
Abstract
The paper summarizes work on the development of the high-accuracy RNAA method for the determination of trace amounts of cobalt in biological materials. The method is based on a combination of neutron activation with selective and quantitative isolation of the analyte in a state of high radiochemical purity by use of column chromatography followed by gamma-ray spectrometric measurements. The method was devised according to a set of rules, which were formulated to obtain high accuracy of the method. The procedure has been also equipped with several criteria as key factors in quality assurance. Qualification of the high-accuracy RNAA method as a primary ratio method has been demonstrated and its usefulness in the certification of the candidate reference materials tea leaves and mixed Polish herbs is presented.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the contemporary world, chemical measurements are the basis for making central decisions to enable effective functioning of society. The areas critically dependent on results from chemical analysis are, e.g., environmental control, health, food safety, trade, crime detection, and support for R&D. Hence, there is a growing need to check the reliability of results from chemical analysis. This is of great importance, especially in trace analysis. One way of checking the accuracy of chemical results is the use of primary methods. According to the definition adopted by CCQM, a primary method of measurement is a method having the highest metrological qualities, whose operation can be completely described and understood, and for which a complete uncertainty statement can be written down in terms of SI units. A primary direct method measures the value of an unknown without reference to a standard of the same quantity. A primary ratio method measures the value of a ratio of an unknown to a standard of the same quantity, its operation must be completely described by a measurement equation [1]. For trace analysis the only recognized primary method so far is isotope-dilution mass spectrometry (IDMS) [2, 3]. It has lately been shown that neutron-activation analysis (NAA) in its instrumental mode (INAA) also has potential as a primary ratio method [4, 5, 6]. In this paper, a high-accuracy radiochemical NAA method (RNAA) for determination of Co in biological materials is presented. It is shown that a high-accuracy RNAA method can meet criteria for a primary ratio method.
Experimental
Sample preparation and irradiation
In this study six plant CRM were analysed: tomato leaves NIST 1573a, spinach leaves NIST 1570a, Virginia tobacco leaves CTA-VTL-2, oriental tobacco leaves CTA-OTL-1, tea leaves INCT-TL-1 and mixed Polish herbs INCT-MPH-2. Samples of the biological materials (0.1–0.3 g) were placed in small polyethylene (PE) containers and firmly covered with PE caps. A stock standard solution of Co was prepared from metallic Co of 99.999% purity by weighing an appropriate amount of the metal after removing possible surface contamination and oxide layers by etching with HNO3 and drying, then dissolving in HNO3 (sp. purity), diluting to the appropriate concentration, and weighing the solution obtained. Standards of 0.5 µg Co were obtained by weighing aliquots of freshly prepared standard solution in a PE container and evaporating to dryness before encapsulation. Zn monitors were prepared in the same way as Co standards from stock Zn solutions obtained by use of metallic Zn of 99.999% purity in a manner analogous to that used for Co stock standard solutions.
Calibrated Sartorius MC5, Precisa 40SM 200A, and Sartorius BP221S micro-analytical and analytical balances were used.
To avoid contamination all operations before irradiation were carried out using a laminar flow air cabinet equipped with an HEPA-filter (air class 100).
The package consisting of standards, samples, and empty PE container for determination of residual blank, sandwiched by monitors, was irradiated in the MARIA nuclear reactor for 1 h at a neutron flux of 1×1014 n cm−2 s−1 and cooled for two weeks before processing.
Digestion procedure and radiochemical separation
Samples were digested under pressure in Teflon digestion vessels using a microwave system (Plazmatronika, Poland) and a mixture of 3 cm3 concentrated HNO3, 2 cm3 concentrated (30%) H2O2, and 1 cm3 concentrated (48%) HF. A one-step procedure was performed for 15 min at 100% power (100 W inside digestion vessel); 28 atm was applied. Before digestion, non-active carriers 50 µg Co and 30 µg Fe (sp. purity) were added to the sample. Carrier-free radiotracer 57Co was also added for evaluation of chemical yield. After decomposition the samples were converted into chlorides and subjected to the separation procedure as described elsewhere [7].
To determine the residual blank, the interior of the empty PE container irradiated in the package was washed with the same amount of concentrated HNO3, the washings were then processed in the same way as the samples, including microwave digestion.
Gamma-ray spectrometry
Measurements were made using a gamma-ray spectrometer with HPGe detector (Ortec), active 212 cm 3, resolution 1.8 keV for the 1332.4 keV peak of 60Co, relative efficiency 47%, with an Ortec analogue line and Tukan multichannel analyser (INP, Poland). The same counting vessels were used for the samples and standards. The samples and standards had the same shape and matrix. The distance from the detector was 8 cm.
Results and discussion
High accuracy RNAA method for determination of Co in biological materials
The high-accuracy method for determination of Co in biological materials is based on radiochemical neutron-activation analysis (RNAA) involving selective and precisely quantitative post-irradiation separation of the analyte by column chromatography followed by gamma-ray spectrometric measurements. It was carefully designed and elaborated according to the following set of rules [8] common for a high-accuracy RNAA method.
-
1.
The method should be a single element method, based on neutron activation combined with selective isolation of an element by column chromatography with practically 100% yield as confirmed by tracer experiments.
-
2.
All potential sources of error starting from sampling and sample dissolution up to gamma-spectrometric measurements should be identified at the stage of elaborating the method, and removed or appropriate corrective actions introduced into the procedure.
-
3.
Whenever possible the colour of the ion in question (or its complex) added as a carrier should be used for visual control to safeguard against unexpected failure of the separation procedure.
-
4.
With each set of samples at least two standards should be irradiated, one of which is later processed exactly as are the samples and the other is not. The specific activities of both standards should agree within predetermined limits.
-
5.
Residual blank resulting from the contact of the sample with sample container should be measured in each series of determinations.
-
6.
The method should be universal, i.e. capable of being used without further modifications for the determination of analyte in all kinds of biological material and sensitive enough to ensure a detection limit of the order of a few ng g−1.
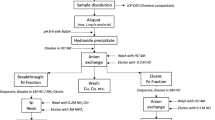
All stages of the method were very carefully studied [9, 10, 11]. Special attention was paid to uncertainty evaluation and reducing uncertainty values at all stages of the method, enabling favourable conditions to be met. The scheme of the elaborated method is presented in Fig. 1. As is normal, the method has been equipped with classification criteria, all of which have to be fulfilled for a result to be accepted. The criteria providing protection against making gross errors were:

Schematic diagram of the high-accuracy RNAA method for determination of Co in biological materials
-
1.
visual control of the correctness of the separation procedure—in the retention stage on both ion-exchange and extraction chromatography columns the blue cobalt band should not travel more than 1/3 of the bed length;
-
2.
small residual blank—the results were accepted if the correction for residual blank determined in a given run was below 5% of the cobalt contents in the sample analysed;
-
3.
agreement of standards—the results for a given series of samples were accepted if the normalized count rates of the two standards, one of which was processed in the same way as the samples and the other of which was measured directly after dissolution, did not differ by more than 5% (after correcting for flux gradient, if any); and
-
4.
the result for the certified material irradiated and analysed with the samples should be in agreement with the certified value.
An important feature of non-destructive NAA is relative freedom from problems related to sample decomposition. Although the above mentioned advantage is lost, the use of radiochemical separation when the indicator nuclides are selectively and quantitatively isolated from the activated samples, significantly improves analytical quality because of reducing the uncertainty of counting statistics and calculation of analytical peak areas in gamma-ray spectra (low background, lack of spectral interference). As post-irradiation separations are carried out, the method is free from contamination compared with separation in non-nuclear methods. The other advantageous features are:
-
performing the chemical operations under controlled conditions by addition of inactive carriers (freedom from trace and ultra-trace concentration behaviour), and
-
accurate yield determination by the use of radiotracer or carrier budgeting.
In RNAA, when the sample is irradiated in the whole reactor neutron spectrum, the mass fraction C m of the element to be determined (x) is given by the equation [12, 13]:
where:
-
A x is the count rate of analytical gamma-ray of indicator nuclide (s−1; A x=N p t c −1, where N p is the net number of counts in the peak corrected for pulse losses and t c is the live counting time);
-
M x is the molar mass of the element determined;
-
W x is the sample mass (g);
-
N A is the Avogadro constant;
-
Θ x is the isotopic abundance of the target nuclei;
-
γ x is the emission probability of the analytical gamma-ray;
-
Φ 0 is the thermal neutron flux (m−2 s−1);
-
Φ e is the epithermal neutron flux (m−2 s−1);
-
σ 0 is the activation cross-section for thermal neutrons;
-
G th is the correction factor for thermal neutron self-shielding;
-
G e is the correction factor for epithermal neutron self-shielding;
-
I 0 is the resonance integral, including 1/ν tail;
-
α is the correction factor for deviation of epithermal neutron flux from the 1/E shape, approximated by a 1/E 1+α function;
-
ε x is the detection efficiency of the analytical γ-ray;
-
Y x is the chemical yield of the separation;
-
S is the saturation factor (S=1−exp(−λt i), where λ is the decay constant and t i is the irradiation time);
-
D is the decay factor (D=exp(−λt d); where t d is the decay time); and
-
C is the measurement factor (C=(1−exp(−λt m)/λt m, where t m is the measurement time).
In the relative standardization method, a standard of known mass of element W st is irradiated with the sample of known mass W x , usually with a neutron-flux monitor and counted under the same geometric conditions using the same HPGe detector. In that case M x =M st, Θ x =Θ st, γ x =γ st, S x =S st, σ x =σ st, and ε x =ε st, and the well known expression is obtained:
This equation is valid when the neutron flux gradient between sample and standard position is negligible or corrected and neutron self-shielding is negligible for sample and standard. In the high-accuracy RNAA method Y x =1, because the determined element is separated quantitatively.
Traceability and uncertainty calculation
The uncertainty budget taking into account all possible sources of uncertainty has been performed using GUM [14] and the Eurachem guide [15]. As has been remarked above, NAA is very well understood and the sources of uncertainty have been established and discussed in the literature [4, 5, 6, 13]. The sources of uncertainty can be grouped into four categories:
-
1.
preparation of the sample, standard, and neutron flux monitor,
-
2.
irradiation,
-
3.
gamma-ray spectrometry measurement, and
-
4.
radiochemical separation.
The standard uncertainties within particular categories connected with individual sources of uncertainty can all be quantitatively evaluated and expressed in SI units [4, 5, 6, 13].
Sample, standard, and neutron flux monitor preparation
In sample, standard, and neutron flux monitor preparation the sources of uncertainty taken into consideration were: mass determination, sample mass changes during weighing, purity and stoichiometry of the chemicals used for preparation of standard and neutron flux monitor, variation of isotopic abundance, and residual blank.
Elemental standards and neutron flux monitors were made from high-purity elements using analytical balances calibrated by national standard weights traceable to the international standard of mass. Their uncertainties are expressed in SI units for mass, kg. A sample of biological material was weighed using a calibrated analytical balance. The uncertainty has been calculated according to producer specification to be at maximum 0.1%. The uncertainties in standard and flux monitor mass come from combining the uncertainties in weighing high-purity metals and standard stock solutions, using a calibrated analytical balance, and standard and flux-monitor solutions (20 mg and 50 mg, respectively), using a calibrated microanalytical balance. The producer specifications were taken into account during combined uncertainty evaluation resulting from the above measurements. This uncertainty has been calculated to be 0.1%. The uncertainties associated with the other sources of uncertainty in the stage of preparation of the sample, standard, and monitor as—change of sample mass during weighing due to moisture changing, variation of isotopic abundance, stoichiometry, and purity—can be neglected. It was found that the analysed plant materials were not hygroscopic. Also, because only CRM with homogeneity guaranteed by the producers were used and the sample masses did not exceed those recommended by the manufacturers, the uncertainty attached to inhomogeneity has been neglected. Co has only one natural isotope and both standard and neutron flux monitor solutions were prepared from metals of 99.999% purity. Before weighing the surface of metal grains was purified. The residual blank was stable and negligible. Uncertainty resulting from residual blank correction was less than 0.1%. Hence, the combined uncertainty attached to sample, standard, and neutron flux monitor preparation was equal to 0.2%.
Irradiation
The uncertainty sources taken into account within the irradiation step are differences in irradiation geometry and neutron spectrum in time and space including neutron self-shielding and scattering, differences in irradiation time, nuclear reaction interferences, and volatilization losses during irradiation.
Differences of the neutron flux caused by the flux gradient in space are determined and corrected for by use of sandwich monitors. The standard uncertainty attached to this correction has been evaluated to be 0.1%. Self-shielding and scattering effect are usually negligible in biological materials. For typical biological, geological, and environmental samples of typical size (250 mg) uncertainty contribution from thermal neutron self-shielding is less than 0.2% [5]. The uncertainty in epithermal neutron self-shielding (the resonance integral for Co I=75.5 b), neutron scattering, and thermalization can be neglected. No uncertainty comes from differences of neutron spectrum in time and duration of irradiation if samples and standards are irradiated together.
In Co determination by the NAA method interfering nuclear reactions must be taken into account: 60Ni (n,p) 60Co, 63Cu (n,α) 60Co and 58Fe (n,γ) 59Fe\( \xrightarrow{\beta } \) 59Co (n,γ) 60Co. The interference contributions of these reactions were measured and appropriate corrections were found to be 2.2×10−6 g Co g−1 Cu, 4.1×10−6 g Co g−1 Ni and 1.0×10−6 g Co g−1 Fe. Taking into consideration the low concentrations of Cu, Ni, and Fe in most biological materials, the uncertainty from the corrections for the interfering nuclear reactions can be neglected. No uncertainty comes from volatilization losses because cobalt is not volatile and does not form easily volatile compounds.
Finally, the combined uncertainty associated with the irradiation step is 0.22%.
Gamma-ray spectrometric measurement
Sources of uncertainty related to gamma-ray spectrometry measurement are: counting statistics, blank correction, differences in counting geometry and time, pulse pile-up losses, cascade summing, effects of dead-time and decay timing, gamma-ray interferences and self-shielding, and peak integration.
The uncertainty introduced due to the counting statistics of induced activity of 60Co in both samples and standards has been calculated using a Poisson distribution: u co=100(N p+2B)1/2/N p, where B is the background. More than 30,000 counts were accumulated when counting each sample and standard. It can easily be realized when the activity of a practically single radionuclide is measured. Moreover, an effect of variability in response from the Compton continuum on the baseline is neglected. The associated uncertainty is less than 0.6%. The uncertainty from differences in counting positioning were evaluated by repeated measurement of the same sample at given geometry with repositioning after each measurement. The observed range was 100%±0.7%. The relative uncertainty is equal to 0.3% assuming triangular distribution. Because the proper hardware was used (fast ADC) and because of the very low dead time of both sample and standard, the uncertainty resulting from pulse pile-up losses is less than 0.1%. Moreover, the uncertainty resulting from differences in dead time between sample and standard can be neglected. Due to long half-live of 60Co (T 1/2=1925±0.5 days) the uncertainties from differences in measurement time between the sample and standard and decay during measurements are negligible. Self-shielding of the 1173.2 keV and 1332.4 keV photons by the PE walls of the measurement flask is negligible. Activity of 60Co in the background was not observed. The uncertainty in the calculation of peak areas in gamma-ray spectra was evaluated from differences between peak areas calculated using TUKAN software and hand integration channel by channel. This was equal to 0.2%, assuming rectangular distribution for both sample and standard.
Combined standard uncertainty in gamma-ray spectrometry measurement amounts to 1.2%.
Radiochemical separation
The method was designed to give precisely quantitative separation of analyte (100% chemical yield, Y x =1 in Eq. 2). The radiochemical yield was confirmed by using the 57Co radiotracer. The recovery was measured by replicate analysis of different biological materials. The yield varies from 99.4 to 100.7%. Assuming a rectangular distribution the uncertainty attached to radiochemical separation is 0.4%.
Combined standard uncertainty
The combined standard uncertainty was calculated according to uncertainty propagation law [12, 13]. Because Eq. (2) contains only multiplication and division of quantities, the combined standard uncertainty can be calculated as the square root of the sum of the squares of the relative standard uncertainties. Under favourable conditions, it amounted to 1.3%.
The budget for calculation of the combined uncertainty for Co determination in the new Polish CRM of biological origin, tea leaves (INCT-TL-1), is presented in Table 1.
The expanded uncertainty (k=2, 95% confidence level) is equal to 2.6%. Hence, the high-accuracy RNAA method can be recognized as a method of the highest metrological quality. The established expanded uncertainty (2.6%) is comparable with values characteristic of IDMS. Hence, the high-accuracy RNAA method can be complementary to IDMS especially for the determination of elements naturally occurring as a single -isotope, for which IDMS cannot be used.
Highly accurate (primary and its predecessors—absolute and definitive) methods play an important role in quality assurance. They can be used for checking the accuracy of routine analytical methods and may be a valuable tool in the process of certification of reference materials.
In our laboratory the certification of the candidate reference materials is performed on the basis of world-wide interlaboratory comparison and proven data evaluation methodology [16, 17, 18]. The uncertainty associated with the central value combines the uncertainty due to dispersion of analytical results obtained by various methods (after rejection of outliers), i.e. analytical variance, and the uncertainty due to estimation of long term stability of the CRM [17, 18].
In Table 2 the results for Co determination from the certification campaign of the two Polish CRM, tea leaves (INCT-TL-1) and mixed Polish herbs (INCT-MPH-2), are shown together with results from the primary ratio method described above. In that case the uncertainty arising from moisture correction has been included in the combined uncertainty. The uncertainty due to moisture correction has been experimentally found to be 1.0% for INCT-TL-1 and 1.2% for INCT-MPH-2. These values were established by replicate determination of moisture carried out according to the procedures given in the certificates provided by the producers. In fact, these procedures (drying in an oven at a given temperature for fixed time) enable establishment of the common mass base only. They are not involved in the radiochemical separation procedure itself but should be included when giving total uncertainty for a given biological material. As can be seen, for both materials the results from the primary method together with their uncertainties are well within the uncertainties assigned to the central values during the certification campaign. Thus, the primary method, traceable to SI units, confirmed the correctness of the data-evaluation procedure used in our Laboratory.
The uncertainty associated with the result obtained by use of a primary method is usually very low, as has been demonstrated in this paper. If, however, one wants to use such a method alone for certification of the Co content of the natural matrix CRM, additional sources of uncertainty would have to be taken into account. These include uncertainties associated with long-term stability and moisture-content determination.
Conclusions
This paper has demonstrated the possibility of using the high-accuracy RNAA method as a primary ratio method. The method satisfies the traceability requirements of a CCQM definition and provides results with very low levels of uncertainty. The method of Co determination in biological materials has been presented as an example. The expanded uncertainty of the analytical procedure itself amounts to 2.6% which is characteristic of a method of the highest metrological quality. The method can be recommended for very accurate determination of Co in biological materials, including certification of CRM. In this case some additional uncertainty associated with the determination of moisture content must be included when calculating the expanded uncertainty.
This and similar RNAA methods, if devised, could become complementary to IDMS methods, being perhaps the ideal choice for monoisotopic elements.
References
BIPM (1998) Proc 4th meeting of CCQM. Bureau International des Poids et Mesures, Paris, France, p 71
Catteric T, Craston D, King B, Walker RF, Webb KS (1999) Accred Qual Assur 4:3–13
De Bievre P (1994) Fresenius J Anal Chem 350:277–2883
Tian W, Ni B, Wang P, Cao L, Zhang Y (2001) Accred Qual Assur 6:488–492
Tian W, Ni B, Wang P, Cao L, Zhang Y (2002) Accred Qual Assur 7:7–12
Bode P, De Nadai Fernandes EA, Greenberg RR (2000) J Radioanal Nucl Chem 245:109–114
Dybczyński R, Danko B, Maleszewska H (1994) J Anal Chem (Moscow) 49:37–44
Dybczyński R, Danko B (1994) J Radioanal Nucl Chem 181:43–59
Polkowska-Motrenko H, Dybczyński R, Danko B, Becker DA (1996) J Radioanal Nucl Chem 207:401–412
Polkowska-Motrenko H, Danko B, Dybczyński R, Koster-Ammerlaan A, Bode P (2000) Anal Chim Acta 408:89–95
Danko B, Polkowska-Motrenko H, Dybczyński R (2000) J Radioanal Nucl Chem 246:279–283
Simonits A, Moens L, De Corte F, De Wispelaere A, Elek A, Hoste J (1980) J Radioanal Nucl Chem, 60:461–516
Kučera J, Bode P, Štěpánek V (2000) J Radioanal Nucl Chem 245:115–122
ISO (1993) Guide to the expression of uncertainty in measurements. International Organization for Standardization, Geneva
Eurachem–CITAC (2000) Quantifying uncertainty in analytical measurement, 2nd edn. Eurachem, Teddington, UK
Dybczyński R, Polkowska-Motrenko H, Samczyński Z, Szopa Z (1998) Fresenius J Anal Chem 360:384–387
Dybczyński R, Danko B, Kulisa K, Maleszewska E, Polkowska-Motrenko H, Samczyński Z, Szopa Z (2002) Preparation and certification of the Polish reference material: tea leaves (INCT-TL-1) for inorganic trace analysis, Raporty IChTJ, Seria A, nr 3/2002. Institute of Nuclear Chemistry and Technology, Warsaw
Dybczyński R, Danko B, Kulisa K, Maleszewska E, Polkowska-Motrenko H, Samczyński Z, Szopa Z (2002) Preparation and certification of the Polish reference material: mixed polish herbs (INCT-MPH-2) for inorganic trace analysis, Raporty IChTJ, Seria A, nr 4/2002. Institute of Nuclear Chemistry and Technology, Warsaw
Acknowledgements
This work was financially supported in part by the Polish State Committee for Scientific Research grant No. 3T0900199C/4265.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Polkowska-Motrenko, H., Danko, B. & Dybczyński, R. Metrological assessment of the high-accuracy RNAA method for determination of cobalt in biological materials. Anal Bioanal Chem 379, 221–226 (2004). https://doi.org/10.1007/s00216-003-2429-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-003-2429-5