd.)
Even though other tannins, e.g. with other sugar units and other aromatic acid substituents as building blocks, such as in ellagitannins do differ in the actual masses of the signals observed, the mass spectral pattern discussed for poylgalloylglucose is transferable [107].
Quantitative aspects of LC-MS analysis of organic dyestuffs
From many other fields of application, LC-MS is known to be a highly sensitive analytical technique, allowing the detection even of trace and ultratrace amounts of analytes, particularly when using the atmospheric pressure ionisation techniques. This seems to be rather different in the case of natural organic dyestuff analysis where sensitivities are reported to be relatively poor [63]. Even in SIM mode, the detection limits of individual dyestuffs with LC-ESI-MS rarely are lower by more than one order of magnitude in comparison with LC-DAD (Table 5). This may be due to several reasons: First, the various compound classes relevant for natural organic dyestuffs appear to not generally be easily ionisable. Second, many of these compounds are acids, or compounds with acidic character. Although detection in negative ion mode is thus the obvious choice, this is somewhat hampered due to the necessity of using acidic eluents for efficient chromatography. With unbuffered or neutral eluents, specific response may be somewhat enhanced; however, acidic compounds would exhibit a significant peak broadening which more than offsets the gain in sensitivity. It should further be mentioned that with MS detection, acetic acid or acetate buffer are the preferred mobile phase additives owing to their volatility and the absence of ion-pairing effects, although they do not necessarily provide the best chromatographic performance. The addition of trifluoroacetic acid or phosphoric acid would typically provide better chromatographic resolution, but they are problematic with MS detection due to their corrosiveness and ion-pairing effects (TFA) or involatility (H3PO4). With UV/Vis detection, however, this is not a real limitation, and owing to the expected improvement of chromatographic peakwidth (and consequently, sensitivity) this technique may thus appear even more favourable than MS detection in quantitative terms.
Considering this relatively mediocre sensitivity in MS detection, the ability of mass spectral structure elucidation of unknown organic dyestuffs must also be seen in perspective: LC-MS will rather prove useful to confirm the presence of organic dyestuffs where the UV/Vis spectrum does not allow one to differentiate between similar compounds. The identification of natural organic dyestuffs on the basis of their fragmentation pattern will, however, only be possible at high concentrations, or with pure standards.
It should be mentioned, though, that the above discussion refers to liquid chromatography with columns of “normal” dimensions, that is, 4.0- or 4.6-mm ID. For columns of these dimensions which are typically operated at column flows around 1 mL min−1, the DAD cell volume typically is in the order of 10 μL. When the column inner diameter is reduced to 1 mm (narrow bore) or 100–530 μm (capillary format) and the flow rate is downscaled appropriately, detection volumes must proportionally be reduced to avoid detector-related peak braodening. This will at a certain point reduce the sensitivity of DAD in relation to MS detection. It can thus be expected that MS detection will be the more sensitive and advantageous technique in the case of capillary chromatography, capillary electrokinetic chromatography (CEC) and capillary electrophoresis (CE) separations of natural organic dyestuffs.
Conclusions and outlook
Liquid chromatography with mass spectrometric detection has demonstrated its usefulness for the analysis of natural organic dyestuffs in many examples reported in the literature. It is typically used complementary to HPLC-DAD, and allows one to confirm the presence of a certain structure or compound class in natural organic dyestuffs which often represent a complex mixture of compounds. Although mass spectra obtained with the soft atmospheric pressure ionisation techniques are typically less informative due to the limited degree of fragmentation, the patterns typically observed allow one to determine the molecular weight of the compound, which particularly in the case of (flavonoid) glycosides is an important piece of information to propose its actual structure and, derived from this identification, possible sources of origin. In the case of glycosides with an oligoglycan moiety, even conclusions on the glycan sequence can be drawn. Although the information content of the spectra can be increased by collision-induced dissociation in the interface of single-quadrupole or ion trap instruments, this mode of operation may be practical only in a few cases where there are sufficiently high concentrations of dyestuff or pure standard solutions available. For real samples, the sensitivity and selectivity of triple-quadrupole instruments in the selected reaction monitoring (SRM) mode would be necessary to extract improved qualitative and quantitative information.
For quantitative analysis, LC-MS presents only a marginal advantage over LC with diode array detection, except for the fact that it may be more selective in complex matrices. This situation is expected to change, however, with the increased use of capillary columns in favour of MS detection.
MS detection in natural organic dyestuff analysis is furthermore expected to gain more importance in the near future owing to the increased availability and use of more powerful mass spectrometric instrumentation: this includes high resolution instruments (allowing the determination of the exact molecular formula of the ion or fragment), and hybrid instruments (q-TOF, TOF-TOF or IT-TOF instruments) which allow one to obtain higher-order mass spectra [108], sometimes even in combination with high mass resolution. This undoubtedly brings a significant increase in information content.
The successful application of mass spectrometric detection for the identification of natural organic dyestuffs will critically depend on the creation and availability of a dedicated MS library for this class of compounds. Such approaches have—with a considerable degree of success—already been established in other fields of application, such as forensic analysis [109–111], clinical analysis and in the environmental field [112], and there is at least one example of a spectral library that has been established for flavonoids with an ion trap-ESI-MS/MS instrument [113]. When aiming at the creation of a database for general use, comparability of mass spectra from different instrument types and manufacturers, however, becomes an important issue [114, 115]. It has thus been proposed to use thoughtfully selected substances to tune the interface’s parameters to values that would ensure a consistent appearance of the spectra across different types of instruments and instrument makes [116, 117]. It can be assumed that such a procedure would also provide comparability of natural dyestuff mass spectra acquired on different instruments.
Finally, the sample preparation step is an important issue to mention. The delicacy of natural dyestuff analysis typically results from the limited availability of samples from historical objects. The analytical techniques have to be capable of dealing with minute amounts of sample (typically, the weight of a fibre or section of a fabric used for one analysis is less than 1 mg) which have to be extracted quantitatively, but at the same time in a way that still ensures the integrity of the analytes to ensure the validity of conclusions drawn from the analytical results. In this particular respect, we can expect the development of further ‘mild’ extraction techniques that aim particularly at preserving the integrity of glycosidic bonds in conjugated dyestuffs.
With all these improvements in sample preparation, chromatographic separation and mass spectrometric detection, analysis of natural organic dyestuffs still remains a challenging, fascinating and in the literal sense ‘colourful’ task.
Abbreviations
- APCI:
-
atmospheric pressure chemical ionisation
- APPI:
-
atmospheric pressure photoionisation
- CE:
-
capillary electrophoresis
- CEC:
-
capillary electrochromatography
- CID:
-
collision-induced dissociation
- DAD:
-
diode array detection
- ESI:
-
electrospray ionisation
- FAB:
-
fast atom bombardment
- FLD:
-
fluorescence detection
- GC-MS:
-
gas chromatography–mass spectrometry
- HPLC:
-
high-performance liquid chromatography
- ICR-FT-MS:
-
ion cyclotron resonance–Fourier transform mass spectrometry
- IT-TOF-MS:
-
ion trap–time-of-flight mass spectrometry
- LC-MS:
-
liquid chromatography–mass spectrometry
- MAE:
-
microwave-assisted extraction
- q-TOF:
-
quadrupole–time-of-flight MS
- SEM-EDS:
-
scanning electron microscopy–energy dispersive spectroscopy
- SF-MS:
-
sector field mass spectrometer
- SIM:
-
selected ion monitoring (MS)
- SRM:
-
selected reaction monitoring mode (MS)
- TLC:
-
thin-layer chromatography
- TOF-MS:
-
time-of-flight mass spectrometer
- UAE:
-
ultrasonically assisted extraction
- UV:
-
ultraviolet
- Vis:
-
visible
- XRF:
-
x-ray fluorscence
References
Hofenk-de Graaff JH (2004) The colourful past: origins, chemistry and identification of natural dyestuffs. Archetype, London
Ferreira ESB, Hulme AN, McNab H, Quye A (2004) Chem Soc Rev 33:329–336
Cardon D (2007) Natural dyes - sources, tradition, technology, science. Archetype, London
Miliani C, Rosi F, Burnstock A, Brunetti BG, Sgamellotti A (2007) Appl Phys A 89:849–856
Clementi C, Miliani C, Romani A, Favaro G (2006) Spectrochim Acta B 64:906–912
Wetzel DL, LeVine SM (1999) Science 285:1224–1225
Vandenabeele P, Weis TL, Grant ER, Moens LJ (2004) Anal Bioanal Chem 379:137–142
Castro K, Pérez-Alonso M, Rodríguez-Laso M-D, Etxebarria N, Madariaga JM (2007) Anal Bioanal Chem 387:847–860
Verhecken A, Mortsel B (2005) Dyes History Archaeol 20:1–22
Wouters J (1985) Stud Conserv 30:119–128
Niessen WMA (2003) J Chromatogr A 1000:413–436
Tiller PR, Romanyshyn LA, Neue UD (2003) Anal Bioanal Chem 377:788–802
Reemtsma T (2001) Trends Anal Chem 20:500–517
Careri M, Mangia A, Musci M (1998) J Chromatogr A 794:263–297
Sleno L, Volmer DA (2004) J Mass Spectrom 39:1091–1112
Zadrona I, Polec-Pawlak K, Guch I, Ackacha MA, Mojski M, Witowska-Jarosz J, Jarosz M (2003) J Sep Sci 26:996–1004
Schweppe H (1993) Handbuch der Naturfarbstoffe. Nikol, Hamburg
Trojanowicz M, Orska Gawryś J, Surowiec I, Szostek B, Urbaniak-Walczak K, Kehl J, Wróbel M (2004) Stud Conserv 49:115–130
van Bommel M, Geldof M, Hendriks E (2005) Art Matters 3:111–137
Joosten I, van Bommel MR, Hofman-de Keijzer R, Reschreiter H (2006) Microchim Acta 155:169–174
Hochleitner B, Desnica V, Mantler M, Schreiner M (2003) Spectrochim Acta B 58:641–649
Lennard F, Hayward M (eds) (2005) Tapestry conservation: principles and practice. Butterworth-Heinemann, London.
Balfour-Paul J (1998) Indigo. British Museum, London
Baeyer A, Drewson V (1882) Ber Dtsch Chem Ges 14:2856–2864
Haude ME (1998) J Am Chem Inst Art Conserv 37:240–270
Cooksey CJ (2001) Molecules 6:736–769
Fouquet H, Bielig H-J (1971) Angew Chem Int Ed Engl 10:816–817
Elsner O (1991) Dyes Hist Archaeol 10:11–16
Wouters J, Verhecken A (1991) Dryers Colour 107:266–269
Ziderman I (2007) Paper presented at Dyes in History and Archaeology 26 (DHA 26), Vienna, Austria, 8–10 Nov 2007
McGovern PE, Michel RH (1990) Acc Chem Res 23:152–158
Sato M, Sasaki Y (2003) Dyes Hist Archaeol 19:19–23
Wouters J, Verhecken A (1989) Stud Conserv 34:189–200
Derksen GCH, Van Beek TA (2002) Stud Natural Prod Chem 26:629–684
Doğan Y, Başlar S, Hüseyin Mert H, Ay G (2003) Econ Bot 57:442–453
Nowik W, Desposiers S, Surowiecz, Trojanowicz M (2005) Archaeometry 47:835–848
Wouters J (2001) Dyes Hist Archaeol 16/17:145–157
Fieser LF (1930) J Chem Educ 7:2609–2633
Burnett AR, Thomson RH (1968) J Chem Soc C 854–857
Nowik W (2001) Dyes Hist Archaeol (2001) 16/17:129–144
Edwards HGM, de Oliveira LFC, Nesbitt M (2003) Analyst 128:82–87
Takahashi Y, Miyasaka N, Tasaka S, Miura I, Urano S, Ikura M, Hikichi K, Matsumoto T, Wada M (1982) Tetrahedron Lett 23:5163–5166
Takahashi Y, Saito K, Yanagiya M, Ikura M, Hikichi K, Matsumoto T, Wada M (1984) Tetrahedron Lett 25:2471–2474
Cardon D (1994) Dyes Hist Archaeol 13:59–73
Knecht E, Rawson C, Loewenthal R (1933) A manual of dyeing, 9th edn. Griffin, London
Kaiser R (1993) Angew Bot 67:128–131
Karapanagiotis I, Lakka A, Valianou L, Chryssoulakis Y (2007) Microchim Acta. DOI 10.1007/s00604-007-0774-4
Purinton N, Watters M (1991) J Am Inst Art Conserv 30:125–144
Novotná P, Pacáková V, Bosáková Z, Štulik K (1999) J Chromatogr A 863:235–241
Surowiec I, Szostek B, Trojanowicz M (2007) J Sep Sci 30:2070–2079
Koestler RJ, Sheryll R, Indictor N (1985) Stud Conserv 30:58–62
Bitossi G, Giorgi R, Mauro M, Salvadori B, Dei L (2005) Appl Spectrosc Revs 40:187–228
Gilbert KG, Cooke DT (2001) Plant Growth Regul 34:57–69
Kaiser R (1993) Angew Bot 67:128–131
Crozier A, Jensen E, Lean MEJ, McDonald MS (1997) J Chromatogr A 761:315–321
Surowiec I, Quye A, Trojanowicz M (2006) J Chromatogr A 1112:209–217
Dabiri M, Salimi S, Ghassempour A, Rassouli A, Taleb M (2005) J Sep Sci 28:387–396
Sanyova J, Reisse J (2006) J Cult Herit 7:229–235
Zhang X, Laursen RA (2005) Anal Chem 77:2022–2025
Laursen R (2007) Paper presented at Dyes in History and Archaeology 26 (DHA26), Vienna, Autria, 8–10 Nov 2007
Karapanagiotis I, de Villemereuil V, Magiatis P, Polychronopoulos T, Vougougiannopoulou K, Skaltsounis A-L (2006) J Liq Chromatogr 29:1491–1502
Pawlak K, Puchalska M, Miszczak A, Rosłoniec E, Jarosz M (2006) J Mass Spectrom 41:613–622
Surowiec I, Nowik W, Trojanowicz M (2004) J Sep Sci 27:209–216
Surowiec I, Szostek B, Trojanowicz M (2007) J Sep Sci 30:2070–2079
Zhang X, Boytner R, Cabrera J-L, Laursen R (2007) Anal Chem 79:1575–1582
Guiochon G (2007) J Chromatogr A 1168:101–168
Halpine SM (1996) Stud Conserv 41:76–94
Orska-Gawryś J, Surowiec I, Kehl J, Rejniak H, Urbaniak-Walczak K, Trojanowicz M (2003) J Chromatogr A 989:239–248
Karapanagiotis I (2006) Am Lab 38:36–40
Goodner KL (2008) LWT - Food Sci Technol 41:951–958
Reijenga JC (2000) J Chromatogr A 903:41–48
Wouters J (2005) Dyes Hist Archaeol 20:23–29
Verhecken A, Mortsel B (2005) Dyes Hist Archaeol 20:1–22
Yamaoka R, Shibayama N, Yamada T, Sato M (1989) Shitsuryo Bunseki 37:249–253
Yinon J, Saar J (1991) J Chromatogr A 586:73–84
Hiserodt R, Hartmann TG, Chi-Tang H, Rosen RT (1996) J Chromatogr A 740:51–63
Szostek B, Orska-Gawrys J, Surowiec I, Trojanowicz M (2003) J Chromatogr A 1012:179–192
Karapanagiotis I, Chryssoulakis Y (2006) Ann Chim 96:75–84
Rafaëlly L, Héron S, Nowik W, Tchapla A (2008) Dyes Pigm 77:191–203
Puchalska M, Połeć-Pawlak K, Zadrozna I, Hryszko H, Jarosz M (2004) J Mass Spectrom 39:1441–1449
Derksen GCH, Niederländer HAG, van Beek TA (2002) J Chrom A 978:119–127
Hulme AN, McNab H, Peggie DA, Quye A (2005) Phytochem 66:2766–2770
Zhang X, Good I, Laursen R (2007) J Archaeol Sci. DOI 10.1016/j.jas.2007.08.001
Niessen WMA (2003) J Chromatogr A 1000:413–436
Niessen WMA (1999) Liquid chromatography-mass spectrometry, 2nd edn. Marcel Dekker, New York
Herderich M, Richling E, Roscher R, Schneider C, Schwab W, Humpf H-U, Schreier P (1997) Chromatographia 45:127–132
Bos SJ, van Leeuwen SM, Karst U (2006) Anal Bioanal Chem 384:85–99
White R, Kirby J, in: Kirby J, ed. (1999) Dyes History Archaeol 16/17:169–174
Mueller SO, Schmitt M, Dekant W, Stopper H, Schlatter J, Schreier P, Lutz WK (1999) Food Chem Toxicol 37:481–491
Liau B-C, Jong T-T, Lee M-R, Chen S-S (2007) J Pharmaceut Biomed Anal 43:346–351
Gilbert KG, Hill DJ, Crespo C, Mas A, Lewis M, Rudolph B, Cooke DT (2000) Phytochem Anal 11:18–20
Cuyckens F, Claeys M (2004) J Mass Spectrom 39: 1–15 and Errata: Cuyckens F, Claeys M (2004) J Mass Spectrom 39:461
Wu W, Yan C, Li L, Liu Z, Liu S (2004) J Chromatogr A 1047:213–220
Stobiecki M (2000) Phytochem 54:237–256
Careri M, Elviri L, Mangia A (1999) Rapid Commun Mass Spectrom 13:2399–2405
Häkkinen S, Auriola S (1998) J Chrom A 829:91–100
Stobiecki M, Malosse C, Kerhoas L, Wojlaszek P, Einhorn J (1999) Phytochem Anal 10:198–207
Ma YL, Li QM, Van den Heuvel H, Claeys M (1997) Rapid Commun Mass Spectrom 11:1357–1364
Ma Y-L, Van den Heuvel H, Claeys M (1999) Rapid Commun Mass Spectrom 13:1932–1942
Ma Y-L, Cuyckens F, Van den Heuvel H, Claeys M (2001) Phytochem Anal 12:159–165
Domon B, Costello CE (1988) Glycoconj J 5:397–409
Li QM, Claeys M (1994) Biol Mass Spectrom 23:406–416
Li QM, Van den Heuvel H, Dillen L, Claeys M (1992) Biol Mass Spectrom 21:213–221
Quideau S, Feldman KS (1966) Chem Rev 96:475–503
Hardouin J, Lange CM (2005) Curr Org Chem 9:317–324
Mueller-Harvey I (2001) Anim Feed Sci Technol 91:3–20
Cai Y-Z, Xing J, Sun M, Zhan Z-Q, Corke H (2005) J Agric Food Chem 53:9940–9948
Ferreira ESB, Quye A, Hulme AN, McNab H (2003) Dyes Histor Anal 19:13–18
Dresen S, Kempf J, Weinmann W (2006) Forensic Sci Int 161:86–91
Pihlainen K, Sippola E, Kostiainen R (2003) J Chromatogr A 994:93–102
Marquet P, Venisse N, Lacassie É, Lachâtre G (2000) Analusis 28:925–934
Baumann C, Cintora MA, Eichler M, Lifante E, Cooke M, Przyborowska A, Halket JM (2000) Rapid Comm Mass Spectrom 14:349–356
Jong SL, Dong HK, Liu K-H, Tae KO, Lee CH (2005) Rapid Comm Mass Spectrom 19:3539–3548
Josephs JL, Sanders M (2004) Rapid Comm Mass Spectrom 18:743–759
Bristow AWT, Nichols WF, Webb KS, Conway B (2002) Rapid Comm Mass Spectrom 16:2374–2386
Weinmann W, Stoertzel M, Vogt S, Wendt J (2001) J Chromatogr A 926:199–209
Weinmann W, Stoertzel M, Vogt S, Svoboda M, Schreiber A (2001) J Mass Spectrom 36:1013–1023
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rosenberg, E. Characterisation of historical organic dyestuffs by liquid chromatography–mass spectrometry. Anal Bioanal Chem 391, 33–57 (2008). https://doi.org/10.1007/s00216-008-1977-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-1977-0