
Abstract
Determination of low molecular weight organic acids in soils and plants by capillary zone electrophoresis was accomplished using a phthalate buffer and indirect UV detection mode. The influence of some crucial parameters, such as pH, buffer concentration and surfactant were investigated. A good separation of seven organic acids was achieved within 5 min using an electrolyte containing 15 mmol L−1 potassium hydrogen phthalate, 0.5 mmol L−1 myristyltrimethylammonium bromide (MTAB), and 5% methanol (MeOH) (v/v) at pH 5.60, separation voltage −20 kV, and temperature 25 °C. The relative standard deviation (n=5) of the method was found to be in range 0.18–0.56% for migration time and 3.2–4.8% for peak area. The limit of detection ranged between 0.5 µmol L−1 to 6 µmol L−1 at a signal-to-noise ratio of 3. The recovery of standard organic acids added to real samples ranged from 87 to 119%. This method was simple, rapid and reproducible, and could be applied to the simultaneous determination of organic acids in environmental samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Low molecular weight organic acids present in water, soils, and plants play important roles due to their capacity to complex with metals and release protons. The low molecular weight organic acids found in the environment comprise mono-, di-, and tricarboxylic acids including compounds containing unsaturated carbon and hydroxyl groups. The low-molecular-weight organic acids function as ligands increasing the total amount of dissolved cations such as aluminium and iron in soil solutions by complexing of metal cations [1, 2, 3]. Chemical conditions in the rhizosphere soils are often different from the bulk soil as plant roots exude organic compounds including low-molecular-weight organic acids [4, 5].
The common methods for determination of low-molecular-weight organic acids include gas chromatography (GC) [6, 7], ion-exclusion chromatography (IEC) [8, 9] and high-performance liquid chromatography (HPLC) [10, 11]. Traditionally, organic acids have been analyzed by using GC methods with derivatization steps to increase their volatility before GC separation. The derivatization process is often quite tedious, and retards the reproducibility of the analysis. Ion exclusion chromatography with various detection techniques was developed for the analysis of weak and medium strength acids with a long run time (at least 30 min), leading to large volumes of eluents required. Additionally, the IEC column used was expensive and would last for up to a few thousand injections. For the last two decades, HPLC has been a common technique for the determination of organic acids. The major disadvantages are clogging problems with sample and complex pretreatment with HPLC. Thus, the methods above mentioned lack specificity and ruggedness for routine analyses.
In recent years, capillary zone electrophoresis (CZE) has become a powerful separation technique and is used more and more as a standard analytical tool for organic acids [12, 13, 14]. Advantages of the CZE technique include small sample volume requirement, the robustness of the capillary to samples containing humic substances, and the low expense of replacement of old or damaged capillaries. In previous papers, excellent separations were reported with different background electrolytes (BGEs) including chromate [15, 16], phthalate [12, 17, 18], benzoic [19], trimellitic [13, 20] and pyromellitic [21] acids used for indirect UV detection and borate [22], phosphate [23], carbonate [24] for direct UV detection. The limitation of direct UV detection in CZE is that the organic acids have poor chromophore properties or none at all. Therefore, much attention was focused on CZE analysis with indirect UV detection. The indirect detection technique relied on the presence of a UV-active buffer component with the same charge as the analytes. Since the introduction of indirect UV detection by Hjertén et al. [25], chromate [15, 16], phthalate [12, 17, 18], benzoic [19], etc., have been successfully applied to detect organic acids as chromophoric electrolytes. Indirect UV detection has become a popular detection mode for all kinds of analytes in electrophoresis, due to its higher sensitivity [17, 18, 19, 20, 21, 26]. In real samples, many interfering peaks can be observed due to the complicated sample matrix. Pretreatments such as ultrafiltration and centrifugation are needed to eliminate contaminants. When the concentrations of solutes are relatively low, organic acids are extracted from the sample solution by adding water-immiscible organic solvents such as ethyl acetate or by solid-phase extraction (SPE) procedures, adsorbing and eluting through a solid-phase C18 column [27]. However, SPE procedures prolonged the overall analysis time and contributed to additional errors to the results. The detection sensitivity is relatively low. In our experiment, the pretreatment is relatively simple due to the use of a sensitive method with a lower LOD.
This paper describes CZE separation of organic acids using phthalate, MTAB and MeOH as buffer. In our study an efficient method was developed for the qualitative and quantitative determination of low molecular weight organic acids in environmental samples, with minimal sample preparation and analysis time. The experimental results show great potential for wide application in the future.
Experimental
Instrument
The instrument used was a P/ACE MDQ capillary electrophoresis system from Beckman (Fullerton, CA, USA), equipped with a UV detector. The electrophoretograms were recorded and integrated by an IBM personal computer with 32 Karat software version 4.0 (Beckman). All separations were performed in a fused-silica capillary of 75 μm i.d. and total length of 57 cm (YongNian, China). A detection window was created 50 cm from the capillary inlet by removing the polyimide coating. The UV detector was set at 254 nm.
Electrophoretic procedures
A new capillary was firstly pretreated by passage of 0.1 mol L−1 HCl for 10 min, and deionized water for 5 min. Before each run the capillary was rinsed according to the following cycle: 0.1 mol L−1 NaOH for 5 min, deionized water for 3 min, and then running background electrolyte for 5 min. The hydrostatic injection mode (5 s, 20 psig) was used for the standard solution and sample injection. The capillary was thermostatted at 25 °C, and the applied constant voltage was set at –20 kV. Detection was carried out using a UV detector at 254 nm.
Chemicals and sample preparation
All organic acids, including formic, tartaric, malic, citric, succinic, acetic and lactic acids were purchased from Beijing Chemical (China). Potassium hydrogen phthalate was obtained from Shanghai Chemical Factory (China). Myristyltrimethylammonium bromide (MTAB) was purchased from Kanto Chemicals (Tokyo, Japan). All reagents were of analytical-reagent grade. Deionized water obtained from a Milli-Q purification system with a 0.2 μm fiber filter (Barnstead, USA) was used for preparation of sample and buffer solutions.
Soils were shaken with water (2 g/4 mL) on a mechanical shaker for 4 h. Crushed plant roots were extracted with 4 mL water (0.4 g/4 mL) in a water bath held at 50 °C for 1 h [17]. The extracts were then centrifuged at 1760 g for 10 min and the supernatants were filtered through a Millipore 0.45 μm membrane filter before injection into the CZE system.
Results and discussion
Effect of BGE
In CZE analysis, selection of the BGE is important for indirect UV detection. To achieve high separation and detection sensitivity, the mobility of the BGE should match the mobility of the organic acids as close as possible. Phthalate with a medium mobility matched the mobility of most organic acids. Hence phthalate was chosen as the suitable BGE for subsequent studies.
The influence of BGE concentration on the mobility of organic acids is shown in Fig. 1. It could be seen clearly that the mobility decreased when BGE concentration increased from 5 to 25 mmol L−1. This could be attributed to the fact that the electroosmotic flow (EOF) was reduced by the higher ionic strength. When the BGE concentration was 10 mmol L−1 and below, malic acid and citric acid could not be baseline separated. Increasing the BGE concentration improved the separation of organic acids, but at the expense of the baseline noise and longer separation time. Therefore, the optimal BGE concentration chosen was 15 mmol L−1, due to the shorter migration times and better separation.

Effect of BGE concentration on the mobility of organic acids. Conditions: fused-silica capillary, total length 57 cm, length to detector 50 cm; buffer phthalate containing 0.5 mmol L−1 MTAB, 5% MeOH, pH 5.6; voltage: −20 kV; detection at 254 nm; temperature 25 °C; hydrodynamic injection 20 psig for 5 s
Effect of electrolyte pH
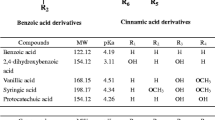
The pH of the electrolyte had a significant impact on the ionization of solutes and BGE. Organic acids have pKa values in the range pH 3 to 6, as shown in Table 1 [28]. So electrolyte pH was tested in this range because the mobility changes of the organic acids were most pronounced between pH 3.6 and 5.8 due to the ionization of the organic acids at their pK a values [14]. In the present experiment the pH was ranged from 4.7 to 5.9, where the mobility of the organic acids was more stable. The influence of pH on the mobility of organic acids is shown in Fig. 2. When the electrolyte pH was below 5.3, the peaks of acetic and lactic acids overlapped. When pH was above 5.7, the mobility of organic acids increased, leading to decrease of resolution. Based on the above study one may conclude that the best resolution and peak shape were attained at the electrolyte pH of 5.6.

Effect of buffer pH on the mobility of organic acids. Conditions: buffer 15 mmol L−1 phthalate containing 0.5 mmol L−1 MTAB, 5% MeOH; other conditions were the same as used in Fig. 1
Effect of surfactant and organic solvent
In CZE separation surfactant and organic solvents are usually added to electrolytes to decrease EOF or reverse the direction of EOF. Previous results were obtained with trimethylammonium bromide (MTAB), hexadecyltrimethylammonium bromide (CTAB) or octadecyltrimethylammonium chloride (OTAC) as surfactant. All of them can successfully reverse the EOF, and similar resolution was obtained. However, the solubility decreased with increasing hydrophobic carbon chain. So MTAB was chosen as the best surfactant. As shown in Fig. 3 the separation of organic acids was little affected by the concentration of MTAB. When MTAB concentration ranged from 0.2 to 0.4 mmol L−1, the electroosmotic mobility increased. Then, the mobility remained almost constant for further higher MTAB concentration. Hence, a 0.5 mmol L−1 MTAB concentration in the electrolyte was chosen, since the sensitivity was affected negatively when the concentration was too high.

Effect of MTAB concentration on the mobility of organic acids. Conditions: buffer 15 mmol L−1 phthalate containing MTAB, 5% MeOH, pH 5.6; other conditions were the same as used in Fig. 1
Generally, organic solvents are used to improve the separation resulting from changes of EOF. In this case, improvement of the separation was studied by adding methanol as organic solvent to the electrolyte. Both resolution and peak shape were improved. To obtain a reasonable migration time, 5% methanol was added to the electrolyte.
CZE separation
The CZE separation of organic acids using indirect UV detection at 254 nm is presented in Fig. 4, where the electrolyte contained 15 mmol L−1 potassium hydrogen phthalate, 0.5 mmol L−1 MTAB, and 5% MeOH at pH 5.6. Seven organic acids were well separated in 5 min with the solutes migrating in the order formic, tartaric, malic, citric, succinic, acetic, and lactic acids.

Electrophoretograms of seven organic acids. Conditions: fused-silica capillary, total length 57 cm, length to detector 50 cm; buffer 15 mmol L−1 phthalate, 0.5 mmol L−1 MTAB, 5% MeOH, pH 5.6; separation voltage: −20 kV; detection 254 nm; temperature 25 °C; electrokinetic injection 5 s, Vinj=−5 kV.Ⅰ. Electrophoretogram of standard sample.Ⅱ. Electrophoretogram of organic acids in real samples. ( A) Heilongjiang soil. ( B) Jiangxi soil. ( C) Chinese cabbage roots. ( D) Chinese garland chrysanthemum roots. Peaks:1. formic acid,2. tartaric acid,3. malic acid,4. citric acid,5. succinic acid,6. acetic acid,7. lactic acid
As shown in Table 2 the peak area was linearly related to the concentration of the tested acids over the range of 0.004–2 mmol L−1. Correlation coefficients (r2) were in the range 0.9960 to 0.9996. Relative standard deviations (R.S.D.) of peak area and migration time were calculated based on five duplicate injections of standard mixtures. The values of reproducibility were between 0.18–0.56% for migration time and between 3.2–4.8% for peak area. The limits of detection (LOD) were obtained at a signal-to-noise ratio of 3, which ranged between 0.5 µmol L−1 and 6 µmol L−1. The high reproducibility and low LOD indicated that the method is reliable for analyses of organic acids.
Analysis of real samples
The proposed method was used to determine the organic acids in different soil, plant and water extracts. Typical electrophoretograms are shown in Fig. 4 and concentrations of organic acids in these samples are listed in Table 3. Acetic and lactic acids were detected in Beijing soil, which is a common soil of northern China. In Heilongjiang and Jiangxi soils, formic, acetic and lactic acids were detected, and the concentrations of acetic acid in these two soils were higher than that in Beijing soil. The LOD of soil samples were 5.2 µmol L−1, 9.7 µmol L−1, and 8.4 µmol L−1 for formic, acetic and lactic acids, respectively. Besides these three acids, malic and succinic acids were also detected in three common kinds of Chinese vegetable roots. Detection limits in plant samples were 5.4 µmol L−1 for formic, 11.7 µmol L−1 for malic, 7.3 µmol L−1 for succinic, 9.6 µmol L−1 for acetic and 8.1 µmol L−1 for lactic acids. Commoelina communis, which is a copper hyperaccumulator, contains much malic acid, possibly due to its particular character. Under hydroponic conditions relatively low contents of formic and lactic acids were detected in Commoelina communis. The LOD of formic and lactic acids in water samples were 5.1 µmol L−1 and 8.4 µmol L−1, respectively. Standard organic acids were added to the real samples before organic acids were extracted from soil and plants. The recovery was found to be 87–119%. It should be pointed out that the migration time of organic acids in real samples differed slightly from the standard organic acids because different matrices in samples might influence the separation. The migration times of organic acids in standards were often different from that of organic acids in real samples. The potential reasons were ascribed to:
-
1.
the surfactants cationic activation decreased; and/or
-
2.
ion concentrations in the buffer changed continuously during the electrophoretic separation process.
All these changes could influence the ionization of solutes, thus mobility of BGEs and migration time could be altered. However, identification of organic acids could be achieved by addition of standard organic acids to the real samples. In addition, a reproducible migration time and peak area could be obtained when the same real samples were successively injected to the CZE system under the operating conditions defined above. Therefore, quantitative analysis could be achieved.
Conclusion
The method presented in this paper is rapid, simple and reliable for the determination of organic acids in environmental samples. In the present experiments five organic acids, including formic, malic, succinic, acetic and lactic acids were quantitatively determined in eight real samples without complicated sample preparation. Compared to other methods of separating organic acids, the recommended method is more suitable for quick determination of organic acids in environmental samples.
References
Hue NV, Craddock GR, Adams F (1986) Soil Sci Soc Am J 50:28–34
Pohlman AA, McColl JG (1988) Soil Sci Soc Am J 52:265–271
Drever JI, Stillings LL (1997) Colloids Surf A 120:167–181
Griffiths RP, Baham JE, Caldwell BA (1994) Soil Biol Biochem 26:331–337
Jones DL (1998) Plant Soil 70:273–286
Morval M, Molnar-Perl I, Knausz D (1991) J Chromatogr A 552:337–344
Limbeck A, Puxbaum H (1999) Int J Environ Anal Chem 73:329–343
Chen J (1996) J Chromatogr 739:273–280
Chen ZL, Glod BK, Adams MA (1998) J Chromatogr A 818:61–68
Qiu JS (1999) J Chromatogr A 859:153–158
Patrick AW, Hees V, Dahlen J, Lundstrom US, Boren H, Allard B (1999) Talanta 48:173–179
Sam PDL, Vindevogel J, Sandra P (1993) J Chromatogr A 652:563–569
Westergaard B, Hansen HCB, Borggaard OK (1998) Analyst 123:721–724
Soga T, Ross GA (1999) J Chromatogr A 834:65–71
Romano J, Jandik P, Jones WR, Jackson PE (1991) J Chromatogr 546:411–421
Krivácsy Z, Molnár A, Tarjányi E, Gelencsér A, Kiss G, Hlavay J (1997) J Chromatogr A 781:223–231
Chen ZL, Tang C, Yu JC (1999) J High Resol Chromatogr 22:379–385
Chen J, Preston BP, Zimmerman MJ (1997) J Chromatogr A 781:205–213
Fung YS, Lau KM (1998) Talanta 45:641–656
Hagberg J, Dahlén J, Karlsson S, Allard B (2000) Int J Environ Anal Chem 78:385–396
Arellano M, Andrianary J, Dedieu F, Couderc F, Puig Ph (1997) J Chromatogr A 765:321–328
Shirao M, Furuta R, Suzuki S, Nakazawa H, Fujita S, Maruyama T (1994) J Chromatogr A 680:247–251
Saavedra L, Gracía A, Barbas C (2000) J Chromatogr A 881:395–401
Cartoni G, Coccioli F, Jasionowska R (1995) J Chromatogr A 709:209–214
Hjertén S, Elentring K, Kilar E, Liao JL, Chen AJC, Sietert J, Zhu MD (1987) J Chromatogr 403:47–61
Kitagishi K, Shintani H (1998) J Chromatogr B 717:327-339
Deng YW, Fan XF, Delgado A, Nolan C, Furton K, Zuo YG, Jones RD (1998) J Chromatogr A 817:145–152
Lide DR, Frederikse HPR (1994) Handbook of Chemistry and Physics, 75th edn. CRC Press, Boca Raton, FL
Acknowledgement
This study was financially supported by the National Natural Science Foundation of China under Grant Nos 20237010, 20177030 and 40171086.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, Yh., Huang, Bx. & Shan, Xq. Determination of low molecular weight organic acids in soil, plants, and water by capillary zone electrophoresis. Anal Bioanal Chem 375, 775–780 (2003). https://doi.org/10.1007/s00216-003-1789-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-003-1789-1