
Abstract
The O-linked β-N-acetylglucosamine (O-GlcNAc) modification is an abundant post-translational modification in eukaryotic cells. This dynamic glycosylation plays a fundamental role in the activity of many nuclear and cytoplasmic proteins and is associated with pathologies like type II diabetes, Alzheimer’s disease or some cancers. However the exact link between O-GlcNAc-modified proteins and their function in cells is largely undefined for most cases. Here we report a strategy based on the 1,3-dipolar cycloaddition, called click chemistry, between unnatural N-acetylglucosamine (GlcNAc) analogues (substituted with an azido or alkyne group) and the corresponding biotinylated probe to specifically detect, enrich and identify O-GlcNAc-modified proteins. This bio-orthogonal conjugation confirms that only azido analogue of GlcNAc is metabolized by the cell. Thanks to the biotin probe, affinity purification on streptavidin beads allowed us to identify 32 O-GlcNAc-azido-tagged proteins by LC-MS/MS analysis in an MCF-7 cellular model, 14 of which were previously unreported. This work illustrates the use of the click-chemistry-based strategy combined with a proteomic approach to get further insight into the pattern of O-GlcNAc-modified proteins and the biological significance of this post-translational modification.

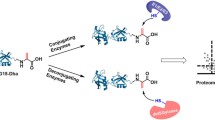
Detection of biotinylated O-GlcNAz proteins in MCF-7 cells
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The O-linked β-N-acetylglucosamine (O-GlcNAc) modification of serine or threonine residues of nuclear and cytosolic proteins is an abundant and dynamic post-translational modification [1, 2]. The enzymes that catalyse the dynamic addition and removal of O-GlcNAc on proteins are respectively the O-GlcNAc transferase (OGT) and a specific β-N-acetylglucosaminidase named O-GlcNAse [3]. This glycosylation plays a fundamental role in the modulation of the activity of proteins, including transcription factors, cytoskeletal proteins, signalling proteins and kinases. Although these roles are not fully understood, an increase of the cellular O-GlcNAc level has been tightly associated with type II diabetes phenotypes, while in the case of Alzheimer’s disease, the hyperphosphorylation of Tau protein is linked to a decrease in O-GlcNAc modification [4, 5]. The specific detection and identification of such modified proteins is consequently of great interest in order to decipher the regulating functions of O-GlcNAc modification under physiological conditions and in pathological situations. For this purpose, Bertozzi and coworkers developed a strategy based on the Staudinger ligation which involves the highly specific reaction between a phosphine and an azide group [6]. This reaction has the feature that neither phosphine nor azide occur naturally in biomolecules, which makes the approach particularly suitable for biological applications. They have demonstrated that the enzymes implicated in the hexosamine salvage pathway tolerate and metabolize the azido analogue (GlcNAz) of their natural substrate N-acetylglucosamine. Indeed, a fraction of the extracted protein was detected using a fluorescent phosphine probe, demonstrating the presence of O-linked GlcNAz on the proteins. Another bio-orthogonal reaction, click chemistry, has proven to be effective in detecting and purifying proteins targeted with azide-substituted affinity labels. This activity-based protein profiling (ABPP) developed by Cravat and coworkers uses the CuI-catalysed azide–alkyne cycloaddition reaction [7, 8]. This reaction is a variant of Huisgen’s 1,3-dipolar cycloaddition between an azide group and a terminal alkyne to afford stable 1,2,3-triazoles. This chemical method represents a powerful tool in the field of biology research as the cycloaddition reaction operates in aqueous buffers compatible with biological investigations using copper(I) as catalyst [9, 10]. Like the azide group, alkynes are essentially inert towards most of the chemical groups encountered in biological molecules [11, 12]. This bio-orthogonality makes the click chemistry reaction suitable for functional proteomic studies. Moreover, comparative studies between the Staudinger ligation, the strain-promoted and the click chemistry cycloadditions have shown that the last of these brings the highest sensitivity of protein labelling in a complex protein extract [13].
Given these data we wondered whether a click-chemistry-based strategy could not only detect proteins modified by O-GlcNAc moieties but also enable their specific purification for subsequent identification. To address this question, two analogues of N-acetylglucosamine modified with an alkyne or azide group and their corresponding reactive biotinylated probes were synthesized. The reactivity of the two pairs of reacting partners in terms of click chemistry cycloaddition was first measured in vitro by mass spectrometry. The efficiency of in cellulo metabolic incorporation of these analogues was then carried out in the breast cancer cell line MCF-7. The specificity of the click chemistry coupling between the modified proteins and their probes was assessed by western blotting prior to their purification on a streptavidin column for further proteomic analysis by mass spectrometry. This strategy allowed us to identify 32 O-GlcNAz-modified proteins in MCF-7 cells.
Materials and methods
Synthesis of GlcNAc analogues and biotin probes
See Electronic supplementary material
In vitro assessment of cycloaddition between the modified GlcNAc and the appropriate probe
Modified GlcNAc (5 mM) and the appropriate probe (5 mM, stock solution in DMSO) were mixed in phosphate buffer pH 8 at room temperature. The ligand (tris(triazolyl)amine) (2 mM, stock solution in DMSO/tBuOH 1:4, v/v) and freshly prepared reducing agent (2 mM) (tris(2-carboxyethyl)phosphine (TCEP) or sodium ascorbate) were added, followed by CuSO4 (1 mM) in a final volume of 1 mL. As soon as CuSO4 was added, the time course of the click chemistry reaction was followed by LC-MS. LC-MS analyses were performed on a triple quadrupole mass spectrometer (API 2000, Applied Biosystems) equipped with an electrospray source and operating in single ion monitoring (SIM) mode. The mass spectrometer was coupled to a high pressure liquid chromatography system (Agilent) equipped with a C18 HPLC column (50 mm × 2.1 mm, dp = 3.5 μm, Waters Symmetry C18).
Cell culture and metabolic protein labelling
MCF-7 cells were routinely cultured in DMEM (4.5 g L−1 Glc) supplemented with 10% fetal calf serum (FCS) and 1% penicillin–streptomycin at 37 °C in a humidified atmosphere of 5% CO2. For labelling experiments, cells were seeded in 6-well dishes (Nunc, Fisher Bioblock Scientific, France) in Complete medium and when 80% confluency was obtained, the culture medium was replaced with low-glucose-containing DMEM (2 g L−1 Glc) supplemented with 10% FCS and containing either DMSO (1:1,000) or peracetylated GlcNAc (GlcNAc4), peracetylated azido-GlcNAc (2, GlcNAz) or peracetylated alkyne-GlcNAc (4, GlcNAk) prepared at 250 mM in DMSO and diluted to the indicated concentrations (50–250 μM). Cells were labelled for 24 h, or for the indicated times.
Preparation of cytosolic and nuclear protein fractions from MCF-7 cells
Two hours before the end of the labelling time, 50 μM O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenyl carbamate (PUGNAc)/well was added into the medium. Cells were then washed twice with cold PBS and lysed for 30 min on ice in 200 μL well−1 of lysis buffer I (20 mM Tris/HCl pH 7.6, 150 mM NaCl, 5 mM MgCl2, 0.5% Triton X-100) containing inhibitors of de-O-glucosaminylation and dephosphorylation (100 μM PUGNAc, 5 mM NaF, 1 mM orthovanadate) and a cocktail of protease inhibitors (Complete protease inhibitors cocktail tablets, Roche Diagnostics). Samples were collected and each well was rinsed once with 100 μL of buffer I. The lysates were centrifuged at 800 g for 10 min at 4 °C. The supernatant was clarified by centrifugation (20,000 g, 20 min, 4 °C) and the resulting supernatant was considered as the cytosolic fraction. The pellet from the first centrifugation was rinsed once with 100 μL of buffer I, centrifugated one more time (800 g, 10 min, 4 °C) and then lysed for 30 min on ice in 100 μL of lysis buffer II (20 mM Tris/HCl pH 7.6, 50 mM KCl, 400 mM NaCl, 1 mM EDTA, 1% Triton X-100, 20% glycerol, containing the same inhibitors as buffer I). After centrifugation (800 g, 10 min, 4 °C) the final supernatant was considered as the nuclear fraction.
Click reaction on labelled proteins and detection by western blotting
Cytosolic or nuclear fractions (50 μL) were treated with 100 μM of the appropriate biotin probe, freshly prepared 1 mM sodium ascorbate and 100 μM ligand. After gentle homogenisation, 1 mM CuSO4 was added and the reaction mixture was gently homogenised. Click reaction was performed for 1 h at room temperature and then stopped by the addition of 15 μL sample−1 of 5× Laemmli buffer (50 mM Tris pH 6.8, 5% SDS, 5% beta-mercaptoethanol, 40% glycerol) and heating at 100 °C for 10 min. Denaturated proteins were separated by 10% SDS-PAGE and transferred onto a nitrocellulose membrane (Hybond-C Extra, Amersham Biosciences). Membranes were then incubated for 1 h in blocking buffer (3% bovine serum albumin, BSA, fraction V, Sigma) prepared in TBST (50 mM Tris pH 8.0, 140 mM NaCl, 0.05% Tween-20) followed by incubation with horseradish peroxidase conjugated to avidin D in blocking buffer (HRP-avidin, Vector, France). After extensive washes in TBST, labelled proteins were revealed using the ECL system according to the manufacturer’s instructions (Immobilon Western, Millipore, France).
Affinity purification of biotinylated GlcNAz-modified proteins
Cytosolic proteins from MCF-7 cells cultured for 24 h with or without 250 μM GlcNAz were submitted to affinity purification on streptavidin beads (Novagen) after click chemistry reaction which was stopped by chloroform/methanol precipitation. After centrifugation, the protein pellet was resuspended in the equilibration buffer (PBS with 1% SDS, 150 mM NaCl, 1 mM DTT and protease inhibitors cocktail), heated at 80 °C and sonicated to solubilise proteins. After centrifugation (to clarify the protein solution), sample was incubated on streptavidin beads for 2 h at room temperature. The flow-through was discarded, beads were washed successively in equilibration buffer, PBS, 8 M HCl guanidium pH 3.5, and labelled proteins were eluted with the elution buffer (8 M urea, 2% SDS, 30 mM biotin in PBS) by vortexing the beads vigorously for 15 min at room temperature followed by incubation for 15 min at 96 °C [14]. For 10% SDS-PAGE separation, 5× Laemmli buffer was added into the eluted fractions. After migration, the gel was silver-stained.
Identification of proteins by nano-HPLC–tandem mass spectrometry
Bands of interest were cut and submitted to in-gel trypsin digestion overnight at 37 °C in 25 mM ammonium bicarbonate buffer (Sequencing modified grade trypsin, Promega, France). Tryptic peptides were sequentially extracted with 45% acetonitrile/45% water/10% trifluoroacetic acid (TFA) (v/v/v) and 95% acetonitrile/5% TFA. The peptide samples were then dried in a speed-vac, dissolved in 5 μL water/ 5% HCOOH (v/v) and submitted to nano-HPLC–tandem mass spectrometry analysis.
Nano-HPLC-nanoESI-MS/MS analyses were performed on an ion trap mass spectrometer (LCQ Deca XP + , Thermo-electron, San Jose, CA) equipped with a nano-electrospray ion source coupled to a nano flow high-pressure liquid chromatography system (LC Packings Dionex, Amsterdam, The Netherlands). Peptides were loaded in buffer A (95% water/5% acetonitrile/0.08% HCOOH (v/v/v)) onto a C18 HPLC column (15-cm length, 75-μm ID, C18 Pepmap column, Dionex) and were eluted from the column with a linear gradient from 5 to 100% solvent B (20% water/80% acetonitrile/0.08% HCOOH (v/v/v)) in buffer A for 45 min. The eluted peptides were directly electrosprayed into the LCQ mass spectrometer operating in a data-dependent mode. Fragment ion spectra were searched against the NCBInr Homo sapiens database using the Mascot software (Matrix Science, London, UK). The search parameters were 1.5-Da tolerance for the parent ion mass and 0.8 Da for the MS/MS fragment ions, one missed cleavage allowed, carbamidomethylcysteine as fixed modification, and methionine oxidation as possible modification. Only the proteins with a significant Mascot score (>41) were considered and reported after manual verification of the fragmentation spectra.
Results and discussion
Herein, we report a click-chemistry-based strategy devoted to the purification and identification of proteins modified by the post-translational O-GlcNAc glycosylation. For this purpose, two analogues of N-acetylglucosamine i.e. the azido (GlcNAz) (compounds 1 and 2) and the alkyne (GlcNAk) analogues (compounds 3 and 4), and their corresponding reactive biotinylated probes bearing respectively an alkyne or an azido group (compounds 5 and 6) were synthesized (Scheme 1). The reactivity of the two pairs of reacting partners was assessed by the time course of the click chemistry cycloaddition which was followed in vitro by reversed-phase liquid chromatography coupled to electrospray ionisation mass spectrometry. The relative signal intensities of the following ions were monitored in single ion monitoring: [M+H] + and [M+Na] + corresponding to the biotinylated probes (5 and 6) (respectively at m/z 457.6, 479.3, 511.3, 533.2) and to the formed triazole-containing cycloadducts (compounds 8 and 7) (respectively at m/z 717.4, 739.4, 773.4, 795.4). The signals corresponding to the GlcNAc analogues (1 or 3) were not monitored since both compounds eluted in the column void volume and were consequently subjected to ion suppression phenomena. The GlcNAc analogue was mixed in an equimolar ratio with the biotinylated probe, followed by 0.2 equivalents of tris(triazolyl)amine (ligand), 0.2 equivalents of sodium ascorbate and 0.1 equivalents of CuSO4 as catalytic reagents [7, 8, 15–17]. Under these experimental conditions, the cycloaddition reaction proceeded to apparent completion in less than 20 min for both pairs of reacting partners (Fig. 1). Indeed, adding CuSO4 and/or ascorbate after 30 min of reaction did not result in any further evolution of the reaction mixture (data not shown). These results suggest that both pairs could be appropriate for the proteomic investigation of proteins modified by O-GlcNAc.

1,3-Dipolar cycloaddition (click chemistry) between a modified GlcNAc analogue and the appropriate biotinylated probe. Synthesized molecules used for the click-chemistry-based detection of proteins modified by O-linked GlcNAc are 1 GlcNAz, 2 peracetylated GlcNAz, 3 GlcNAk, 4 peracetylated GlcNAk, 5 azido-biotin probe, 6 alkyne-biotin probe

a 1,3-Dipolar cycloaddition between a GlcNAc analogue (1 or 3) and the appropriate probe (5 or 6). b Time course of the click chemistry reaction followed by liquid chromatography coupled to electrospray mass spectrometry operating in single ion monitoring mode: • disappearance of the reacting probe 6, ▪ final product 7 of the click chemistry reaction between 1 and 6, ▴ disappearance of the reacting probe 5,  final product 8 of the click chemistry reaction between 3 and 5
final product 8 of the click chemistry reaction between 3 and 5
In order to evaluate the ability of the click chemistry reaction to specifically detect the nucleocytoplasmic fraction of glycosylated proteins, breast cancer MCF-7 cells were incubated for 24 h either with GlcNAz (2) or GlcNAk (4) at several concentrations (50, 125, 250 μM). A low-glucose-containing culture medium (2 g L−1) was used to favour incorporation of the modified GlcNAc into the proteins. The GlcNAz (2) has been previously demonstrated to be a substrate of the hexosamine salvage pathway and to be subsequently incorporated into proteins [6, 18]. The coupling between the azido compound and the alkyne-biotinylated probe (6) was thus tested first. After cell lysis, the cycloaddition reaction was carried out both on the cytosolic and nuclear fractions obtained from MCF-7 cells. Proteins were then separated by 10% SDS-PAGE. Figures 2a and b show the western blot analysis of these fractions when cells were cultured either with peracetylated GlcNAc (control), peracetylated GlcNAz (2) or peracetylated GlcNAk (4). As illustrated, the GlcNAz-modified proteins can be selectively detected after conjugation with the alkynyl-biotinylated probe (6) and western blot detection by avidin peroxidase. Indeed, no chemiluminescence signal was observed when cycloaddition was performed with the same biotinylated probe on protein extracts from cells cultured with peracetylated GlcNAc. This feature underlines the lack of cross reactivity between the alkyne group and the chemical diversity of the amino acid side chains of proteins. Similarly, omitting the alkyne probe during the click chemistry step fully abolished the chemiluminescent signal despite the presence of O-linked GlcNAz proteins. This observation highlights the specificity of the HRP-avidin detection towards the biotinylated probe. No signal was observed with the other reacting pair i.e. GlcNAk (4) and the azido probe (5). Since we have previously shown in Fig. 1 that the two pairs of reacting partners exhibit similar reactivity, we can postulate that the alkyne analogue of GlcNAc is not metabolized by the MCF-7 cells. It is unlikely that O-linked GlcNAk does not react with the azido probe since it has been recently shown that an alkyne analogue of fucose allowed in vivo fluorescence imaging of glycans based on a click chemistry reaction [19].

Detection of metabolically labelled O-GlcNAz proteins in MCF-7 cells. a Cytosolic proteins and b nuclear proteins extracted from cells cultured for 24 h in the presence of increasing concentrations of peracetylated analogue of GlcNAc were conjugated to the corresponding biotinylated probe. Peracetylated GlcNAc (GlcNAc4, 250 μM) was used as a control. The resulting tagged proteins were separated by SDS-PAGE and detected by western blotting using the HRP-conjugated avidin. c For the time course experiment, cells were labelled with 250 μM GlcNAz for 2–48 h, followed by conjugation with the alkynyl-biotinylated probe on the cytoplasmic fractions. Protein detection was performed as described above. d PUGNAc (100 μM) or alloxan (5 mM) were added respectively in the last 2 h and 20 h of the labelling period (24 h) in the presence of 250 μM GlcNAz. Labelled proteins were detected as previously described. Molecular masses of prestained protein standards are indicated in kDa
For both the cytosolic and nuclear subcellular fractions, a concentration of 250 μM of GlcNAz was optimum to get an intense and specific signal, without affecting the cellular viability compared with control cells (i.e. culture medium with or without peracetylated GlcNAc). Moreover, a time course of GlcNAz incorporation into cellular proteins was performed and revealed that this GlcNAc analogue is metabolized in MCF-7 cells as rapidly as with 2 h, with the most intense signal obtained after 24 h of incorporation (Fig. 2c). After 48 h of incorporation, a decrease of chemiluminescence intensity was noted, which may reflect the natural turnover of proteins during this time and the possible degradation of the GlcNAz in the culture medium at 37 °C. O-GlcNAc dynamics are controlled by the interplay between O-GlcNAc transferase and O-GlcNAcase. Moreover it has been demonstrated that both enzymes tolerate the azido group in their respective catalytic activity [6]. The effect of alloxan, an inhibitor of O-GlcNAc transferase and O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenyl carbamate (PUGNAc), an O-GlcNAcase inhibitor [20], were both tested towards the incorporation rate of GlcNAz in MCF-7 cells. Western blotting (Fig. 2d) shows that the addition of PUGNAc in the last 2 h of the 24-h incorporation significantly increases the detection of O-GlcNAz-modified proteins, indicating that O-GlcNAz-labelled proteins can be a substrate for the O-GlcNAcase. In contrast, the lack of signal in the presence of alloxan indicates that the functionality of OGT is required for the metabolic incorporation of the azido analogue in living cells.
The demonstration that proteins bearing O-GlcNAz moieties are specifically detected by western blotting opens the way for affinity purification on a streptavidin column for their subsequent identification by mass spectrometry. A cytosolic protein extract treated with GlcNAz for 24 h was purified on streptavidin beads after tagging with the alkyne probe. In order to limit the nonspecific binding, the streptavidin affinity column was preferred to monomeric avidin as it exhibits a stronger affinity (K d of 10–15 versus 10–7 M) therefore permitting harsh washes before the elution step [14].
The protein fractions specifically retained by streptavidin were separated by SDS-PAGE. After silver-staining, the protein bands were subjected to trypsin digestion and the resulting peptide mixtures were analysed by nano-LC–tandem mass spectrometry. Protein identification was performed using the Mascot search engine against the NCBInr protein database which allowed the identification of 32 proteins (Table 1). Control experiments towards affinity purification were carried out in parallel in order to eliminate the possible contaminants resulting from residual unspecific interaction. As a control, cytosolic proteins from MCF-7 cells treated with peracetylated GlcNAc (and tagging with the alkyne-biotinylated probe) or GlcNAz (without tagging with the alkyne-biotinylated probe) were subjected to similar affinity chromatography. Two proteins were detected in all purified eluted fractions: they were identified as fatty acid synthase (accession number NP_004095), a multienzymatic complex involved in fatty acid biosynthesis, and pyruvate carboxylase which is involved in gluconeogenesis (accession number AAB31500). They are both biotin-dependent enzymes [21, 22], that might explain why they are present in the eluted fraction of control samples. Nonetheless, we cannot exclude that they might also be OGT targets such as many metabolic enzymes which have been reported already.
As reported in Table 1 the 32 identified proteins have been classified according to their cellular function i.e. protein synthesis (7), signal transduction (3), protein folding (4), cytoskeletal and structural proteins (7), metabolism (8) and other functions (3). This classification reflects the wide regulatory role of this nucleocytoplasmic post-translational glycosylation and underlines the necessity to dispose of reliable and robust tools to study the dynamics of O-GlcNAc modification. Among these proteins, 18 have already been reported as O-GlcNAc-modified proteins, using either the tagging via substrate strategy (Staudinger ligation) or usual biochemical techniques based on 3H-Gal transfer or immunoprecipitation/immunorevelation [19, 23–26]. Fourteen proteins are new identified targets of OGT, 11 of which belong to protein families from whom some members are already known to be O-glycosylated. This is the case, for instance, for the proteins of the 14–3–3 family (14–3–3 zeta and theta), the ribosomal proteins (S2 and L8) or TCP1 chaperons [23, 27].
Conclusion
The present work demonstrates that the nucleocytoplasmic cellular proteins bearing O-linked GlcNAc residues are able to be specifically detected using a click-chemistry-based strategy that constitutes an alternative to the commonly used immunodetection with monoclonal antibodies. In our hands, only the azido analogue of GlcNAc seems to be efficiently metabolized by the enzymatic pathway driving this post-translational modification. Furthermore, after specific purification on a streptavidin column and subsequent analysis by LC coupling to tandem mass spectrometry we identified 32 O-GlcNAc-substituted proteins among which 14 were not previously reported to be a target of OGT. In the meantime, a commercial kit based on a similar click chemistry reaction (ClickIT™ by Invitrogen) has been proposed for western blot analysis of such glycosylated proteins. Nevertheless, the structure and accurate mass of the commercial biotinylated probe has up to now been unknown, thus preventing identification of the peptides modified by an O-GlcNAc residue in the database. Considering the biology community’s growing interest in deciphering and understanding the cellular events governed by this dynamic glycosylation, we believe that it is of great interest to provide access to easily synthesised and cheaper reagents than those commercially available.
The accurate localization of the O-GlcNAc modification site is still challenging. We are currently developing a chemical alternative to the streptavidin affinity purification technique based on covalent tagging of O-GlcNAc-modified peptides. Enriching and selecting only the modified peptides would thus facilitate not only the localization of the O-GlcNAc on the peptide sequence but also the dynamics of this post-translational modification at each site.
References
Torres CR, Hart GW (1984) J Biol Chem 259:3308–3317
Wells L, Vosseller K, Hart GW (2001) Science 291:2376–2378
Iyer SP, Hart GW (2003) Biochemistry 42:2493–2499
Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX (2004) Proc Natl Acad Sci USA 101:10804–10809
Konrad RJ, Kudlow JE (2002) Int J Mol Med 10:535–539
Vocadlo DJ, Hang HC, Kim EJ, Hanover JA, Bertozzi CR (2003) Proc Natl Acad Sci USA 100:9116–9121
Speers AE, Adam GC, Cravatt BF (2003) J Am Chem Soc 125:4686–4687
Speers AE, Cravatt BF (2004) Chem Biol 11:535–546
Kolb HC, Finn MG, Sharpless KB (2001) Angew Chem Int Ed Engl 40:2004–2021
Bock DV, Hiemstra H, van Maarseveen JH (2006) Eur J Org Chem 2006:51–68
Kolb HC, Sharpless KB (2003) Drug Discov Today 8:1128–1137
Rostovtsev VV, Green LG, Fokin VV, Sharpless KB (2002) Angew Chem Int Ed Engl 41:2596–2599
Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR (2006) ASC Chem Biol 1:644–648
Rybak JN, Scheurer SB, Neri D, Elia G (2004) Proteomics 4:2296–2299
Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG (2003) J Am Chem Soc 125:3192–3193
Link AJ, Tirrell DA (2003) J Am Chem Soc 125:11164–11165
Gupta SS, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG (2005) Bioconjugate Chem 16:1572–1579
Sprung R, Nandi A, Chen Y, Kim SC, Barma D, Falck JR, Zhao Y (2005) J Proteome Res 4:950–957
Sawa M, Hsu TL, Itoh T, Sujiyama M, Hanson SR, Vogt PK, Wong CH (2006) Proc Natl Acad Sci USA 103:12371–12376
Perreira M, Kim EJ, Thomas CJ, Hanover JA (2006) Bioorg Med Chem 14:837–846
Jitrapakdee S, Wallace JC (1999) Biochem J 340:1–16
Smith S, Witkowski A, Joshi AK (2003) Prog Lipid Res 42:289–317
Nandi A, Sprung R, Barma DK, Zhao Y, Kim SC, Falck JR, Zhao Y (2006) Anal Chem 78:452–458
Ku NO, Omary MB (1995) J Biol Chem 270:11820–11827
Walgren JL, Vincent LS, Schey KL, Buse MG (2003) Am J Physiol Endocrinol Metab 284:E424–E434
Wells L, Vosseler K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW (2002) Mol Cell Proteomics 1:791–804
Zachara NE, Hart GW (2004) Biochim Biophys Acta 1673:13–28
Acknowledgements
The authors are grateful to Sébastien Vidal, Philippe Belmont and Tony Lefebvre for helpful discussions, to Peter Goekjian for critical reading of the manuscript and to Adeline Page for nano-HPLC-MS/MS experiments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Caroline Gurcel and Anne-Sophie Vercoutter-Edouart contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 96.2 kb)
Rights and permissions
About this article
Cite this article
Gurcel, C., Vercoutter-Edouart, AS., Fonbonne, C. et al. Identification of new O-GlcNAc modified proteins using a click-chemistry-based tagging. Anal Bioanal Chem 390, 2089–2097 (2008). https://doi.org/10.1007/s00216-008-1950-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-1950-y