
Abstract
Quantitative structure–retention relationship (QSRR) models were constructed for the GC×GC–TOFMS retention time of 209 polychlorinated biphenyl (PCB) congeners. Principal component analysis (PCA) was used to recognize groups of samples with similar behavior and assist the separation of the data into training and test sets. The best multi-linear regression (BMLR) method was used for the systematic development of multi-linear regression equations; the best regression model involved four descriptors which were related to GC×GC–TOFMS chromatographic retention of PCBs. The obtained model has good predictive ability. For the test set, it gave a predictive correlation coefficient (R) of 0.988 and an average absolute relative deviation (AARD) of 3.08%. Results of a six-fold cross-validation procedure, which were in accordance with those from validation of training and test sets, demonstrated that this model was reliable. Additionally, this paper provides a simple, practical, and effective method for analytical chemists to predict the retention times of PCBs in GC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polychlorinated biphenyls (PCBs) are considered as persistent organic pollutants (POPs) because of their biological and chemical stability and their historical widespread use in the power-generation industry [1]. As is widely known, PCBs are toxic, lipophilic, and tend to be bioaccumulated. Toxicological effects of exposure to PCBs include hepatotoxicity, immunotoxicity, and reproductive problems, as well as respiratory, mutagenic, and carcinogenic effects [1].
The chromatographic separation of PCBs has been a great challenge for analytical chemists. Despite the broad range of gas chromatography (GC) stationary phases available, none can separate all 209 PCBs from each other. Various techniques have been used based on the combination of commercially available GC columns to improve the separation efficiency of PCBs, e.g., two-dimensional GC×GC [2], GC×GC–FID (flame ionization detector) [3], GC×GC–μECD (microelectron capture detector) [4], and GC×GC–TOFMS (time-of-flight mass spectrometry) combined with the HT-8/BPX-50 column set [5], which has successfully separated 192 congeners from 209 PCBs with the remaining 17 forming eight coelutions.
The study of the quantitative structure–retention relationship (QSRR) of solutes is an important topic in chromatographic thermodynamics. QSRR studies are widely investigated in gas chromatography (GC) [6], HPLC [7], and gas–liquid chromatography (GLC) [8], etc. By the construction of mathematic models correlating the gas chromatographic retention indices and molecular parameters, QSRR models can provide significant information on the effect of the molecular structure on retention time and on the possible mechanism of absorption and elution.
In the present investigation, the best multi-linear regression method (BMLR) was used to study of the retention time of 192 PCB congeners in Ref. [5] using descriptors calculated by the CODESSA software package [9]. The aim was to develop a reliable and accurate QSRR model to predict the retention time of PCB congeners, and to investigate the structural factors affecting the congeners’ retention behavior on the thermally stable stationary phase of HT-8/BPX-50.
Experimental
Descriptor generation
The GC×GC–TOFMS retention time was taken from Ref. [5]. The structures of the PCBs together with their retention times are listed in Table 1. The molecular structures were drawn and optimized with the semi-empirical AM1 method in HyperChem 6.0 [10]. All calculations were carried out at restricted Hartree–Fock level using the Polar–Ribiere algorithm until rms gradient was less than 0.01 kcal Å mol−1. The resulting geometries were transferred into the MOPAC 6.0 software package [11] to get files readable by CODESSA in order to calculate constitutional, topological, geometrical, electrostatic, and quantum chemical descriptors. These descriptors contain the information about the connections between atoms, shape, branching, symmetry, distribution of charge, and quantum-chemical properties of the molecule. Following the above process, the structures of all the PCBs studied in our work were represented using the various types of molecular descriptors calculated from molecular structures alone.
Best multi-linear regression
CODESSA includes two advanced approaches, i.e., the heuristic method (HM) and best multi-linear regression (BMLR) method, for systematical development of multi-linear QSAR/QSPR equations: preselection of descriptors by eliminating the descriptors that are not available for every structure or those having a small variation in magnitude or the descriptors having F-test or t-values less than the predetermined values. The two correlation approaches differ in their method of arriving at the best possible combination of descriptor contributions among the remaining descriptors. For HM all two-parameter regression models with remaining descriptors were subsequently developed and ranked by the regression correlation coefficient. A stepwise addition of further descriptor scales was performed to find the best multi-parameter regression models with the optimum values of statistical criteria (highest values of R 2, the cross-validated \( R^{2}_{{CV}} \), and the F-value). In the case of BMLR, it implements the following strategy to search for the multi-parameter regression with the maximum predicting ability. It commences by correlating the given property employing two-parameter regression with pairs of orthogonal descriptors (default value R 2 of the inter-correlation less than 0.1). The descriptors sets with highest correlation coefficients are chosen to perform higher-order regression. Further inclusion of non-collinear descriptors (default value R 2 < 0.6) in the regression is made, one descriptor after another, on the basis of the improved Fisher criterion F at a given probability level upon successive addition of descriptors [12].
Model performance evaluation
Model performance can be measured by different metrics. The squared coefficient of correlation (R 2), which can be interpreted mathematically as the proportionate reduction of total variation associated with the independent variable, and the average absolute relative deviations (AARD, %), which gives the bias in the prediction, were used to evaluate the correlation results. The closer the R 2 value is to unity, the greater the degree of association of the models is. On the other hand, the lower the average absolute relative deviations (AARD, %), the better the correlation precision is. AARD can be calculated as below
where k represents the k-th molecule, y ke is the desired output (experimental property), y kp is the actual output of the models, and n is the number of compounds in the analyzed set. AARD and R are used to give information about the predictive ability for the test set and indicate how well the model fits the training set.
In this investigation, model performance was validated by two methods: the results of the test set and the results of six-fold cross-validation procedure.
Results and discussion
Principal component analysis (PCA) of the data set
A principal component analysis (PCA) has been performed using the calculated structure descriptors for the whole data with two aims in mind: first, to detect the homogeneities in the data set, i.e., to identify possible outliers and clusters and, second, to show spatial location of samples to assist the separation of the data into training and test sets. Principal component analysis (PCA) is an unsupervised multivariate statistical technique in which new variables (called principal components, PCs) are calculated as linear combinations of the old ones. The PCs obtained (usually the first two or three components) are used to derive scores which can be used to display most of the original variations in a smaller number of dimensions. These scores can also allow us to recognize groups of samples with similar behavior. The design of the training set is performed by selecting a subset of representative compounds that are most efficient in describing the distribution of the compounds in the principal components space. When the number of principal components is less than three, it is possible to select representative samples manually from visual inspection of the score plots. This method was used in the present study to select the training set compounds.
A total of 367 descriptors were generated by the CODESSA program. Among these descriptors, 131 were deleted because of zero variance. PCA analysis on the remaining 236 descriptors gives three significant PCs (eigenvalues > 1), which explains 66.29% of the variation in the data, 42.51%, 13.22%, and 10.56%, respectively). The score plots are shown in Fig. 1. As shown in Fig. 1a (plot of PC1 and PC2), nine clusters of PCB congeners can be observed clearly, corresponding to the different degrees of chlorination, from mono (left) to deca (right). Only compound 207 (i.e., 2,2′,3,3′,4,4′,5,6,6′-chlorination) falls into the space of octachlorination groups. While in the case of Fig. 1b, the score plot of the PC1 and PC3, all the compounds clustered very well according to their degree of chlorination.

a, b Principal component analysis of the structural descriptors for the data set
In this investigation, the 192 well-separated congeners were further studied in our QSRR model. The data sets were split into 80% for the training set (155 compounds) and 20% for the test set (37 compounds). This separation ratio of training and test set compounds is often used in QSAR studies [13]. The training set was used to develop the model and the test set was used to evaluate the model performance. The training set was guaranteed to cover the entire range of the retention times and, at the same time, contain the most dissimilar structures as much as possible. As shown in Fig. 1, the data set was split into two representative sets in which the training set consists of representatives of the more dissimilar structures, thus confirming the applicability of data set splitting.
Results of best multi-linear regression
When adding another descriptor did not significantly improve the statistics of a model, it was determined that the optimum subset size had been achieved. The best multi-linear regression performed for the 155 congeners in the training set afforded seven models containing two to eight parameters. To avoid the “over-parametrization” of the model, an increase of the R 2 value of less than 0.02 was chosen as the breakpoint criterion [14]. Comparing the respective statistical characteristics of these models, it can be found that four descriptors, i.e., the third-order Kier & Hall index (\( ^{3} \chi ^{\nu } \)), atomic charge weighted PPSA [Quantum-Chemical PC] (3PPSA), max valency of a C atom (\( V^{{\max }}_{C} \)) and principal moment of inertia C/# of atoms (#IC), appear to be sufficient for a successful regression model (Fig. 2). The leave-one-out cross-validated procedure gives the squared correlation coefficient \( R^{2}_{{CV}} \) as 0.9815. This indicates that the obtained model is stable and has good prediction ability. Multicolinearity between the above four descriptors was detected by calculating their variation inflation factors (VIF), which can be calculated as
where r is the correlation coefficient of multiple regression between one variable and the others in the model. If VIF equals unity, no intercorrelation exists for each variable; if VIF falls into the range 1.0–5.0, the related model is acceptable; and if VIF is larger than 10.0, the related model is unstable [15]. The r 2 values of these descriptors are 0.762, 0.626, 0.314, and 0.792 for \( ^{3} \chi ^{\nu } \), 3PPSA, \( V^{{\max }}_{C} \) and #IC, respectively. Accordingly, their respective VIF values are 4.202, 2.674, 1.458, and 4.808 (Table 2). All VIF values are less than 5, indicating that the obtained model has obvious statistic significance. The multi-linear regression (MLR) model based on these four descriptors is also presented in detail in Table 2. From the positive and negative symbols of the respective coefficient in Table 2, one can evaluate the effects of each descriptor on the GC×GC–TOFMS retention times of PCB congeners (also demonstrated in Fig. 3).

Correlation coefficients (R 2, \( R^{2}_{{CV}} \)) versus number of descriptors

a–d Relationship between the selected descriptors and experimental retention times (t R)
The first descriptor is a topological one, the third-order Kier & Hall index (\( ^{3} \chi ^{\nu } \)). This descriptor belongs to the well-known molecular connectivity indices, which are the contribution of one molecule to the bimolecular interactions arising from encounters of bonds among two molecules [16]. \( ^{3} \chi ^{\nu } \) can be calculated as follows [17]
where N SB is the number of skeleton bonds and δ i corresponds to the values of the atomic connectivity for the i-th atom in the molecular skeleton, which is calculated by the formula
where Z i is the total number of electrons in the i-th atom, \( Z^{\nu }_{i} \) is the number of valence electrons, and H i is the number of hydrogens directly attached to the i-th atom. The introduction of this descriptor in the model is not surprising as, on the one hand, it can describe molecular structure information such as the degree of branching, the position and influence of heteroatoms, and the degree of cyclicity [17, 18], and on the other hand, the interaction between the HT-8 column and PCB π-electrons is related to the degree of ortho-substitution and to the planarity of the molecules [19]. The high correlation coefficient of 0.942 indicates that there is a positive strong linear relationship between \( ^{3} \chi ^{\nu } \) and the chromatographic retention times of PCBs; shown in Fig. 3a. Increasing \( ^{3} \chi ^{\nu } \) values of the PCBs leads to high affinity of the HT-8 phase towards PCBs.
There are also three quantum-chemical descriptors included in the correlation model. These were calculated from the output results of the MOPAC program. 3PPSA means atomic charge weighted partial positive surface area. This descriptor depends directly on the quantum-chemically calculated charge distribution in the molecule, and therefore encodes features responsible for polar interactions between molecules and the hydrogen-bond interactions as well. Max valency of a C atom (\( V^{{\max }}_{C} \)) relates to the strength of intramolecular bonding interactions and characterizes the stability of the molecule, their conformational flexibility, and other valency-related properties [20]. The moments of inertia characterize the mass distribution in the molecule. Principal moment of inertia C/# of atoms (#IC) is useful for distinguishing between the isomers [21].
From the above discussion, it can be seen that the above four descriptors can account for the factors influencing the chromatographic retention behavior of PCB congeners, i.e., the molecular size, polar interaction, and hydrogen bonding, etc. As indicated by the t-test values in Table 2, \( ^{3} \chi ^{\nu } \) is the most relevant descriptor related to the retention times. In this sense, the chromatographic retention behavior of PCB congeners is preferentially determined by the molecular shape (molecular size and branching) and, less importantly, by the polar interaction between the PCBs and the stationary phase.
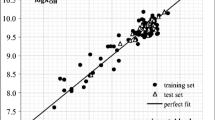
Based on the above regression model, the GC×GC–TOFMS retention times of the 192 congeners are calculated and listed in the Table 1. For the test set, this model gave the high predictive squared correlation coefficient as 0.975 and small AARD as 3.08%, respectively, thus confirming the good predictive ability of the model. Figure 4 is the plot of calculated versus experimental retention times for the 192 congeners by the MLR model. The small scatter of the individual points about the regression line of Fig. 4 and the overall linear correlation coefficients of 0.988 prove that this model is statistically stable and can fit the data well. Using this model, most molecules have relative errors less than 10% except for congeners 1, 8, 19, and 30 in the training set that show large relative errors of −12.08%, 11.05%, −10.44%, and 11.14%, respectively.

Predicted vs. experimental t R by MLR
Six-fold cross-validation
In order to consider the differences between groups in the training set, a six-fold cross-validation procedure based on the whole 192 congeners was performed [22, 23]. The data set was randomly divided into six subsets of equal size, each containing 32 congeners. In addition, it was verified that each set contained exactly the same percentage of same homologue groups. Five of them were used to construct the model, the sixth to evaluate the prediction ability. This procedure was repeated six times, each time with one of the six subsets treated as a test set. During the six-fold cross-validation procedure each of the data sets was used once for the prediction and five times for the construction of the model. Average predictive accuracy obtained with the cross-validation procedure was used for the evaluation of prediction capabilities of individual models. The average predictive AARD of 3.21% for the models (shown in Table 3), which was almost exactly equal to the results obtained based on one training set/test set (AARD = 3.08%), demonstrated that the MLR model we proposed is reliable.
Analysis of the residuals between the predicted versus the experimental retention time for the training and test sets indicates that no evident systematic error exists in the construction of the MLR model. Using this model, the GC×GC–TOFMS retention times for 17 coeluting congeners were also predicted and these are listed in the Table 1 and shown in Fig. 4. As can be seen, the results are very good with all coeluting congeners differentiated from each other.
Conclusion
In this investigation, the CODESSA program was used to calculate constitutional, topological, geometrical, electrostatic, and quantum-chemical descriptors from molecular structures of polychlorinated biphenyls (PCBs). Principal component analysis was used to ensure the reasonable separation of the data set into training and test sets. Four descriptors selected by the best multi-linear regression (BMLR) method can describe the structural features influencing the GC×GC–TOFMS retention of PCBs. The results from a six-fold cross-validation procedure proved that this model was reliable and were used to predict the retention indices for 17 coeluting PCBs.
References
Giesy JP, Kannan K (1998) Crit Rev Toxicol 28:511–569
Liu Z, Philips JB (1991) J Chromatogr Sci 29:227–231
Philips JB, Xu J (1995) J Chromatogr A 703:327–334
Haglund P, Harju M, Ong R, Marriott P (2001) J Microcol Sep 13:306–311
Focant JF, Sjödin A, Patterson DG (2004) J Chromatogr A 1040:227–238
Yao XJ, Zhang XY, Zhang RS, Liu MC, Hu ZD, Fan BT (2002) Talanta 57:297–306
Al-Haj MA, Kaliszan R, Nasal A (1999) Anal Chem 71:2976–2985
Ivanciuc O, Ivanciuc T, Cabrol-Bass D, Balaban AT (2000) J Chem Inf Comput Sci 40:732–743
Katritzky AR, Lobanov VS, Karelson M (1995–1997) CODESSA version 2.0 reference manual
HyperChem (2000) Release 6.0 for Windows. Hypercube, Inc
Stewart JPP (1989) MOPAC 6.0, quantum chemistry program exchange (QCPE). Indiana University, Bloomington, IN, Program 455
Katritzky AR, Perumal S, Petrukhin R, Kleinpeter E (2001) J Chem Inf Comput Sci 41:569–574
Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A (2003) J Comput Aided Mol Des 17:241–253
Katritzky AR, Kuanar M, Fara DC, Karelson M, Acree WE Jr (2004) J Bioorgan Med Chem 12:4735–4748
Famini GR, Penski CA, Wilson LY (1992) J Phy Org Chem 5:395–408
Kier LB, Hall LH (2000) J Chem Inf Comput Sci 40:792–795
Kier LB, Hall LH (1976) Molecular connectivity in chemistry and drug research. Academic Press, New York
Kier LB, Hall LH (1986) Molecular connectivity in structure-activity analysis. Research Studies, New York
Bolgar M, Cunningham J, Cooper R, Kozloski R, Hubball J, Miller DP, Crone T, Kimball H, Janooby A, Miller B, Fairless B (1995) Chemosphere 31:2687–2705
Sannigrahi AB (1992) Adv Quant Chem 23:301–351
Jalali-Heravi M, Garkani-Nejad Z (2002) J Chromatogr A 971:207–215
Geman S, Bienenstock E, Doursat R (1992) Neural Comput 4:1–58
Efron B, Tibshirani RJ (1993) An introduction to the Bootstrap. Chapman and Hall, New York, pp 239–241
Acknowledgement
The authors wish to thank Dr. Huanling Wang for informative discussions about the descriptors. Thanks also to the editors and unknown referees for reviewing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ren, Y., Liu, H., Yao, X. et al. An accurate QSRR model for the prediction of the GC×GC–TOFMS retention time of polychlorinated biphenyl (PCB) congeners. Anal Bioanal Chem 388, 165–172 (2007). https://doi.org/10.1007/s00216-007-1188-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-007-1188-0