
Abstract
Liquid chromatography (LC) hyphenated with both elemental and molecular mass spectrometry has been used for Se speciation in Se-enriched garlic. Different species were separated by ion-pair liquid chromatography–inductively coupled plasma mass spectrometry (LC–ICP–MS) after hot-water extraction. They were identified by on-line reversed-phase liquid chromatography–electrospray ionization tandem mass spectrometry (RPLC–ESI–MS–MS). Se-methionine and Se-methylselenocysteine were determined by monitoring their product ions. Another compound, γ-glutamyl-Se-methylselenocysteine, shown to be the most abundant form of Se in the garlic, was determined without any additional sample pre-treatment after extraction and without the need for a synthesized standard. Product ions for this dipeptide were detected by LC–ESI–MS–MS for three isotopes of Se—78 Se, 80Se: and 82Se. The method was extended to the species extracted during in-vitro gastrointestinal digestion. Because both Se-methylselenocysteine and γ-glutamyl-Se-methylselenocysteine have anticarcinogenic properties, their extractability and stability during human digestion are very important. Garlic was also treated with saliva, to enable detection and analysis of species extracted during mastication. Detailed information on the extractability of selenium species by both simulated gastric and intestinal fluid are given, and variation of the distribution of Se among the different species with time is discussed. Although the main species in garlic is the dipeptide γ-glutamyl-Se-methylselenocysteine, Se-methylselenocysteine is the main compound present in the extracts after treatment with gastrointestinal fluids. Two more, so far unknown compounds were observed in the chromatogram. The extracted species and their transformations were analysed by combining LC–ICP–MS and LC–ESI–MS–MS. In both the simulated gastric and intestinal digests, Se-methionine, Se-methylselenocysteine, and γ-glutamyl-Se-methylselenocysteine could be determined by LC–ESI–MS–MS by measuring their typical product ions.

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Garlic (Allium sativum) has acquired a reputation in Asiatic and Western cultures as a prophylactic and therapeutic medical agent [1]. Nowadays, it is renown for its anticarcinogenic properties and there is evidence that this is because of the presence of Se compounds. The idea of Se species being responsible for these anticarcinogenic effects finds its origin in the fact that garlic is a source rich of sulfur. When garlic is grown on selenized soil, selenium tends to replace sulfur. The allyl sulfides present in garlic were known to have anticarcinogenic effects and by substituting the sulfur in these compounds with selenium it was correctly believed that even more powerful anticarcinogenic agents would be produced [2]. A study by Ip et al. revealed that the anticarcinogenic power of garlic is really because of the Se compounds it contains [3–7]. Similar, although not equal, anticarcinogenic effects were observed when Se-MeSeCys standard solution and a water extract of the garlic were used [8]. Later it was stated that the anticarcinogenic effects of Se-MeSeCys were because it was metabolized to methylselenenic acid by the action of β-lyase. In vivo and in vitro studies provided strong evidence for the hypothesis that monomethylated selenium plays a key role in the prevention of cancer [6]. Apart from Se-MeSeCys, γ-glutamyl-Se-methylselenocysteine played a key role. This dipeptide serves primarily as a carrier of Se-MeSeCys. Research revealed that γ-glutamyl-Se-MeSeCys is an effective anticarcinogenic agent and that its action is very much like that of Se-MeSeCys. Both of the compounds are well absorbed and the major route of excretion is via the urinary tract [9].
The state of the art of analytical methodology used for identification of the Se species in garlic extracts is briefly summarized below. Because garlic contains many volatile species, gas chromatography (GC) in combination with atomic emission detection (AED) and mass spectrometry (MS) have been extensively applied in several studies. Eight compounds were detected by GC–AED and identified by GC–MS [10]. The same approach was used for speciation of Se in human breath after the consumption of Se-enriched garlic [11]. Another research group separated and detected dimethyl selenide and dimethyl diselenide by using solid-phase micro extraction (SPME) in combination with inductively coupled plasma mass spectrometry (ICP–MS) [12]. Other compounds were detected by this group by use of SPME combined with moderate-temperature multicapillary GC and microwave-induced plasma-emission spectrometry [13]. Headspace GC–AED has been used for the determination of Se-containing amino acids in highly enriched garlic (1355 μg g−1 dry weight). Se-MeSeCys was shown to be the major Se-containing amino acid present, with minor amounts of Se-Cys and Se-Met [14].
Most analyses of Se species in garlic have been performed by combining liquid chromatography (LC) with elemental and molecular mass spectrometric detection. SeMeSeCys and γ-glu-SeMeSeCys were identified on the basis of retention time matching with commercially available and synthesized Se standards [15]. The presence of a compound of mass 326 u was attributed to γ-glutamyl-Se-methylselenomethionine without use of a standard and without any further molecular information [16]. Another method based on ion-exchange liquid chromatography combined with elemental mass spectrometry revealed the presence of Se-Cys, Se-MeSeCys, Se-Met and two inorganic Se species, selenite and selenate. Two other, so far unknown, compounds were also observed [17].
However, as stressed recently by Francesconi and Sperling, use of liquid chromatography in combination with element detection and retention-time matching with standards does not provide sufficient evidence for unequivocal characterization of a Se species [18]. By hyphenation of an ion-pair liquid chromatographic method with ICP–MS, Ip et al. [19] were able to detect selenate, Se-cystine (Se-(Cys)2), selenocystathionine, Se-MeSeCys, Se-Met, γ-glutamyl Se-methylselenocysteine (γ-glu-SeMeSeCys) and γ-glutamyl-Se-methionine in a garlic extract (total Se-content 296 μg g−1dry weight) . By measuring at the m/z of the corresponding species, Se-Met and γ-glu-SeMeSeCys were detected in a garlic extract by LC–ESI–MS; for the other compounds molecular information was lacking [19]. Using the same approach, Se-Met Se-MeSeCys and γ-glu-Se-MeSeCys were detected in a highly enriched garlic extract (containing 1355 μg Se g−1 dry weight) [20]. The species readily extracted from garlic depend on the amount of Se present in the garlic. Garlic samples with Se concentrations less than 333 μg g−1 contain more γ-glutamyl-Se-methylselenocysteine than Se-methylselenocysteine. In garlic containing more than 333 μg g−1 the opposite is true [21].
Several research groups have used extensive sample-preparation procedures to preconcentrate compounds before ESI–MS(–MS) analysis for the identification of the species. McSheehy et al. [22] developed a very elaborate method based on two-dimensional LC with parallel ICP–MS and off-line ESI–MS–MS detection for speciation of Se in garlic. γ-Glu-Se-MeSeCys was identified on the basis of the isotopic pattern of Se and the product ions formed [22]. Ogra et al. also determined γ-glu-Se-MeSeCys by use of LC–ICP–MS equipped with an octapole reaction system and off-line ESI–MS–MS detection [23]. The procedures of both were, however, very cumbersome and included several clean-up steps.
In this paper we describe a method based on the hyphenation of LC with both elemental and molecular mass spectrometry for characterization of Se species in a hot-water extract of garlic, in an extract of a mixture of saliva and garlic, and in in vitro gastrointestinal digests of garlic. To the best of our knowledge this is the first time Se species in a moderately enriched garlic sample have been detected on-line without the need for additional sample clean-up before analysis by LC–ESI–MS–MS. Se-Met and Se-MeSeCys were identified in the extracts by measuring their characteristic product ions. Another Se compound, γ-glutamyl-Se-methylselenocysteine was identified without the need for a synthesized standard. This compound was characterized by monitoring product ions typical of this dipeptide and by using the characteristic isotope pattern of Se.
Experimental
Chemicals and materials
All chemicals used were of analytical grade. Ultrapure Milli-Q water was obtained from a Millipore (Bedford, MA, USA) system. Se-cystamine (Se-Cya) Se-methionine (Se-Met), Se-methylselenocysteine (Se-MeSeCys), tetraethylammonium chloride (TEACl), pepsin from porcine gastric mucosa, pancreatin from porcine pancreas, and protease XIV from Streptomyces griseus were from Sigma (Bornem, Belgium). Methanol p.a., KH2PO4, and formic acid were purchased from Vel (Leuven, Belgium). NaOH was from Carlo Erba (Milan, Italy). Nitric acid (14 mol L−1) was purified by sub-boiling distillation in quartz equipment. The 5 mL polypropylene test tubes used in the simulation of digestion were from Falcon, Becton Dickinson labware (Meylan, France). The 0.22 μm pore PVDF syringe filters used to filter the samples before analyses were from Millipore.
Instrumentation
Complete mineralization was conducted in Teflon vessels in a Milestone mls 1200 mega microwave digester from Analis (Namur, Belgium). The ICP–MS instrument used was a quadrupole-based Perkin Elmer (Glendale, Ontario, Canada) Sciex Elan 5000. The instrument was equipped with a sample-introduction system consisting of a pneumatic nebulizer and a spray chamber; the sample uptake rate was 0.8 mL min−1. The LC system used for hyphenation with ICP–MS was an Äkta Purifier 10 system (Amersham Biosciences, Roosendaal, The Netherlands). The six-way valve was supplied with a 20 μL sample loop. The outlet of the column (XTerra MS C18 (length 25 cm, i.d. 4.6 mm, 5 μm particles) and XTerra MS C18 guard column (length 20 mm, i.d. 3.9 mm, 5 μm particles), both from Waters, Milford, MA, USA) was connected by PEEK tubing (i.d. 0.1 mm) to the nebulizer of the ICP–MS.
LC–ESI–MS–MS experiments were performed with a Quattro Micro Z-spray source system (Micromass, Manchester, UK) to which a Waters Alliance model 2690 LC equipped with an autosampler was hyphenated. A narrow-bore XTerra column (length 25 cm, i.d. 2.1 mm, 5 μm particles), with an XTerra MS C18 guard column (length 20 mm, i.d. 2.1 mm, 5 μm particles), both from Waters, was used for separation of the species.
Sample description
The garlic (Allium sativum) sample was purchased from Phytoselenium Research Laboratories (Kumamoto, Japan). Barium selenite and barium selenate (500 mg m−2 of each) were added once to the soil on which the garlic was grown. The garlic was planted in the autumn of 2001 and was harvested in the summer of 2002 [24]. The garlic samples were kept at −20°C until further treatment/analysis. For speciation analysis the garlic cloves were lyophilized and ground to powder in a coffee mill. The powder generated from seven different cloves was thoroughly mixed to homogenize the sample.
Determination of the total Se concentration in garlic
The total Se content was determined in both the defrosted garlic and in the lyophilized sample. To determine the total Se content of fresh garlic the cloves were chopped into small pieces, a 0.5 g sample was placed in a Teflon vessel, and 5 mL 14 mol L−1 HNO3 and 1 mL H2O2 were added. Ga (final concentration 50 μg L−1) was used as internal standard for ICP–MS detection. The microwave program has been described elsewhere [25]. The digests obtained were diluted with Milli-Q water and analysed by ICP–MS by the standard addition method by spiking with selenate (final concentration 10 μg L−1). The same procedure was followed for a lyophilized sample; sample size was 0.1 g (final Ga concentration 20 μg L−1, final selenate concentration 10 μg L−1). All digestions were performed five times.
Extraction
Milli-Q water (20 mL) was added to approximately 2.5 g freeze-dried selenized garlic. The mixture was placed in a hot-water bath at 50°C and shaken at 140 rpm for 8 h. The mixture was then centrifuged and the supernatant (extract) filtered through a syringe filter. The filtrate was kept at −20°C until analysis.
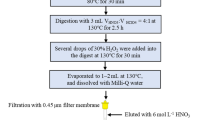
In vitro gastrointestinal digestions
Fresh human saliva (2 mL) was added to 0.2 g freeze-dried garlic. One sample was immediately centrifuged and an other was shaken in a hot-water bath (37°C, 140 rpm) for 15 min before centrifugation. Simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) were prepared according to the US Pharmacopeia, as described elsewhere [25–28] and 2 mL SGF or SIF was added to 0.2 g garlic in a 5 mL propylene test tube. The tubes were shaken in a hot-water bath (37°C, 140 rpm) for 0 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 16 h, or 24 h. Immediately after removal from the bath mixtures were centrifuged and the supernatant was kept in a freezer (−20°C) until analysis. These experiments were performed three times.
Total Se concentration in garlic extracts
The total Se concentration in the hot-water extract, saliva, SGF, and SIF samples was determined by ICP–MS, by standard addition. All samples were diluted tenfold in 0.14 mol L−1 HNO3. Ga was used as internal standard (final concentration 50 μg L−1) and selenate was used for calibration purposes (concentration added 50 μg L−1).
LC–ICP–MS of Se compounds in garlic extracts
For all the extracts, 20 μL was injected on to the XTerra column. The species were monitored at a mass-to-charge ratio of 82 (82Se+) for measurement under interference-free conditions. The ICP–MS settings have been described elsewhere [26]. The mobile phase was 0.01% TEACl–, 2% MeOH and the pH was adjusted to 4.5 with formic acid [5, 25, 26]. These conditions were used because ion-pairing is essential for separation and detection of the inorganic species that might be present in the extracts.
LODs of Se standard solutions with LC–ESI–MS–MS
Because multiple reaction monitoring (MRM) is the most sensitive mode of measurement with ESI–MS–MS, the limits of detection of some commonly detected organic Se species were determined for the separation method applied here, measuring in MRM mode. Optimum ESI–MS–MS conditions depend on the nature of the species and have been described elsewhere [5]. Se-Met, Se-MeSeCys, Se-(Cys)2 and Se-Cya standards were prepared at two concentrations, 1 and 10 mg L−1, in Milli-Q water, and to improve ionisation, formic acid was added to the solution (final concentration 1%). The solutions (10 μL) were then injected on to the XTerra column. Two different mobile phases were used: 0.01% TEACl+2% MeOH and 2% MeOH (the pH of both was adjusted to 4.5 with formic acid). The latter mobile phase was of interest because TEACl hampers the ESI process and, hence, results in a loss of sensitivity. LODs were also measured for garlic samples; commercially available garlic powder (bought at a local supermarket) was used as a blank. Determination of total Se showed that the Se content of this product was below the detection limit of ICP–MS (0.03 μg L−1, which means the Se concentration in this garlic is <0.6 ng g−1 wet weight). Se-Met, Se-Cya, Se-MeSeCys and Se-(Cys)2 were added to separate hot-water extracts of garlic powder at a concentration of 25 mg L−1, each with addition of formic acid (1%). The species were eluted with 2% MeOH at pH 4.5. The same procedure was followed for the Se standards in SGF and in SIF.
LC–ESI–MS–MS of the Se compounds in garlic extracts
The species were separated on the narrow-bore XTerra MS C18 column. The flow was adjusted to 0.16 mL min−1 and the mobile phase was 2% MeOH at pH 4.5. The compounds were detected on the basis of their typical product ions. ESI–MS–MS conditions for Se-Met and Se-MeSeCys were as described elsewhere for standard solutions [5]. Formic acid was added to all samples (final concentration 1%). All samples were filtered through a syringe filter and 5 μL aliquots were injected on to the column.
Results and discussion
Limits of detection for LC–ESI–MS–MS measurements
LC–ICP–MS is known to be much more sensitive for detection of elemental Se than LC–ESI–MS–MS is for detection of molecular species of Se. LC–ESI–MS–MS, however, provides us with molecular information about the compounds eluting from the column. Previous research on Se species in garlic (and other food sources) overcame this problem of too low sensitivity by preconcentrating the sample before ESI–MS-(MS) analysis, at the risk of species transformation and loss of species [22]. Our objective, of course, was identification of the species as originally present in the sample. An attempt was therefore made to measure the species eluting directly from the column by multiple reaction monitoring (MRM). Limits of detection with LC–ESI–MS–MS were determined by measuring Se-Met via the five transitions MRM 198–181, MRM 198–152, MRM 198–135, MRM 198–109, and MRM 198–102. Se-MeSeCys was measured on its four product ions resulting from the transitions MRM 184–167, MRM 184–149, MRM 184–123, and MRM 184–95. Se-(Cys)2 and Se-Cystamine give rise to one product ion only and hence were measured via MRM 337–248 and 249–204, respectively. The LODs for measurement by ion pairing LC and reversed-phase LC are given in Table 1. From this table it is clear that reversed-phase conditions are favourable and that the use of TEACl should be avoided if possible, especially when the concentration of the compounds is low. Depending on the compound and on the transition measured, LODs can be a factor 3–50 higher when using TEACl. It is also obvious from this table that LODs are very much compound and transition-dependent. The LODs for Se-Met for instance may differ by a factor of 10 (MRM 198–135 compared with MRM 198–102). This table shows that of these four compounds Se-(Cys)2 is the biggest challenge. The fact that this was also one of the minor compounds in the LC–ICP–MS chromatogram, made it impossible to detect this compound by LC–ESI–MS–MS in any of the extracts at this low level.
Another aspect of speciation analysis which causes severe deterioration of LODs is the effect of the sample matrix. Therefore, LODs of the four compounds were determined in the presence of a garlic extract (that referred to above as the blank matrix) with a Se concentration below the detection limit of the ICP–MS and consequently of ESI–MS(–MS) also, and in the presence of SGF and SIF. These experiments were performed under reversed-phase conditions only, to present a clear and fair picture of the matrix effect. The results are shown in Table 2. Two transitions for Se-Met are not mentioned, MRM 198–181 and MRM 198–152. When measuring these MRMs in a blank sample (containing only garlic extract and formic acid) these transitions could be measured, although their intensity was negligible compared with the intensity measured for a garlic sample spiked with Se-Met. The signals measured are from biomolecules giving rise to the same product ions but lacking Se. The blank garlic powder, on the other hand, is only a substitute for the real sample. The garlic used for speciation analysis is grown under different conditions and its matrix might be slightly different from that of the blank and hence may lack the above mentioned interfering compounds. The effect of the garlic extract on the detectability of the species leads to a LOD which is 3–10 times worse for Se-Met, depending on the transition measured. For Se-(Cys)2 the LOD differs by a factor of two and for Se-Cya, no harmful effect is observed. For Se-MeSeCys, the effect is highly dependent on the transition. When measuring the MRM 184–167 the LOD is a factor of ten better in garlic; for the other transitions, in contrast, the LOD is worse in garlic. In Table 2 the effect of SGF and SIF is also given. For Se-Met the effect is highly dependent on the transition measured and the LOD can be up to a factor of ten higher in SGF and SIF. The Se-(Cys)2 and Se-Cya signals are not quenched when measured in SGF or SIF solutions. Depending on the transition, the LODs for Se-MeSeCys can be up to a factor of five higher. Measurements of Se species in any type of garlic preparation seem to be susceptible to the SGF or SIF matrix and the garlic matrix.
Total Se concentration in garlic and its extracts
The total concentration of Se in the fresh Se-garlic was 28.33 μg Se g−1 wet weight ±21% (n=5). The total Se content of the lyophilized selenized garlic was 95.95 μg Se g−1 ±17% (n=5). The large biases might be because of the inhomogeneity of the product (a garlic clove consists of several parts and the composition of the middle part, from where a new shoot starts, is substantially different from that of the outer parts). The spread in Se over a single clove and between cloves has never been documented. Substantial inhomogeneity in Se distribution is evident, notwithstanding the thorough mixing of the material before analysis. The Se concentration in the hot-water extract of the lyophilized garlic was 102 μg Se g−1±27% (n=3). The concentration of Se in lyophilized garlic after saliva treatment was 63 μg Se g−1±10% (n=3). The results for extractable Se when treated in vitro with simulated gastrointestinal fluids, SGF and SIF, are given in Fig. 1 for 0 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 16 h, and 24 h of treatment. There are large fluctuations (mean values between 80 and 125 μg L−1) between the amounts of Se extracted, but the values fall within the experimental uncertainty of the individual data. Large scatter was to be expected, because the inhomogeneous distribution of the Se was already evident from the total concentration of Se in the garlic (RSD 17%). No trend in the extraction efficiency can be observed. Most of the Se species are extracted very quickly on contact with both SGF and SIF.

Amounts of Se extracted from freeze-dried garlic after treatment with simulated gastric and intestinal fluid
LC–ICP–MS of the Se compounds in a garlic hot-water extract
On monitoring of the Se species in the extract several compounds could be distinguished, of which some co-eluted with Se standards. The resulting chromatogram is shown in Fig. 2 (upper chromatogram). A compound eluting close to the dead volume co-eluted with Se-(Cys)2, a compound eluting at t R=300 s co-eluted with Se-MeSeCys, a third compound eluted at t R=420 s, the retention time of Se-Met. A major compound eluted later in the chromatogram and its behaviour under our chromatographic conditions was not consistent. Severe peak tailing was observed and the peak tended to shift from sample to sample; peak splitting was another concern. This led to the supposition that either several compounds were extracted or only one compound was present but in two different forms. As there was evidence this compound was the dipeptide γ-glutamyl-Se-methylselenocysteine (γ-glu-Se-MeSeCys) [22], its behaviour might be because of charge differences. The dipeptide was present in two different forms because of its pI and the pH value of the extract. By using TEACl as a positively charged ion-pairing reagent, one form was affected by the ion pairing agent and the other one was not. This behaviour has previously been observed by Kotrebai et al. for a γ-glu-Se-MeSeCys standard solution [15]. This problem was now solved by adding formic acid to the sample solutions (final concentration 1%); a reagent commonly added in the reversed-phase separation of peptides. Under these conditions the compound eluted at t R=620 s and its chromatographic profile was acceptable. The last eluting compound was the major compound in the extract and contributed for 49.7% of the total Se in the hot-water extract. Se-MeSeCys, Se-Met, and Se-(Cys)2 contributed for 28.8%, 15.5% and 6.0%, respectively.

LC–ICP–MS chromatogram obtained from a hot-water extract of garlic
LC–ESI–MS–MS analysis of the Se compounds in a hot-water extract of garlic
Detection of γ-glu-SeMeSeCys
γ-Glu-SeMeSeCys is a dipeptide in which the glutamyl moiety is linked to Se-MeSeCys via its carboxylic acid group in the γ-position. Because no standard was available for γ-glu-SeMeSeCys, ESI–MS–MS conditions cannot be optimized for analysis of this specific compound. Because of the Se-MeSeCys moiety in the dipeptide, conditions shown to be optimum for this compound [5] were also used for measurement of the dipeptide. These conditions were: capillary voltage 3.2 kV; cone voltage 18 V; collision energy 20 eV; cone gas flow rate 50 L h−1; desolvation gas flow rate 650 L h−1, photomultiplier voltage 650 V; source temperature 120°C; and desolvation temperature 350°C. Originally, the mobile phase consisted of 2% MeOH at pH 4.5. This was changed into 5% MeOH at pH 4.5 without any change on the order of elution of the compounds but substantial reduction in peak tailing. γ-Glu-SeMeSeCys (m/z 313) is characterized by some typical fragments produced after collision-induced dissociation; some of these are common to Se-MeSeCys also. Product ions are expected after loss of NH3, CO, H2O, the glutamyl moiety, or the Se-MeSeCys moiety. These were also the fragments observed by McSheehy et al. during analysis of a garlic sample after extensive sample clean-up [22]. The transitions to be measured were MRM 313–296, loss of NH3; MRM 313–250, loss of NH3 and COOH, MRM 313–224, loss of COOHCH(NH2)CH2; MRM 313–184, loss of the glutamyl moiety; MRM 313–167, loss of glutamyl and NH3; MRM 313–130, loss of the Se-MeSeCys moiety; and MRM 313–84, loss of CO and H2O from the glutamyl moiety. The transitions starting from m/z 313 for the compounds containing 80Se (theoretical isotopic abundance 49.61%) are shown in Fig. 3a. All transitions can be measured. To be completely sure of the identity of the compound, the isotopic pattern of Se can assist in the identification, because Se is part of the molecule. Se has six naturally occurring isotopes: 74Se, 76Se, 77Se, 78Se, 80Se, and 82Se. The product ions typical of a Se compound can thus be measured on different isotopes. The same transitions were therefore measured for fragmentation of the molecular ion with m/z 311 (78Se isotopic abundance 23.77%) by collision-induced dissociation. The transitions to be measured were: MRM 311–294, MRM 311–248, MRM 311–222, MRM 311–182, MRM 311–165, MRM 311–130, and MRM 311–84. The last two MRMs have the same product ions as for m/z 313, because these product ions do not contain Se. The results from these measurements are shown in Fig. 3b; MRM 311–294 and MRM 311–84 could not be detected in the sample. Both transitions were already of low intensity when focussing on 80Se and the abundance of the ions is now only half that, as a result of the lower isotopic abundance of 78Se. The transitions for measurement of the product ions starting from the molecular ion with m/z 315 (82Se) are shown in Fig. 3c: MRM 315–169, MRM 315–130, MRM 315–186, and MRM 315–226 could be measured. It was impossible to measure the other transitions because of lack of sensitivity. The presence of all these product ions for three different isotopes of Se leads to the conclusion that the last eluting compound in the LC–ICP–MS chromatogram is γ-glu-SeMeSeCys. This dipeptide was determined here without any need for sample pre-treatment or preconcentration and without the use of a Se standard.

LC–ESI–MS–MS chromatograms obtained from a hot-water extract of garlic: MRM measurements for Se isotopes 80 (a), 78 (b), and 82 (c)
Detection of Se-MeSeCys
Optimum conditions for detection of Se-MeSeCys were described previously and are the same as those used for detection of γ-glu-SeMeSeCys. Se-MeSeCys is characterized by four product ions. The corresponding transitions are: MRM 184–167, loss of NH3; MRM 184–149, loss of NH3 and H2O; MRM 184–123, loss of CO2 and NH3; and MRM 184–95 resulting in the +SeCH3 ion. Results from measurements on garlic are shown in Fig. 4a. Two peaks can be observed in all the chromatograms. One small peak is observed at t R=4.7 min; a second major peak is detected for all transitions at t R=7.2 min. The Se-MeSeCys standard elutes at 4.7 min and it can thus be concluded that the minor peak is from Se-MeSeCys. There is no need for measurements using different isotopes because a Se standard of this compound is available and elutes at the same retention time (4.7 min). If no Se standard had been available the isotopic pattern of Se would have revealed that the peak at t R=4.7 min contained Se. The peak at t R=7.2 min is another biomolecule giving rise to the same product ions, with no Se present, as was observed from the LC–ICP–MS measurements.

LC–ESI–MS–MS chromatograms obtained from a hot-water extract of garlic: determination of Se-MeSeCys (a) and Se-Met (b)
Detection of Se-Met
Se-Met has previously been detected by this method in several yeast-based Se supplements [6] and in Brazil nuts [25]. In both these food sources, Se-Met was the major selenium compound present in the extracts and, hence, its concentration was much higher. In the hot-water extract of garlic Se-Met accounts for only 15.5% of total Se. The concentration of Se-Met in the garlic is estimated to be 15.5 μg g−1 dry weight; the concentration of Se-Met in the garlic extracts was calculated to be approximately 2 mg L−1. When looking at the detection limits, one can see that because of this low concentration, it is absolutely necessary to use the most sensitive approach for the LC–ESI–MS–MS determination, because LOD values in this mode are only one tenth the estimated concentration present in the garlic extracts. The transitions were all measured on the 80Se isotope: MRM 198–181, loss of NH3; MRM 198–152, loss of CO and H2O; MRM 198–135, loss of CO, NH3, and H2O; MRM 198–109 corresponds to formation of the ion CH3SeCH2 + and MRM 198–102 corresponds to formation of the ion NH3CH(COOH)CH2CH2 +. The resulting chromatograms are shown in Fig. 4b. All product ions could be measured with sufficient sensitivity, the compound eluting at t R=420 s in the LC–ICP–MS chromatogram is assigned to Se-Met.
LC–ICP–MS of the saliva extract of garlic
Saliva contains a lubricating substance, known as mucus, buffers maintaining the acid–base balance, and salivary amylase, an enzyme that initiates hydrolysis of carbohydrates [28]. On the basis of this composition it can be assumed unlikely saliva will transform the Se species, but the question remains whether these Se compounds will be extracted during mastication and mixing with saliva. The resulting chromatogram is shown in Fig. 5. From total Se determination it was clear only part of the Se was present in this extract. The chromatogram shows the presence of several species. Four elute in the same order as for the hot-water extract. For a proper profile of the compound eluting at t R 620 s, addition of formic acid was needed. The retention times of the peaks matched those of Se-(Cys)2, Se-MeSeCys, Se-Met, and γ-glu-SeMeSeCys (compared with the hot-water extract), respectively. Two unknown compounds, eluting at t R 960 s and 1120 s , were also observed in the chromatogram. They did not co-elute with any of the commercially available standards and their concentrations were too low for characterization by LC–ESI–MS–MS. Although the main species correspond to those observed in the hot-water extract, the relative distribution is different. The main species here are Se-MeSeCys, Se-Met, Se-(Cys)2, γ-glu-SeMeSeCys, and two unknown compounds U1 and U2, accounting, respectively, for 40.6%, 10.0%, 5.1%, 37.1%, 3.9%, and 3.3% of the Se.

LC–ICP–MS chromatogram obtained from a saliva extract of garlic
LC–ICP–MS of the in vitro gastrointestinal digests of garlic
In contrast with saliva treatment, treatment of garlic with simulated gastric fluid involves more severe and extreme conditions. Species stability was tested in the presence of the enzyme pepsin, which is partly responsible for the digestion of proteins, and the presence of HCl and the low pH. There was special interest in the behaviour of the dipeptide under these conditions. Standard solutions of Se-Met, Se-MeSeCys, and Se-(Cys)2 had previously been shown to be stable in this medium. Se-Met and Se-(Cys)2 were extractable from yeast supplements and Brazil nuts [25, 26], the only difference in the garlic is that they are present in the free form and hence not bound to proteins. The compounds could be extracted by hot water at 37°C and there was no need for enzymatic hydrolysis to liberate them, in contrast with the Se compounds in Se-yeast and Brazil nuts. They are expected to be more readily bioavailable, because no protein bonds must be broken before liberation of the Se compounds. Addition of formic acid was of the utmost importance for proper separation of, especially, the later eluting compounds. Chromatograms obtained from garlic treated with SGF in the time-related experiment are shown in Fig. 6a. Nine chromatograms obtained throughout the duration of the experiment are shown. The same Se profiles are observed in all the chromatograms. The main species elute at the retention times of Se-Met, Se-(Cys)2, Se-MeSeCys, and γ-glu-SeMeSeCys and there are also two unknown compounds, U1 and U2. No changes can be observed in the species extracted after treatment for 0 min to 24 h. This indicates that γ-glu-SeMeSeCys remains intact during treatment. Even after extensive periods of SGF treatment the presence of γ-glu-SeMeSeCys is not altered. Because the gastric transit time of food is a few hours or less, γ-glu-SeMeSeCys will outlast SGF digestion. The species distribution is shown in Fig. 7a. The amounts of Se-(Cys)2, Se-Met, U1, and U2 remain more or less the same and account for ±5%, 7−9%, 2–3%, and ±2.5% of the Se, respectively. When the amount of γ-glu-SeMeSeCys diminishes, the amount of Se-MeSeCys increases. Because γ-glu-SeMeSeCys serves as a carrier for Se-MeSeCys, it might be that during treatment with SGF γ-glu-SeMeSeCys loses its glutamyl moiety, forming the resulting seleno amino acid. Because Se-MeSeCys accounted for only 28.8% in the hot-water extract and accounts for 40–50% in the SGF digestion fluid, here it is most likely that part of the γ-glu-SeMeSeCys is transformed to Se-MeSeCys during gastric human digestion.

LC–ICP–MS chromatograms of Se in garlic treated with simulated gastric fluid (a) and simulated intestinal fluid (b) for periods from 0 min to 24 h

Species distribution of the Se compounds in garlic treated with SGF (a) and SIF (b) for periods from 0 min to 24 h
The major function of the intestinal tract is to continue breakdown of food and absorb the products formed. In this part of the work the action of the enzyme pancreatin and the effect of changes in pH on the Se species in garlic were investigated. Garlic was subjected to in vitro intestinal digestion for different times. The chromatograms are shown in Fig. 6b. The same compounds are observed: Se-Met, Se-(Cys)2, Se-MeSeCys, γ-glu-SeMeSeCys, and the two unknown compounds U1 and U2. Differences between species distribution are shown in Fig. 7b. The amounts of Se-(Cys)2, Se-Met, U1, and U2 extracted are ±5%, 10–15%, 3–4% and ±2.5%, respectively, of the total Se-content. This is basically the same as for the SGF experiments, except for Se-Met. During SIF treatment a trend can be observed which shows that the longer the garlic is treated with SIF the more Se-Met is extracted. The main compound extracted during SIF treatment is Se-MeSeCys (45–60%). The amounts of Se-MeSeCys and γ-glu-SeMeSeCys are again related. Here another trend can be observed—the longer the garlic is treated, the less γ-glu-SeMeSeCys is present in the extracts and the more Se-MeSeCys is observed. This suggests that γ-glu-SeMeSeCys is converted to Se-MeSeCys as the fraction of the former decreases from 30 to 18% and that of the latter increases from 45 to 57%. That the dipeptide loses its glutamyl moiety is of interest for cancer research, because this typical form is thought to be responsible for the major anticarcinogenic properties of garlic [8].
LC–ESI–MS–MS for detection of the Se compounds in the in vitro gastrointestinal digests
Detection of γ-glu-Se-MeSeCys
Because γ-glu-Se-MeSeCys is a dipeptide and peptide bonds are easily hydrolysed, it is of interest to mimic the conditions and determine the species by molecular mass spectrometry. Because the peptide bond is of the γ-type, it is unlikely this bond will be acted on by aminopeptidases, which liberate amino acids by scission of the peptide bond adjacent to the free amino acid [9]. The γ-glu-Se-MeSeCys can be determined as described for the hot-water extract. Because the retention time of the compound is now known, it is no longer necessary to measure on different isotopes. Because of the lower abundance of the γ-glu-SeMeSeCys compound in the SGF and SIF extracts, problems with the detectability of the different transitions might be a problem. MRM 313–296 could not be measured, the other MRMs are shown in Fig. 8a and b for the SGF experiments. Figure 8A shows MRM 313–167, MRM 313–130, MRM 313–184, MRM 313–224, and MRM 313–250; MRM 313–84 is shown in a separate chromatogram (Fig. 8b) because, although this compound could be detected, its peak was not visible on the scale used to present the other chromatograms. The results from the SIF experiments are presented in Fig. 8c and d. The γ-glu-SeMeSeCys could be detected on the basis of its retention time and the presence of six product ions. The chromatograms shown are for detection of the product ions in samples treated for 1 h. Similar results were obtained from all the other samples. From these results, it can be concluded that the statements made on the basis of the LC–ICP–MS measurements are valid. The same compounds can be detected in the SGF and SIF extracts as in a hot-water extract, although two more compounds were observed after SGF and SIF treatment. The identity of these compounds remains unknown and it is unclear whether they are original garlic species or transformations from Se species formed during SGF and SIF treatment.

LC–ESI–MS–MS chromatograms: detection of the 80Se isotope of γ-glu-Se-MeSeCys in extracts of garlic treated with SGF (a and b) and with SIF (c and d)
Detection of Se-MeSeCys and Se-Met
All MRMs typical of Se-MeSeCys were measured under the conditions described above. Fragments with m/z 167, 149, 123, and 95 were measured on-line for SGF and SIF samples. The results were the same as those for the hot-water extract and hence are not shown here.
The major route of excretion of Se-MeSeCys and γ-glu SeMeSeCys is in the urine [9]. Because both compounds are typically found in Se-accumulating plants and are responsible for the huge build-up of Se in those plants, without any signs of toxicity [29], they might be metabolized differently. The metabolic pathways for both compounds through the human body remain unclear and is of major interest for further research.
Se-Met was analysed by the same LC–ESI–MS–MS method as the hot-water extract. Chromatograms based on the typical Se-Met transitions, measured after treatment with SGF and SIF, were the same as for the hot-water extracts. The MRM 198–135 transition could not be measured in the SGF extract; this is also the transition with the worst LOD. The other transitions could be measured at the retention time of Se-Met. For the SIF extracts all the MRMs could be measured.
Conclusion
This method, based on ion-pair LC–ICP–MS in combination with RPLC–ESI–MS–MS, has proven very powerful for Se-speciation in food and for tracking the species during in vitro simulated human digestion. The main species determined in a hot-water extract of garlic were γ-glu-SeMeSeCys, Se-Met, and Se-MeSeCys. Determination was based on the on-line monitoring of their typical product ions and, hence, these species could be detected without the need for extensive sample clean-up. In saliva, simulated gastric fluid, and simulated intestinal fluid, the same species were observed and additional unknown compounds were detected. The distribution of the species differed from that in the hot-water extract. It is most probable that γ-glu-Se-MeSeCys is converted to Se-MeSeCys during human digestion. The method enables rapid screening of Se species in garlic and would be of interest in the analysis of new “functional food”.
References
Song K, Milner JA (2001) J Nutr 131:1054S–1057S
Ip C, Lisk DJ (1993) Nutr Cancer 20:129–137
Ip C, Lisk DJ (1995) Carcinogenesis 16:2649–2652
Ip C, Lisk DJ, Thompson HJ (1996) Carcinogenesis 17:1979–1982
Dumont E, De Cremer K, Van Hulle M, Chéry CC, Vanhaecke F, Cornelis R (2005) J Chromatogr A 1071:191–196
Goenaga Infante H, OConnor G, Rayman M, Wahlen R, Entwisle J, Norris P, Hearn P, Catterick P (2004) J Anal At Spectrom 19:1529–1538
Goenaga Infante H, O’Connor G, Rayman M, Wahlen R, Spallholz JE, Hearn R, Catterick T (2005) J Anal At Spectrom DOI:10.1039/b503895b
Lu J, Pei H, Ip C, Lisk DJ, Ganther H, Thompson HJ (1996) Carcinogenesis 17:1903–1907
Dong Y, Lisk D J, Block E, Ip C (2001) Cancer Res 61:2923–2928
Cai X, Uden PC, Block E, Zhang X, Quimby BD, Sullivan JJ (1994) J Agric Food Chem 42:2081–2084
Cai X, Block E, Uden PC, Quimby BD, Sullivan JJ (1995) J Agric Food Chem 43:1751–1753
Dietz C, Pérez-Corona T, Madrid-Albarrán Y, Cámara C (2003) J Anal At Spectrom 18:467–473
Dietz C, Sanz Landaluze J, Ximénez-Embún P, Madrid-Albarrán Y, Cámara C (2004) 501:157–167
Block E, Cai XJ, Uden PC, Zhang X, Quimby BD, Sullivan JJ (1996) Pure Appl Chem 68:937–944
Kotrebai M, Tyson JF, Block E, Uden PC (2000) J Chromatogr A 866:51–63
Kotrebai M, Birringer M, Tyson JF, Block E, Uden PC (1999) Anal Commun 36:249–252
Ge H, Cai X, Tyson JF, Uden PC, Denoyer ER, Block E (1996) Anal Commun 33:279–281
Francesconi K, Sperling M (2005) Analyst 130:998–1001
Ip C, Birringer M, Block E, Kotrebai M, Tyson JF, Uden PC, Lisk DJ (2000) J Agric Food Chem 48:2062–2070
Kotrebai M, Birringer M, Tyson JF, Block E, Uden PC (2000) Analyst 125:71–78
Uden PC, Hafezi R, Kotrebai M, Nolibos P, Tyson J, Block E (2001) Phosphorus Sulfur Silicon 171:31–56
McSheehy S, Yang W, Pannier F, Szpunar J, Lobinski R, Auger J, Potin-Gautier M (2000) Anal Chim Acta 421:147–153
Ogra Y, Ishiwata K, Iwashita Y, Suzuki KT (2005) J Chromatogr A 1093:118–125
Iwashita Y, Nishi K (2004) Biomed Res Trace Elem 15:72–75
Dumont E, De Pauw L, Vanhaecke F, Cornelis R (2006) Food Chem 95:634–692
Dumont E, Vanhaecke F, Cornelis R (2004) Anal Bioanal Chem 379:504–511
US Pharmacopeia XXIV and national formulary 19 (2000) The United States pharmacopeial convention Rockville 2235–2236
Avila VL (1995) (Ed) Biology investigating life on earth. Jones and Bartlett Publishers, Boston 550
Neuhierl B, Thanbichler M, Lottspeich F, Block A (1999) J Biol Chem 274:5407–5414
Acknowledgements
E. Dumont is a grant holder of the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT). The Fund for Scientific Research (FWO) is thanked for financial support through grant G.0026.01.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dumont, E., Ogra, Y., Vanhaecke, F. et al. Liquid chromatography–mass spectrometry (LC–MS): a powerful combination for selenium speciation in garlic (Allium sativum). Anal Bioanal Chem 384, 1196–1206 (2006). https://doi.org/10.1007/s00216-005-0272-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-005-0272-6