
Abstract
A chitosan resin possessing a phenylarsonic acid moiety (phenylarsonic acid type chitosan resin) was developed for the collection and concentration of trace uranium prior to inductively coupled plasma (ICP) atomic emission spectrometry (AES) measurement. The adsorption behavior of 52 elements was systematically examined by packing it in a minicolumn and measuring the elements in the effluent by ICP mass spectrometry. The resin could adsorb several cationic species by a chelating mechanism, and several oxo acids, such as Ti(IV), V(V), Mo(VI), and W(VI), by an anion-exchange mechanism and/or a chelating mechanism. Especially, U(VI) could be adsorbed almost 100% over a wide pH region from pH 4 to 8. Uranium adsorbed was easily eluted with 1 M nitric acid (10 mL), and the 25-fold preconcentration of uranium was achieved by using a proposed column procedure, which could be applied to the determination of trace uranium in seawater by ICP-AES. The limit of detection was 0.1 ng mL−1 for measurement by ICP-AES coupled with 25-fold column preconcentration.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
According to World Health Organization guidelines, Health Canada, the National Health and Medical Research Council of Australia, and the US Environmental Protection Agency, the maximum concentration level of uranium in drinking waters has been regulated to be below 15, 20, 20, 30 ng mL−1, respectively. Seawater contains about 3 ng mL−1 uranium, which should be accurately determined for evaluating the uranium behavior. Although some spectrophotometric methods have been developed, they cannot be applied to the trace analysis of uranium, whose limits of detection (LODs) are above the parts-per-million level (1 ppm is equivalent to 1 μg mL−1) [1, 2]. An inductively coupled plasma (ICP) atomic emission spectrometry (AES) measurement is one of the powerful analytical methods, whose LOD is 3 ng mL−1, whereas the preconcentration technique is necessary for the accurate determination of uranium at several parts-per-billion levels (1 ppb is equivalent to 1 ng mL−1) in seawater [3].
In order to overcome these problems, solid-phase extraction methods have been used as preconcentration techniques prior to trace analysis [4–13]. Some commercially available resins, such as UTEVA and Dowex 1-X8, have been used for the uranium collection, but these resins have some problems [5–8]. They adsorb U(VI) only in high concentrations of acidic solution, and then produce a large amount of acidic waste. Some researchers have reported on solid materials modified with succinic acid, maleic acid, o-vaniline semicarbazone, and 5-aminoquinoline-8-ol moieties, which adsorb uranium only over narrow pH regions [9–13].
Chitosan resins are considered to be suitable for the collection and concentration of trace elements in aquatic media, because the adsorption rate of ions on chitosan resins is faster than that on synthetic base materials, such as polystyrene–divinylbenzene, polyethylene, and polyurethane, which is due to the hydrophilicity of chitosan itself [14]. The authors have synthesized chitosan resin possessing an iminodi(methylphosphonic acid) moiety for uranium collection. The resin can adsorb trace uranium over a wide pH region, whereas uranium adsorbed on it is difficult to elute even with high concentrations of acid, such as 6 M HNO3 or 6 M HCl [15]. We must overcome such problems as the adsorption and elution of uranium.
In this study, we newly and simply synthesized a novel chelating resin possessing a phenylarsonic acid moiety, which can form a stable complex with U(VI) in aquatic media [16], for the concentration of trace uranium, and then examined its preconcentration for the ICP-AES measurement.
Experimental
Apparatus
The ICP mass spectrometry (MS) system was a model SPQ 8000H (Seiko Instruments, Chiba, Japan), and the ICP-AES system was a model Vista-PRO (Seiko Instruments, Chiba, Japan). The optimized operating conditions are summarized in Table 1. An automated titration system (model AT-310; Kyoto Electronics Manufacturing, Kyoto, Japan) was used for the acid–base titration to estimate the pK a values of the resin. IR spectra were recorded with the KBr pellet method using an FT/IR-4100 spectrometer (JASCO, Tokyo, Japan).
Reagents
Chitosan flake (deacetylated degree, about 80%) was purchased from Tokyo Kasei (Tokyo, Japan). All other reagents used for the synthesis of a phenylarsonic acid type chitosan resin were of analytical reagent grade.
The stock solution of an analytical standard for metal ions was prepared by diluting several kinds of a single-element standard solution for atomic absorption spectrometry (1,000 μg mL−1; Wako Pure Chemicals, Osaka, Japan) and a multielement standard solution for ICP-MS provided by Spex CertiPrep (Metuchen, NJ, USA). This stock solution was diluted by weight just before the column treatment with 0.1 M nitric acid to give 10 ng mL−1 of each metal.
Ultrapure grade nitric acid (60%, density 1.38 g mL−1, Kanto Chemicals, Tokyo, Japan) was diluted with ultrapure water. The acetic acid (minimum 96%) and ammonia–water (29%) used for the preparation of ammonium acetate solution were of an electronic industrial reagent grade purchased from Kanto Chemicals (Tokyo, Japan).
Ultrapure water (18.3 MΩ cm−1 resistivity) prepared by an Elix 3/Milli−Q Element system (Nihon Millipore, Tokyo, Japan) was used throughout.
Synthesis of phenylarsonic acid type chitosan resin
The cross-linked chitosan resin as a base material was synthesized in a similar manner to that described in our previous work as shown in Fig. 1, scheme 1 [17]. Chitosan flake (20 g), which was ground to fine pieces and sieved to obtain chitosan particles of diameter 100–300 μm, was suspended in ethanol (200 mL). Benzaldehyde (80 g) was then added to the suspension. The mixture was stirred at room temperature for 12 h to protect the amino groups of chitosan as a Schiff base. After the reaction, the product was filtered off and washed with ethanol and water, to remove unreacted benzaldehyde. The chitosan derivative protected amino groups with benzaldehyde was refluxed with ethylene glycol diglycidyl ether (30 g) in dioxane (300 mL) and 1 M NaOH (40 mL) for 6 h. The product was filtered off and washed with ethanol and water. The Schiff base was cleaved to an amino compound by twice stirring the product in 0.5 M HCl (1,000 mL) at room temperature for 12 h, followed by filtration and washing with ethanol and water, respectively.

Scheme for the synthesis of phenylarsonic acid type chitosan resin. EGDE ethylene glycol diglycidyl ether, r.t. room temperature
A phenylarsonic acid moiety was introduced in two steps as shown in Fig. 1, scheme 2. First, the cross-linked chitosan resin (5 g) was suspended in a mixture of water (50 mL) and ethanol (50 mL), then chloromethyloxirane (10 g) was added to the suspension, and the mixture was refluxed for 6 h. The product was filtered off and washed with ethanol and water to remove the residual reagents. Second, the cross-linked chitosan resin with the arm of chloropropanol and p-aminophenylarsonic acid (20 g) were suspended in dioxane (100 mL). After 1 M NaOH (40 mL) had been added to the suspension, the mixture was refluxed for 6 h in order to couple the amino group of p-aminophenylarsonic acid with the terminal chloro group of the arm of the cross-linked chitosan resin. The product was filtered off and washed with ethanol and water. The IR adsorption frequency assignment to the phenylarsonic acid type chitosan resin was 826 cm−1, whose frequency depicted the additional bands compared with cross-linked chitosan itself. This band is due to the benzene ring of phenylarsonic acid introduced into the cross-linked chitosan resin as a base material.
Column procedures
Before packing into columns, the phenylarsonic acid type chitosan resin was cleaned up to remove residual metallic impurities in the resin as follows. A 10-mL aliquot of the wet resin was transferred to a 100-mL plastic beaker, containing 2 M nitric acid (80 mL). The mixture was stirred carefully at a low speed for 6 h. The resin was then filtered off, and rinsed with ultrapure water.
The column procedure is similar to that described in our previous work [14, 17]. One milliliter of the resin was packed in small polypropylene columns (5.0-mm inner diameter × 50 mm; Muromachi Chemical, Kyoto, Japan). Each 10-mL aliquot of 1 M nitric acid and ultrapure water was passed thorough the column for washing. Then, a 5-mL aliquot of a conditioning solution (pH 1–2, 0.1 or 0.01 M nitric acid; pH 3–9, 0.5 M ammonia–acetate solution) was passed through the column to adjust the pH of each column. A sample solution (10 mL), whose pH was adjusted, was passed through the column. Then, a 5-mL portion of a rinsing solution (pH 1–2, 0.1 or 0.01 M nitric acid; pH 3–9, 0.2 M ammonia–acetate solution) was passed through the column to remove matrix ions remaining on the resin, as alkali and alkaline-earth metal ions. In order to rinse the remaining components of the rinsing solution in the column, a 5-mL portion of the ultrapure water was passed through the column. Finally, 1 M nitric acid (10 mL) was passed through it to recover the elements adsorbed on the resin. The elements in these eluates were determined by ICP-MS to examine the adsorption behavior. In order to determine trace uranium with 25-fold preconcentration by ICP-AES, 50 mL of the sample solution was used, and then the eluent (10 mL) was evaporated to dryness, and the residue was dissolved in 2 mL of 1 M nitric acid, and measurement was made by ICP-AES. Throughout all the column procedure, the flow rate was maintained at about 1 mL min−1.
Procedures for the acid–base titration
The phenylarsonic acid type chitosan resin (wet volume, 1 mL) was suspended in a mixture of 2 mL of 0.1 M hydrochloric acid and 28 mL of the ultrapure water. The mixture was stirred and titrated with 0.10 M NaOH solution. The titration rate was maintained at about 0.05 mL min−1.
Results and discussion
Fundamental characteristics of phenylarsonic acid type chitosan resin
The pK a values obtained by acid–base titration for the phenylarsonic acid type chitosan resin were about 4.2 and 9.1, which might be attributed to the phenylarsonic acid moiety introduced into the resin, as the pK a values of phenylarsonic acid are reported as 4.1 and 9.2, respectively [18]. To neutralize the phenylarsonic acid moiety, 0.8 mL of 0.10 M NaOH is required, which means that 0.08 mmol of the arsonate group exists in 1 mL (wet volume) of the resin. Figure 2 shows the adsorption equilibrium of U(VI) and Cu(II) on the resin. The resin (1 mL) was equilibrated with each metal ion for 5 h at pH 5 in the presence of an excess amount of each metal ion. The adsorption capacities for U(VI) and Cu(II) were 0.046 mmol mL−1 (equivalent to 83 mg g−1) and 0.020 mmol mL−1, respectively. The capacity of uranium on the phenylarsonic acid type chitosan resin is higher than that on the other chelating resin reported so far as shown in Table 2, and uranium could be adsorbed over a wider pH region. Considering the quantities (millimoles per milliliter) of U(VI) and Cu(II) adsorbed on the resin and the quantity (millimoles per milliliter) of the phenylarsonic acid moiety in the resin, U(VI) and Cu(II) form chelates of 1:2 and 1:4 (metal to phenylarsonate group), respectively. Uranium can be adsorbed more effectively than Cu(II). Phenylarsonic acid and its derivatives are known to form a stable complex with U(VI) [16]. Therefore, uranium might be adsorbed on the resin by forming the chelate with the phenylarsonic acid groups introduced into the resin.

Relationship between the adsorption time and the amount of U(VI) and Cu(II) adsorbed on phenylarsonic acid type chitosan resin at pH 5. Resin, 1 mL (wet volume); concentration of metal ions, 0.01 M U(VI) or Cu(II); metal ions solutions, 100 mL
Adsorption behavior of 52 elements on phenylarsonic acid type chitosan resin
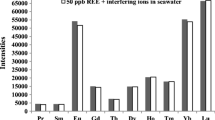
The adsorption behavior of 52 elements on the phenylarsonic acid type chitosan resin was examined by using the column procedure. Figure 3 shows the results obtained for the adsorption and recovery of each 10 ng mL−1 of 52 elements in the pH region from 1 to 9. Most of the elements adsorbed on the resin were thoroughly recovered with 10 mL of 1 M nitric acid as an eluent. The phenylarsonic acid type chitosan resin could adsorb Cu(II) at pH 5–6 and U(VI) at pH 4–8 effectively, and the adsorption behaviors were similar. These metal ions may be adsorbed fundamentally by a chelating mechanism with the phenylarsonic acid moiety in the resin. The adsorption properties of other cationic species except for Cu(II) and U(VI) were similar to that on cross-linked chitosan resin. Therefore, the cationic species might be adsorbed by forming chelates with the residual amino groups of chitosan itself. On the other hand, oxo acids, such as Ti(IV), V(V), Mo(VI), and W(VI), exist as neutral or cationic species in the acidic pH region and as anionic species around the neutral pH region. Such oxo acids might be adsorbed on the resin by a chelating and/or an anion-exchange mechanism. Especially, the phenylarsonic acid type chitosan resin possess good adsorption ability and selectivity for U(VI) at pH 5, which is due to formation of the stable chelate with the phenylarsonic acid moiety in the resin.

Adsorption behavior of trace elements at various pH values with phenylarsonic acid type chitosan resin. Sample, 10 mL; concentration of each element in the samples, 10 ng mL−1; column, 1 mL of the resin (wet volume). The oxidation states of some specific elements in a sample solution are as follows; Be(II), B(III), Al(III), Sc(III), Ti(IV), V(V), Cr(VI), Mn(II), Fe(III), Co(II), Ni(II), Cu(II), Zn(II), Ga(III), Ge(IV), As(III), Se(IV), Y(III), Mo(VI), Ru(III), Rh(III), Ag(I), Cd(II), In(III), Sn(II), Sb(III), Te(IV), W(VI), Hg(II), Tl(I), Pb(II), Bi(III), Ln(III), Th(IV), U(VI). Elements with small atomic symbols were the ones tested and each small scale is represented by the example of uranium near the top
Application of phenylarsonic acid type chitosan resin to the collection and concentration of uranium in seawater
Seawater contains high concentrations of matrices, such as Na, K, Mg, and Ca, which interfere with the determination of trace elements, because the deposition of such matrix constituents on the torch and nebulizer affect the accuracy of ICP-AES measurement. Also, uranium is present at the several parts-per-billion level in seawater, which is difficult to determine directly by ICP-AES (LOD about 3 ng mL−1). Therefore, the preconcentration of trace uranium and the separation of matrices are necessary for ICP-AES measurement.
Table 3 also shows the results of U(VI) recovery in the concentration range from 1 to 10 ng mL−1 and the results obtained from the examination of the effect of cationic matrices, such as Na, K, Mg, and Ca, on the recovery of uranium. Twenty-five-fold preconcentration of uranium in the presence of matrices was examined for the preconcentration of uranium at several parts-per-billion levels and the elimination of matrices by using the proposed column procedure. These results in Table 3 show good recovery, and also indicate that the proposed technique can be applied to the preconcentration of trace uranium in real seawater by ICP-AES measurement. With 25-fold preconcentration, the LOD by ICP-AES was 0.1 ng mL−1. Uranium adsorbed on the resin could be recovered with 10 mL of 1 M nitric acid, and the column procedure was effective for removing high concentrations of matrices in sample solutions.
Table 4 shows the accuracy of this method for the determination of trace uranium in seawater is assessed by analyzing real seawater samples. A trace amount of uranium at a similar concentration to uranium in real seawater was added to each real seawater sample before the column procedure. The recovery tests for U(VI) added are given in Table 4, which shows a sufficient recovery and a high reproducibility. The proposed procedure is suitable for the elimination of matrices and the concentration of trace uranium in real seawater for the ICP-AES measurement.
Conclusions
A novel chelating resin, phenylarsonic acid type chitosan resin, was developed for the concentration of trace uranium and the elimination of matrices in seawater. The main advantages of the resin were as follows: (1) U(VI) was adsorbed over a wide pH region from 4 to 8, (2) matrices were eliminated with the column procedure, and (3) U(VI) in seawater was concentrated by 25-fold with the minicolumn (wet volume of the resin, 1 mL). Also, the proposed technique could be applied to the determination of trace uranium in seawater by ICP-AES measurement.
References
Gupta KK, Kulkarni PG, Thomas G, Varadarajan N, Singh RK, Nair MKT (1993) Talanta 40:507–510
Agnihotri NK, Singh VK, Singh HB (1993) Talanta 40:1851–1859
Haraguchi H (1999) Bull Chem Soc Jpn 72:1163–1186
Zougagh M, Cano Pavon JM, Garcia de Torres A (2005) Anal Bioanal Chem 381:1103–1113
Goodall P, Lythgoe C (1999) Analyst 124:263–269
Carter HE, Warwick P, Cobb J, Longworth G (1999) Analyst 124:271–274
Perna L, Betti M, Moreno JMB, Fuoco R (2001) J Anal At Spectrom 16:26–31
Kato K, Ito M, Watanabe K (2000) Fresenius J Anal Chem 366:54–58
Prabhakaran D, Subramanian MS (2004) Anal Bioanal Chem 379:519–525
Metilda P, Sanghamitra K, Mary Gladis J, Naidu GRK, Prasada Rao T (2005) Talanta 65:192–200
Caykara T, Inam R, Ozyurek C (2001) J Polym Sci Polym Chem 39:277–283
Mary Gladis J, Prasada Rao T (2002) Anal Bioanal Chem 373:867–872
Jain VK, Handa A, Sait SS, Shrivastav P, Agrawal YK (2001) Anal Chim Acta 429:237–246
Gao Y, Oshita K, Lee KH, Oshima M, Motomizu S (2002) Analyst 127:1713–1719
Yamakawa S, Oshita K, Sabarudin A, Oshima M, Motomizu S (2004) Bunseki Kagaku 9:1039–1043
Nakata H, Kusaka Y, Kikkawa S (1962) Bull Chem Soc Jpn 1611–1614
Oshita K, Gao Y, Oshima M, Motomizu S (2001) Anal Sci 17(Suppl):a317–a320
Gil PE, Ostapczuk P, Emons H (1999) Anal Chim Acta 389:9–19
Acknowledgement
The present work was partially supported by the Grant for Environmental Research from the Yakumo Foundation for Environmental Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oshita, K., Seo, K., Sabarudin, A. et al. Synthesis of chitosan resin possessing a phenylarsonic acid moiety for collection/concentration of uranium and its determination by ICP-AES. Anal Bioanal Chem 390, 1927–1932 (2008). https://doi.org/10.1007/s00216-008-1931-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-1931-1