
Abstract
The implementation of quality systems in analytical laboratories has now, in general, been achieved. While this requirement significantly modified the way that the laboratories were run, it has also improved the quality of the results. The key idea is to use analytical procedures which produce results that fulfil the users’ needs and actually help when making decisions. This paper presents the implications of quality systems on the conception and development of an analytical procedure. It introduces the concept of the lifecycle of a method as a model that can be used to organize the selection, development, validation and routine application of a method. It underlines the importance of method validation, and presents a recent approach based on the accuracy profile to illustrate how validation must be fully integrated into the basic design of the method. Thanks to the β-expectation tolerance interval introduced by Mee (Technometrics (1984) 26(3):251–253), it is possible to unambiguously demonstrate the fitness for purpose of a new method. Remembering that it is also a requirement for accredited laboratories to express the measurement uncertainty, the authors show that uncertainty can be easily related to the trueness and precision of the data collected when building the method accuracy profile.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The quality of measurements produced by analytical laboratories during the last decade has significantly improved, and laboratories have been requested to control their procedures and organisation more effectively. The quality of an analytical data can be defined at two main levels:
-
1.
Metrological requirements. The end-users of chemical data expect that measurements are close to the true value for the sample submitted to an analysis. This is known as the “accuracy”. The accuracy depends on the precision and the bias of the method, and there are several factors which significantly influence the uncertainty of a measurement. An important goal is to evaluate this uncertainty and express it in such a way that end-users are able to adequately use the result produced by the laboratory.
-
2.
Socio-economic requirements. While metrological requirements are implicit and cannot be directly controlled by the end-user, the costs involved in a measurement can be controlled to some degree. There is always a strong pressure on laboratories to reduce costs and propose cost-effective services.
The goal of this paper is to describe two complementary approaches set up by laboratories: (1) to develop and validate methods fit for a given purpose; (2) to express the uncertainty in the chemical measurements. These approaches are not independent, and we will try to demonstrate how they can be combined. This paper presents synthetic results, but all of the practical details required for computation are available in the literature cited.
Method lifecycle
In order to clearly understand the place of validation, it is interesting to introduce a concept that underlies the recent standard ISO 17025 [1], namely the lifecycle of a method. The basic idea is that a method of analysis is not static; it is a dynamic entity that goes through several dependent steps. Too often a method is described as an unchangeable and frozen procedure; this is the impression many manuals or standards give. However, like all production processes, methods of analysis are created, they evolve, and they die. Different schemes are proposed to describe this specific lifecycle but we think that the most convenient way is summarised in Fig. 1 because it illustrates that it is a continuous cycle. In this diagram, the various steps of the cycle appear as grey boxes with bold letters; the procedures or criteria that will be used, handled or computed during a given step are placed in rectangular boxes. The main tools or techniques to be used during the step are written in italics.

The lifecycle of a method of analysis
The first step in the method lifecycle is the selection of the method. In Sect. 5.4.2 of ISO 17025 [1] on method selection, it is stated that:
The laboratory shall use test and/or calibration methods, including methods for sampling, which meet the needs of the client and which are appropriate for the tests and/or calibration, it undertakes preferably those published as international, regional or national standards.
This statement is clearly consistent with the reasoning of the new ISO 9000:2000 standards, but it over-simplifies. For an analyst, method selection can be a difficult process. It often means trying to transform a given problem into chemical measurements. For instance, controlling air pollution requires the combination of several analytes, each needing a specific method of analysis, and meteorological data in order to create a composite index which can be easily understood by end-users. In any case, the expertise of the analytical chemist is the basic tool for selecting the most “adequate” method.
Once the method is selected, it often necessary to perform several experiments in order to either adapt to the laboratory conditions or fully develop the method. The development of the method can be simple when starting from a standardised method but much more complex with an original procedure; this step is often called “optimisation”, although this term is vaguely defined. It is regrettable that analytical chemists do not use more extensively experimental designs and response surface methodology to achieve this task. As method development proceeds, regular reviews should be carried out to verify that the needs of the client are still being fulfilled and that the method is still fit for its purpose. Changing requirements to the development plan should be approved and authorised.
When the development phase is finished, the draft of the standard operating procedure (SOP) can be written.
Method validation is “... the confirmation by examination and the provision of objective evidence that the particular requirements for a specific intended use are fulfilled” [1 in Sect. 5.4.5.1], and validation can only occur at this moment. It is inadequate to try to merge development and validation in the same step. It is now accepted that we can make a distinction between intra-laboratory (or in-house) and inter-laboratory (or collaborative) validation. The first is universal and compulsory, the second is mainly applicable to methods that will be used by several laboratories or where the results can be used in economic decisions. For example, in the pharmaceutical industry, it is useless or impossible to perform a collaborative study for a new molecule under development. On the other hand, all methods used for food safety control must be inter-laboratory validated. Verification may occur at the end of the validation procedure, as proposed in the definition.
This clearly means that specific objectives must be defined before starting any validation; a method must be fit for a given purpose, as described in the next chapter. According to this statement, when modifications or new requirements occur, one must perform a revalidation. It is difficult to define the exact extent of this revalidation in respect to the modifications: this appreciation can be left to the analyst’s know-how.
If the validation proves to be compliant, the next step in the lifecycle is the use of the method in a routine. After a certain amount of time, the method may be abandoned because it is obsolescent, and another lifecycle begins.
The framework of method validation is now well defined by several reference texts and can be summarised by the following statements [1, 2, 3, 4]:
-
1.
Analytical measurements should be made to satisfy an agreed requirement (a defined objective).
-
2.
Analytical measurements should be made using methods and equipment that have been tested to ensure they are fit for purpose.
-
3.
Staff making analytical measurements should be qualified for and competent at undertaking the task (and should demonstrate that they can perform the analysis properly).
-
4.
There should be a regular independent assessment of the technical performance of a laboratory.
-
5.
Analytical measurements made in one location should be consistent with those made elsewhere.
-
6.
Organisations making analytical measurements should have well defined quality control and quality assurance procedures.
-
7.
Validation is the process of establishing the performance characteristics and limitations of a method, as well as identifying the influences that may change these characteristics (and to what extent they may change them).
-
8.
Validation is also the process of verifying that a method is suitable for its intended purpose (to solve a particular analytical problem).
Accuracy profiles
Objectives of validation
Members of the SFSTP (Société Française des Sciences et Techniques Pharmaceutiques) have contributed to the development of consensus validation procedures since 1992 [5, 6, 7]. More recently Boulanger et al [8, 9] introduced the concept of accuracy profiles to circumvent some of the fundamental drawbacks of validation procedures currently available. The basic idea behind this concept is that analysts expect an analytical procedure to return a result X which differs from the unknown “true value” μ of the analysed sample less than an acceptability limit λ. This requirement can be expressed by Eq. 1:
The acceptability limit λ fully depends on the objectives of the analytical procedure and is the responsibility of the analyst. For instance, when expressed in percent, it can be 1% on bulk materials, 5% on pharmaceutical specialities, 15% for biological samples, and so on. The analytical method can be characterised by systematic error or “true bias” δM and a random error or “true precision” σM. Both of these figures of merit are unknown, like the true value of the sample μ, but estimates can be obtained from measurements made during validation. The reliability of the estimates depends on the adequacy of the measurements made on known samples, called validation standards (VS), the experimental design and the number of replicates. However, obtaining estimates for bias and precision is not the objective of the validation per se; it is a necessary step toward evaluating the ability of the analytical procedure to satisfy its objective, although this alone is not a sufficient step, as we will see. The ultimate objective is to guarantee that most of measurements the procedure will provide in the future are accurate enough—close enough to the unknown true value of the sample assayed.
Figure 2 illustrates Eq. 1 graphically by showing the distributions of 95% of the measurements given by four different analytical procedures with different true bias values δM and precisions σM. The relative acceptability limit λ is set to be identical (±15%) for all four methods. This value was chosen since it is required by the Conference of Washington for bio-analytical procedures [10, 11]. Figure 2 shows that procedure 3 does not fulfil the given objective, since its bias is null but its precision is 20% (expressed as a relative standard deviation or RSD). Similarly, procedure 4 with a bias of 7% but with a precision of 12% will also be rejected because the expected proportion of measurements outside the limits of acceptability is too great. In contrast, procedures 1 and 2 fulfil the objective in the same figure. They can possibly be declared to be valid procedures, depending on the maximum risk that is to be accepted, since the analyst can expect that at least 95% (maximum risk 5%) and 80% (maximum risk 20%) respectively of future measurements will be inside the acceptability limits. Procedure 1 has a larger bias (+7%) but a better precision (3%), than procedure 2 (respectively +1 and 10% RSD). The differences between these two procedures are not relevant here, since the results obtained in both cases are never too far from the true values of the sample to quantify, so they are within the acceptability limits. The quality of each result sets is therefore the criterion used to evaluate the validity of a procedure in terms of bias or precision, since high quality is the objective when providing accurate results.

Examples of four procedures with the same acceptability limits λ = ±15%. Bias is expressed in percentage of difference from the true value, and the precision is expressed as a relative standard deviation (RSD). Curves indicate 95% data intervals
Another possible illustration consists of representing the domain of acceptable analytical procedures—the acceptability region—characterised by the true bias and precision, as illustrated in Fig. 3. Inside the triangles, acceptable procedures are those for which a given proportion of measurements, for instance 95, 80 or 66%, are likely to fall within the ±15% acceptability limits. Therefore, it is in these domains that “valid” analytical procedures are located with respect to the proportion of measurements that the analyst would like to have within the acceptability limits. The triangles correspond to proportions of 95, 80 and 66% of measurements included within the acceptability limits (the last being a proportion that doesn’t correspond to any known regulatory requirement). These triangles were built thanks to Eqs. 6 and 8 (as explained in the section “Decision rules and accuracy profiles“) using a range of values for the true precision variance and β risk, while the true bias was kept constant and equal to 0. However, it’s important to realise that for a procedure characterised by a null true bias and a true precision of 15%, about 66% will fall within the acceptability limits. Indeed, according to the normal distribution, 66% (2/3) of the observations are expected to fall within ±1 standard deviation (in this case ±15% since the bias is null). This proportion reaches 95% when the precision is increased to 8% (with null bias), as apparent from Fig. 3 at the top of the 95% acceptability region triangle. This proportion of 66% is nevertheless a direct consequence, as opposed to being intended, of the rule recommended by the Conference of Washington for routine quality control [10, 11], which states that at least four control samples out of six must fall within the acceptability limits of ±15% (4-6-15 rule). This rule is equivalent to accepting that only 2/3 or 66% of measurements are within the acceptability limits. This illustrates the gap that exists between requirements in the validation phase and those required during routine work to guarantee the quality of the results. This gap is paradoxical, since the goal of the validation of an analytical procedure is to demonstrate that the analytical procedure will be able to fulfil its intended objectives.

The interiors of the triangles of the left graphic represent the acceptability regions for analytical procedures that give at least 66, 80 and 95% of their measurements within acceptability limits respectively, as a function of their theoretical true bias (%) and true precision (RSD %). The performance of an analytical procedure is represented by a point in this graphic. The expected distributions of future measurements for the procedures are represented in the small graphics on the right, corresponding to the four procedures of Fig. 2. The acceptability limit of [−15%, +15%] is just an example of a frequent recommendation for bioanalytical procedures. See [9] for more computational details
For illustrative purposes, the four procedures of Fig. 2 are inserted into Fig. 3 as a function of their performances. One can observe that procedures 1 and 2 are located inside the acceptability regions that guarantee that at least 95 and 80% respectively of their results will be within the acceptability limits.
Finally, a procedure can be validated if it is very likely (in other words, there is only limited risk) that the difference between every measurement X of a sample and the true value μ is inside the acceptability limits that the analyst has predefined. This can be translated into Eq. 2:
where β is the probability that a measurement falls inside the acceptability limits defined by λ (15%, say).
It seems reasonable to claim that the objectives of the validation are to guarantee that every single measurement that will later be performed routinely will be close enough to the unknown true value of the sample. Consequently, the objectives of validation are not simply to obtain estimates of bias and precision; it is also to evaluate this risk or confidence. In this respect, trueness, precision, linearity and other validation criteria are no longer sufficient to make these guarantees. In fact, adapted decision tools are necessary to give guarantees that a reasonable proportion of future measurements will fall inside the acceptability limits.
Decision rules and accuracy profiles
Considering the proposals published on method validation, the decision rules used in the validation phase are mostly based on the use of the null hypothesis:
A procedure is therefore declared adequate when the 95% confidence interval of the average bias includes the value 0. The decision is based on the computation of the rejection criterion of the Student’s t-test:
An obvious consequence of this is that the smaller the variance (the better the precision), the less likely the confidence interval is to contain the 0 bias value, leading to a rejection of the procedure. On the other hand, the worse the precision, the more likely the confidence interval is to contain the 0 bias value and so it is more likely that the procedure will be declared valid! This paradoxical situation is illustrated by Fig. 4a, showing how the four procedures are accepted or rejected. For instance, procedure 1 presents a reduced bias (+7%) and a small measurement dispersion (3% RSD) and is therefore rejected by this rule, while procedure 4, which has the same bias but is much less precise (12% RSD instead of 3% RSD), is accepted!

Graphical illustration of acceptability regions (shaded area) of analytical procedures, that depend on the statistical decision rules, for the four procedures of Fig. 2 as illustrated by a dot. a Acceptability region when the decision is based on the classical null hypothesis test, H0: Bias = 0, using a t-distribution at 5% with 12 degrees of freedom. b Acceptability region when decision is based on the β-expectation tolerance limits that should be included within the acceptability limits. In this example, the acceptability region in grey is defined for β > 66%, with 12 degrees of freedom and acceptability limits at [+15%, −15%]
According to the proposal of Mee [12], it is possible to compute the so-called “β-expectation tolerance interval”. It defines an interval which contains an expected proportion β of future results. Following a classical notation in statistics, β represents the second error type probability—the probability of accepting the null hypothesis when it is wrong; in this context it corresponds to the error of concluding that the result is conforming when it is actually not conforming. The ^ symbol is used for the estimate of the statistic. This tolerance interval obeys the following property:
where E is the “expected value” of the result for a given bias and standard deviation. In this case, the calculation of the β-expectation tolerance interval involves estimating the bias and the standard deviation of intermediate precision of the method, denoted as \( \hat{\delta }_{{\text{M}}} ,\hat{\sigma }_{{\text{M}}} . \) respectively. Developing Eq. 5 as proposed by Mee in [12], it can be demonstrated that the β-expectation tolerance interval is equal to:
where the estimate of the intermediate precision variance (or reproducibility, depending on the conditions) is:
In Eq. (7), \( \hat{\sigma }^{2}_{{\text{B}}} \;{\text{and}}\;\hat{\sigma }^{2}_{{\text{W}}} \) are the estimates of the variance between series and within series respectively. They can be obtained ideally by maximum likelihood methodology [9] or by means of the classical sum of squares methods, as in [7]. The “conditions” are here defined in a large sense to indicate standardised experimental conditions or the set-up used to obtain measurements. Depending on the experimental design and the intent, or the stage of the validation phase, conditions can be either be days within a laboratory, or the laboratories themselves. Depending on the definition given, variance components analysis will lead to an estimate of the intermediate or reproducibility variance. These variances can only be computed if data are collected under various conditions (or intermediate precision); the measurements are made when varying at least one factor such as the laboratory, the day or the operator. The example of the application proposed in the paper uses between day and series conditions. In this context p is the number of series and n the number of replicate measurements per series.
In Eq. 6, Q t is the β quantile of the Student’s t-distribution with ν degrees of freedom; ν is computed according to the correction method proposed by Satterthwaite [13]; ks an expansion factor that takes into account the variability of the mean bias as estimated from the experimental design and, as demonstrated and derived in Mee [12], is obtained as follows:
Figure 4b shows the acceptability regions (in grey) of the procedures accepted as valid using the β-expectation tolerance intervals. Here, the triangle corresponds to the set of procedures for which, according to the bias and precision observed during the validation experiments, the proportion of measurements inside the acceptability limits is greater or equal to β, the proportion chosen a priori (66%, say). This decision rule appears to be more sensible than the previous one, since all procedures that have good precisions are accepted, while the procedures that have large standard deviations are rejected. In addition, if a procedure has a bias, it must have a small variance to be accepted. Symmetrically, a procedure with a high variance with respect to the acceptability limits has very little chance of being accepted.
An interesting extension of this method consists in constructing the β-expectation tolerance intervals for the whole range of expected measurements, as illustrated by Fig. 5. It produces an easy and visual method called the accuracy profile of the analytical procedure. The accuracy profile is constructed for a given set of concentration levels C1, C2... For each level it is possible to obtain estimates of bias and precision and calculate the β-expectation tolerance limits according to Eq. 5. The upper and lower tolerance limits are then connected by straight lines in order to interpolate the behaviour of the limits between the levels at which measurements have been made: these are the interpolating lines. In Fig. 5, for both concentration levels C1 and C4, the tolerance interval is wider than the acceptance limits. The limits of quantification are therefore at the intersection between the interpolating lines and the acceptance limits. It is possible to then define two experimental limits, which correspond to lower and upper quantification limits. Below the lower limit of quantification (LLQ) or above the upper limit of quantification (ULQ) it is unacceptable to say that the procedure accurately quantifies the analyte. This is in agreement with the definition of the limits of quantification: the smallest/highest quantity of the substance to be analysed that can be measured with a given trueness and precision [4]. In Fig. 5, the grey area represents the range where the procedure is expected to quantify at least a proportion β of the samples with a predefined accuracy. If the analyst wants to take no more than a 5% risk, he will be able, at the end of the validation, to expect that at least 95 out of 100 future measurements will fall within the acceptability limits, which are fixed according to the requirements of the field (pharmaceutical, environment, food analysis, and so on).

Illustration of an accuracy profile based on four levels of VS, used as a decision tool to characterise method performances. LLQ is the lower limit of quantification, ULQ is the upper limit of quantification
The use of the accuracy profile as a single decision tool allows the objectives of the procedure to be brought into line with those of the validation, and also allows us to visually grasp the ability of the procedure to fit its purpose.
It is also important to recall that there is no global consensus among the various standardised documents (ISO, ICH, AFNOR, SANCO, FDA, Washington conference,...) for the definition of the criteria to be tested during the validation step. For example, the linearity criterion can appear or not and its interpretation can be different from one document to another. This is also the case for the accuracy that can be merged with the trueness, as in the ICH Q2A document.
We consider it preferable to establish validation criteria, as much as possible, in the same matrix as that routinely sampled. Each analytical procedure should be validated for each type of matrix [14]. Nevertheless, the definition of a matrix is the responsibility of the analyst. Moreover, each modification of a previously validated method automatically involves a revalidation, the extent of which depends on the modifications done and their potential influence on the specific validation criteria.
Using the accuracy profile: an example
In the validation phase, all results obtained must be reported. At the end of the validation phase and before the routine stage, the analytical procedure must be completely described in the form of a standardised operational procedure (SOP).
It is mandatory to prepare calibration standards (CS) using the same procedure that will be applied routinely—the same operation mode, the same number of concentration levels (calibration points), and the same number of replicates by level [9].
The VS should be prepared independently in the matrix, if applicable [9, 14]. When reference materials (certified or internal) are available, they represent one of the most interesting ways to have the VS; however it is also possible to use spiked samples. Each VS is prepared and fully analysed as an unknown sample. Independence is critical to a good estimation of the between-series variance. In practice, the effects of “day” and/or “operator” are most often used as different conditions for experiments. It is not necessary to have consecutive days. Any rejected data must be documented.
According to the harmonised procedure recently published by SFSTP [9], several experimental designs are available. Table 1 presents some of these experimental designs: they all consist of a set of CS and VS; additional spiked calibration standards (SS) can also be added. It is noticeable that the total number of experiments may vary; this is due to regulatory constraints. For instance, the minimal number of VS required can be set to 6, or the number of concentration levels for these VS can vary from 3 to 5 in order to be compliant with the ICH or FDA requirements.
The example presented here consisted of validating a method of determining the sotalol in plasma by HPLC using atenolol as an internal standard [6, 15]. Sotalol concentrations are expressed in ng/ml, and instrumental response as the ratio between the peak areas of sotalol and atenolol. All of the data are shown in Table 2. The calibration experimental design is 3×5×2 and consisted of preparing duplicate standard solutions at five concentration levels ranging from 25 to about 1,000 ng/ml. This was replicated three times over three days (or three conditions). The VS were prepared in the matrix, and consisted of three days with three concentration levels, each replicated three times: a 3×3×3 experimental design. Therefore, the same experimental design for calibration and validation is not necessary.
Once all data were collected, they were processed according to the following procedure:
-
1.
The “best-adapted” response function was obtained using the calibration standards, relating the response and the concentration. Several mathematical models can be fitted, and the best was selected at the end of the procedure, according to its accuracy profile (in other words, the model that gives the most accurate results over the range). Mathematical transformations, such as logarithmic or square root, can also be applied. Finally, weighted regression algorithms can also be used if the response variance obviously varies as a function of the concentration. In this example, accurate results were obtained when fitting a weighed linear regression model with 1/X2 as weight, a classical and convenient model.
-
2.
An inverse prediction equation was used to predict the actual concentrations of the VS. These data are reported in Table 3 as predicted values and relative bias, using the theoretical concentration introduced in each VS as a reference value. The precision data corresponding to the weighed linear model are reported in Table 4. These data were used to compute the accuracy profile.
-
3.
Thereafter, for each concentration level, trueness and precision were estimated and used to compute the accuracy profile according to Eqs. 6, 7 and 8. For each regression model, the limits of the β-expectation tolerance intervals were calculated and are illustrated in Fig. 6. These profiles are the visual decision tools that allow the analyst to evaluate the ability of each procedure. For this determination, the acceptability limits were set to 20%, as illustrated in the figures. This value was used because the method is a bioanalytical technique [10, 11].
-
4.
The acceptable accuracy profiles were selected. In this example they correspond to the weighed linear model on the original data, the linear model after square root transformation, and the linear model after log transformation, as illustrated in Figs. 6b, e and f respectively. In the present method, the weighed linear model on the original data was selected simply because it’s commonly accepted and used in analytical chemistry. Several selection rules can be proposed to achieve this selection: the most intuitive is to calculate the limits of quantification as illustrated in Fig. 5. If none of the accuracy profiles falls within the acceptability limits, the analyst can either restrict the application range of the method or extend the acceptability limits. Various techniques, such as residual plots, and the lack-of-fit test, are available to identify the source of the problem(s).

Illustrations of different accuracy profiles obtained if the same calibration and validation data (from Table 2) but different calibration models (expressed as the relative bias of the method vs. the recovered concentration) are used. Range 25–1,000 ng/ml. Computational details are available in [9]. a Quadratic regression. b Weighted linear regression (weights: 1/χ2). c Linear regression. d Linear regression through 0. e Linear regression after square-root transform. f Linear regression after log transform
Therefore, we can validate this method for an acceptability of ± 20%, an application range of [25.35, 838.7] ng/ml, and a weighed linear regression model.
Measurement uncertainty
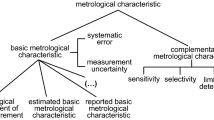
Expressing the measurement uncertainty is now a well known problem for most analytical chemists who want to be accredited according to the ISO 17025 standard. The famous guide published by EURACHEM [16] has been intensively downloaded since it was made available in April 2000 on several websites. This success can be related to the fact that it presented several practical examples of the basic principles of traditional metrology as applied to chemical tests. Actually, two thirds of the document is devoted to examples. The strategy proposed in the ISO Guide for the Expression of the Uncertainty of Measurement (GUM) (type A and type B evaluation of uncertainty) are presented and illustrated in many examples which show how different uncertainty contributions can be combined. However, the general feeling that the reader can get from this document is that the most applicable procedure may be to identify uncertainty sources by a cause-and-effect diagram (sometimes known as the Ishikawa or fishbone diagram) and then combine them. This is likely to be the most convenient procedure for an in-house determination of uncertainty but, in a case where the mathematical formula used to express the measurement result does not exhaustively describe the complete analytical procedure, it can be a reducing procedure. Often many important sources of uncertainty are not taken into account, such as sampling, sample handling, or environmental sources, because they are difficult to estimate.
As clearly identified by Ranson [17] in a recent communication, the traditional cause-and-effect diagram with its five “bones” can be related to some important chapters of the ISO 17025 standard, as summarised in Fig. 7. Therefore, it is clear that most of the examples presented in the EURACHEM guide deal mainly with the influence of the method (Sect. 5.4: Test and calibration methods and method validation) or the equipment (Sect. 5.5: Equipment) and consequently underestimate the uncertainty [18].

Relationship between the cause-and-effect diagram and chapters from the ISO 17025 standard (from [17])
Therefore, there is still a lot of debate among analysts about whether sources of uncertainty that are not taken into account may have a strong influence. The goal at stake is not insignificant, because it is clear that it is tempting for some laboratories to use uncertainty as a commercial argument: the smaller the uncertainty, the better the measurement (or the laboratory). Therefore it is necessary to give an unambiguous response to this question in order to avoid any flaw in the use of uncertainty. The adoption of this new concept can be a great opportunity for analytical laboratories because it gives a real meaning to chemical measurements. This advantage has already been demonstrated for many other industrial activities using different kinds of measurements to physical–chemical, such as weight in packaging, or length for lathe-worked devices.
Considering the problems raised by the expression of uncertainty and the need to have realistic values, new proposals have been made. They consist of using experimental data obtained from precision studies. A recent draft of guide ISO/DTS 21748 [19] suggests using repeatability, reproducibility and trueness estimates to estimate measurement uncertainty. Considering the data collected by the analysts when developing an accuracy profile, it seems sensible to also use these data to estimate uncertainty. Depending on the way the experimental design is envisaged—several conditions within a laboratory, or several laboratories—the accuracy profile corresponds to either an intra-laboratory validation or an inter-laboratory validation (reproducibility). The guide only refers to interlaboratory precision data. Intra-laboratory validation can also precisely reflect the actual capability of the method when applied by a given laboratory. When choosing between the intra-laboratory and inter-laboratory approaches it is important to note that the goal of the validation process must be to reflect the way an analytical procedure will be used in the future. If an analytical procedure is only intended to be used within a laboratory, then the only conditions that will be changed in the experimental design are within the laboratory of interest such as days, runs, operators or batches. The estimated total variance, or intermediate precision \( \hat{\sigma }^{2}_{{\text{M}}} , \) will be the sum of the between-condition variance and the within-condition variance or repeatability \( \hat{\sigma }^{2}_{{\text{W}}} . \) On the other hand, if an analytical procedure is intended for use by many laboratories, then the important conditions to vary in the experimental design are the between-laboratory conditions. In such a design, the between-condition (runs, days, batches) variances are by default integrated into, or more precisely not differentiated from, the within-laboratory variance (the repeatability). With such a design, the total variance, called the “reproducibility”, will be the sum of the between-laboratory variance and the within-laboratory variance (repeatability). To summarise, the only difference between the interlaboratory and the intralaboratory studies is the way the experiments are designed, and the definition that is given to the “condition” when estimating the “between-condition” variance components or uncertainties.
According to the recommendations of the ISO/DTS 21748 guide [19], a basic model for the uncertainty in a measurand Y associated with observations can be (notations are those used in this standard):
where sR is the reproducibility standard deviation, \( u(\hat{\delta }) \) is the uncertainty associated with the bias δ of the method, and \( {\sum {c^{2}_{i} u(x_{i} )^{2} } } \) is the sum of all of the effects due to other deviations. This equation can be simplified if only a statistical estimation approach is used.
According to the same guide [19], the bias uncertainty can be estimated as:
where n is the number of replicates (within-condition), p the number of varied conditions (the number of laboratories when reproducibility is the objective) and \( \gamma = \raise0.7ex\hbox{${s^{2}_{{\text{r}}} }$} \!\mathord{\left/ {\vphantom {{s^{2}_{{\text{r}}} } {s^{2}_{{\text{R}}} }}}\right.\kern-\nulldelimiterspace} \!\lower0.7ex\hbox{${s^{2}_{{\text{R}}} }$}. \) s 2r is an estimate of the repeatability (within-condition) variance, and \( s^{2}_{{\text{R}}} = s^{2}_{{\text{r}}} + s^{2}_{{\text{B}}} \) is the estimate of the reproducibility conditions (the sum of the repeatability and the between-condition variance s 2B components).
Starting from Eq. 6, it is possible to write that the variance used to estimate the β-expectation tolerance interval is equal to:
We can develop this equation using Eqs. 7 and 8 yielding:
From the classic theory of ANOVA models for random effects, we know that \( {{\left( {ns^{2}_{{\text{B}}} + s^{2}_{{\text{W}}} } \right)}} \mathord{\left/ {\vphantom {{{\left( {ns^{2}_{{\text{B}}} + s^{2}_{{\text{W}}} } \right)}} {np}}} \right. \kern-\nulldelimiterspace} {np} \) is an estimator of the uncertainty (variance) of the overall mean (or bias) \( \hat{\delta }_{{\text{M}}} \) when a nested design (with p conditions for experiments and n replicates within each condition) is envisaged as it is here. Equation 13 then can be simplified as follows:
where \( \hat{\sigma }^{2}_{{\hat{\delta }_{{\text{M}}} }} \) is the estimated uncertainty (variance) of the estimated bias \(\hat \delta _{\text{M}} .\) So Eq. 14 clearly shows that the variance used for computing the β-expectation tolerance interval is equal to the sum of the total variance of the method plus the variance of the bias. Therefore, as long as the “between-conditions” are the same (laboratories or days), it is clear that Eqs. 14 and 10 account for the same sources of uncertainty: the estimated total variance plus the estimated variance of the bias, meaning that it is then possible to use the standard deviation of the β-expectation tolerance interval as an estimate of the standard uncertainty in the measurements.
This was calculated for each concentration level and reported in Table 5. The relative expanded uncertainty ranges from 11.0 to 8.7% over the whole application domain of the technique.
Conclusion
The lack of generalisation between different validation protocols has led analysts to work out a harmonised approach. However, although the first initiatives widely contributed to improvements in analytical validations, they have resulted in problems regarding the conclusions of the tests carried out and, consequently, any decisions made based on the validity of the analytical procedures.
The procedure based on the construction of an accuracy profile proposes to examine the objectives of the analytical validation, to review some validation criteria, and to propose a visual tool that distinguishes the diagnosis rules and the decision rules. The latter are based on the use of the accuracy profile and the concept of total error. At the same time, this approach allows us to simplify the validation approach of an analytical procedure and to control the risk associated with its use. The common objective is to rationalise the decision-making, to improve the basis for and the documentation of the choices carried out, and therefore improve quality over the long term. This procedure proposes a sufficient but realistic number of experiments. The gain in quality is not obtained by increasing the total cost of the validation process.
On the other hand, a practical and direct way of using the data collected during the validation step to estimate the uncertainty in the measurements can be deduced. This is an interesting and important issue to tackle while the development of easy and simple rules is still the subject of much debate. Being able to provide a good estimate for the measurement uncertainty represents a crucial goal for the coming year. While ISO 17025 [1] makes “customer satisfaction” central to laboratory activity, uncertainty is presented as the key to this satisfaction. Therefore, it is important to have a common experimental procedure that provides critical information on the validity of the method and uncertainty estimates without any extra effort (or additional experiments).
References
ISO/IEC 17025 (2000) General requirements for the competence of testing and calibration laboratories. ISO, Geneva
UK Department of Trade and Industry (1998) Manager’s guide to VAM, valid analytical measurement programme. LGC, Teddington, UK http://www.vam.org.uk
EURACHEM (1998) The fitness for purpose of analytical methods: a laboratory guide to method validation and related topics, 1st edn. EURACHEM, Budapesthttp://www.eurachem.bam.de
Food and Drug Administration (1995) International conference on harmonization, definitions and terminology, Q2A. Federal Register 60:11260–11262
Caporal-Gautier J, Nivet JM, Algranti P, Guilloteau M, Histe M, Lallier M, N’guyen-Huu JJ, Russoto R (1992) Guide de validation analytique SFSTP. STP Pharma Prat 2:205–226
Chapuzet E, Mercier N, Bervoas-Martin S, Boulanger B, Chevalier P, Chiap P, Grandjean D, Hubert P, Lagorce P, Lallier M, Laparra MC, Laurentie M, Nivet JC (1997) Méthodes chromatographiques de dosage dans les milieux biologiques: stratégie de validation. STP Pharma Prat 7:169–194
Chapuzet E, Mercier N, Bervoas-Martin S, Boulanger B, Chevalier P, Chiap P, Grandjean D, Hubert P, Lagorce P, Lallier M, Laparra MC, Laurentie M, Nivet JC (1997) Méthodes chromatographiques de dosage dans les milieux biologiques: stratégie de validation, exemple d’application de la stratégie de validation. STP Pharma Prat 8:81–107
Boulanger B, Dewe W, Hubert P (2000) Objectives of pre-study validation and decision rules, AAPS conference. APQ Open Forum, Indianapolis, IN
Hubert P, Nguyen-Huu JJ, Boulanger B, Chapuzet E, Chiap P, Cohen N, Compagnon PA, Dewe W, Feinberg M, Lallier M, Laurentie M, Mercier N, Muzard G, Nivet C, Valat L (2003) Validation of quantitative analytical procedure, Harmonization of approaches. STP Pharma Prat 13:101–138
Shah VP, Midha KK, Dighe S, McGilveray I, Skelly JP, Yacobi A, Layloff T, Viswanathan CT, Cook CE, McDowall RD, Pittman KA (1992) J Pharm Sci 81:309–312
Food and Drug Administration (2001) Guidance for industry, bioanalytical methods validation. US Food and Drug Administration, Washington, DC, http://www.fda.gov/cder/guidance
Mee RW (1984) Technometrics 26(3):251–253
Satterthwaite FE (1946) Biometrics Bull 2:110–114
Hubert Ph, Chiap P, Crommen J, Boulanger B, Chapuzet E, Mercier N, Bervoas-Martin S, Chevalier P, Grandjean D, Lagorce P, Lallier M, Laparra MC, Laurentie M, Nivet JC (1999) Anal Chim Acta 391:135–148
Chiap P, Ceccato A, Miralles Buraglia B, Boulanger B, Hubert Ph, Crommen J (2001) J Pharm Biomed Anal 24:801–814
EURACHEM/CITAC (2000) Guide: quantifying uncertainty in analytical measurement, 2nd edn. EURACHEM/CITAC, Budapest, http://www.eurachem.ul.pt
Ranson C (2001) Workshop on the experience with the implementation of ISO/IEC 17025, Eurachem-Eurolab, Paris, 4 October 2001
Feinberg M, Montamat M, Rivier C, Lalère B, Labarraque G (2002) Accred Qual Assur 7:409–411
ISO/DTS 21748 (2003) Guide to the use of repeatability, reproducibility and trueness estimates in measurement uncertainty estimation. ISO, Geneva
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Feinberg, M., Boulanger, B., Dewé, W. et al. New advances in method validation and measurement uncertainty aimed at improving the quality of chemical data. Anal Bioanal Chem 380, 502–514 (2004). https://doi.org/10.1007/s00216-004-2791-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-004-2791-y