
Abstract
The structural properties and relative stabilities of the Na+Nen clusters, with n = 1–16, have been investigated by using pairwise and density functional theory (DFT) methods. The DFT calculations have been carried out using the M06-2X functional combined with the augmented aug-cc-pVTZ basis sets. For both methods, the high stability is obtained for Na+Ne12 cluster with an icosahedral structure, and the twelve Ne atoms complete the first solvation shell around the Na+ cation. Therefore, the icosahedral Na+Ne12 structure will be considered as the core for the larger sizes. The relative stabilities have been studied in terms of the binding energies, second-order difference of energies, fragmentation energies and HOMO–LUMO energy gaps. The Na–Ne interactions within the Na+Nen clusters in relation to the relative stabilities have been discussed through topological atom-in-molecules and reduced density gradient analyses. Finally, the relative stabilities of the Na+Nen clusters have been discussed topologically.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Van der Waals interactions between rare gas and alkali ions have been frequently studied thanks to the closed shell structure of the noble gas atoms. The study of interactions within clusters and their stabilities is of great importance to understand many phenomena such as nucleation, phase transition and solvation. Also, these mixed clusters are helpful to understand the evolution of many optical, chemical and physical properties from the microscopic levels to the bulk [1]. Cationic clusters M+Rgn (where M is a metal atom and Rg is a rare gas) have been studied both experimentally [1,2,3,4,5,6,7,8,9,10] and theoretically [1,2,3,4, 11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48]. Experimentally, the mass spectrometry method is the most used to evaluate the stabilities of these complexes. The mass spectra obtained by Luder et al. [6] for Na+Arn clusters show the following magic number at n = 6, 8, 10, 13, 16, 20, 23, 25 which refer to the most stable clusters. These numbers were analyzed by Velagrakis et al. [1, 4, 6]. The growing pattern of these clusters consists of a self-arrangement in shells around the centered cation M+ to reach the first solvation layer. At this defined size, we obtain the most stable cluster. In this context, five structures have been found: We quote the tetrahedron for n = 4, the octahedron for n = 6, the square antiprism for n = 8, the capped square antiprism for n = 10 and the icosahedron for n = 12 [1, 4, 6]. The change of cluster geometry as a function of size presents an interesting matter. In fact, the crystals of rare gas exhibit a faced centered cubic structure, whereas clusters with size less than 1000 atoms adopt icosahedral structure [9, 10]. Theoretically, various methods have been implemented to investigate the geometries of the different types of clusters. The most successful methods are those of the basin hopping method into the Monte Carlo simulation [11,12,13, 30, 40,41,42,43] and the genetic algorithm [45,46,47,48]. The DFT calculations have been also well used to analyze different cluster systems, particularly in the case of metal clusters such as CuSin [27, 31, 39], AunBe and AunBe2 [33], GenAl [32], FenN [36], AuSin [37] and FenCr [38]. In the present work, the stable geometries of the Na+Nen clusters have been investigated by using pairwise method and DFT calculations. The relative stabilities of Na+Nen clusters are discussed based on different energetic quantities, such as bending energy, second-order energy difference, fragmentation energy, HOMO–LUMO gap energy and dipole moment. Topological AIM analysis of Na–Ne interactions have been reported and discussed in connection with the stabilities of the Na+Nen clusters.
2 Computational methods
The geometry optimizations of the Na+Nen clusters have been performed by using the basin hopping global optimization and DFT methods. For the first method, we have used the pairwise potential to describe the potential energy surface of the clusters. The pairwise potential is an additive model where the total energy is obtained by a simple addition of the Na+–Ne and Ne–Ne interaction potentials:
The VNe–Ne potential is obtained from the Lennard-Jones one:
where ε and σ are the well depth and the equilibrium distance, respectively. These quantities are ε = 0.00011399 a.u. and σ = 5.10 a.u. The V +Na –Ne potential is taken from the numerical form [49] using the analytical form of Tang and Toennies [50]:
The exponential term describes the short-range repulsion interactions, while the remaining terms express the long-range dispersion ones. The parameters Aeff, b, D4, D6, D8 and D10 result from the square fitting of the numerical potential [49]. The αNe = 2.663 a 30 is the neon’s polarizability.
The DFT calculations have been carried out with the M06-2X exchange–correlation functional and the augmented basis sets aug-cc-pVTZ, as implemented in Gaussian 09 package [51]. Before the optimization, we have used the artificial bee colony (ABC) algorithm which is incorporated into ABCluster algorithm [52, 53] to search the lowest energy structures of Na+Nen clusters. The Lennard-Jones potential has been used for the research of global minimum. For the small cluster sizes n≤6, 100 initial configurations were generated, and from n = 7, more than 500 guess structures are generated. For each size, the geometry having the lowest energy is then optimized with DFT calculations. The M06-2X functional is a reliable hybrid functional, and it has been developed by Zhao et al. [54, 55]. In fact, this functional is suitable to describe the non-covalent interactions and it is able to provide accurate binding energies. In order to test the reliability of our DFT calculations, some test calculations are carried out on Na+–Ne and Ne–Ne using B3LYP, B3PW91 and M06-2X functionals with 6-311(d), 6-311++(d,p), aug-cc-pVDZ and aug-cc-pVTZ basis sets. The calculated Na+–Ne and Ne–Ne bond lengths and available experimental data are listed in Table 1. Comparing with the experimental data [56, 57], we can conclude that M06-2X functional combined with aug-cc-pVTZ basis set is more optimal than others.
3 Results and discussion
3.1 Lowest energy structures
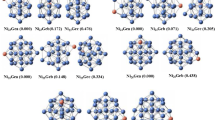
The lowest energy structures of Na+Nen clusters computed by the pairwise method and DFT calculations are depicted in Figs. 1 and 2, respectively. The Na+–Ne and Ne–Ne bond lengths in each cluster are regrouped in Table 2. For the Na+Ne dimer, Na+ and Ne are distant by 2.413 and 2.508 Å for the pairwise and DFT methods, respectively. The trimer Na+Ne2 shows an isosceles triangular geometry (C2v symmetry) with vertex Na+. The tetramer Na+Ne3 cluster has C3v symmetry and exhibits that a pyramidal structure with the Na+ ion is on the summit.

Lowest energy geometries of Na+Nen (n = 1–16) clusters obtained by the pairwise method

Lowest energy geometries of Na+Nen clusters calculated with DFT/M06-2X method
The Na+Ne4 cluster shows a bipyramid structure with the C3v symmetry by using the pairwise method and a tetrahedron structure with Td symmetry with the DFT calculations. The Na+Ne6 cluster shows two distinctly different geometries. We obtained a pentagonal geometry using DFT calculations and an undefined with a pairwise approach. For the Na+Ne8 cluster, both methods have shown a square antiprism structure with D4h symmetry. The Na+Ne10 cluster obtained by the pairwise method presents a double-crown structure; the upper and lower ones possess five and four Ne atoms, respectively. However, the DFT calculations converged to a capped square antiprism structure. The Na+Ne11 cluster is obtained by adding only one Ne atom to the lower side of Na+Ne10 found via the pairwise method.
As clearly seen, in Figs. 1 and 2, the deposition of the neon atoms gradually leads to the formation of the first solvation layer. For both methods, the first solvation layer is obtained for Na+Ne12 cluster, which has an icosahedral structure (C5h). This structure can be considered a core for larger clusters, and then, the number of neon atoms added will gather to build the second solvation layer.
3.2 Relative stabilities
To explore the relative stabilities of the Na+Nen clusters, we have calculated the binding energy per atom Eb, the second-order difference of energies ∆2E and the fragmentation energies ∆E.
For the pairwise method, the binding energy per atom Eb is calculated by using the following formula:
where E(Na+Nen) is the lowest energy corresponding to each cluster.
For the DFT calculations, the binding energy per atom Eb of Na+Nen and Nen clusters is obtained by using the following formulas:
where E(Na+Nen), E(Ne) and E(Na+) are the lowest energies corresponding to each cluster or atom. The binding energies per atom of Na+Nen and Nen are plotted as a function of cluster size as in Fig. 3. For the pairwise method, a pronounced variation is noted for n = 8, which expresses the stability of the Na+Ne8 cluster. We distinguish an increasing linear behavior up to n = 8 which manifests by the domination of Na+–Ne interactions. These clusters continue to gain energy during their growth process. Beyond the first solvation layer, the binding energy per atom decreases progressively with the increase in the cluster size due to the domination of the Ne–Ne interactions. According to the DFT calculations, it can be seen that the binding energy per atom increases with the increase in the cluster size up to n = 9. The binding energy per atom varies slowly for the clusters with n = 8–12, and the curve presents two bumps at n = 9 and 12, implying that these clusters are more stable than their neighbors.

Binding energy per atom of Na+Nen and Nen as a function of the cluster size
For pure neon clusters Nen, one may observe that their binding energy is lower than that for the mixed Na+Nen clusters. Thus, we can conclude that the doping of the Na+ cation to the pure Nen clusters increases its stability. The second-order difference of energy ∆2E is a sensitive quantity that reflects the relative stability of clusters, and the results can be compared with the relative abundances obtained by using mass spectroscopy analysis. ∆2E is defined by the following formula:
where E(Na+Nen−1), E(Na+Nen) and E(Na+Nen+1) represent the total energies of the most stable Na+Nen+1, Na+Nen−1 and Na+Nen clusters. The second-order difference in energies as a function of the cluster size is plotted in Fig. 4. The clusters associated with positive Δ2E values present a high stability compared to their neighboring clusters. For the DFT method, the most stable clusters are found for n = 5, 8, 9, 10, 12, 13 and 15. In the case of the pairwise method we note positive peaks for n = 8, 9, 10 and 12. For both methods, the most pronounced peak of Δ2E is shown for Na+Ne12 cluster implying its high stability, and as a consequence, this cluster is presented at large abundance in mass spectrometry analysis.

Variation of second-order energy difference of Na+Nen as a function of cluster size
Another energetic quantity used to examine the stability of clusters is the fragmentation energy Ef. The fragmentation energy of Na+Nen and Nen clusters is expressed as the following:
The curves are plotted in Fig. 5. As shown in this figure, the most pronounced fragmentation energies are found for Na+Ne8 and Na+Ne12. Hence, these clusters are very difficult to be fragmented and they have a lower reactivity with regard to their neighboring clusters which reflect their high stabilities.

Variation of fragmentation energy of Na+Nen as a function of cluster size
The relative stabilities can be discussed by exploring the energy gaps between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).
Larger HOMO–LUMO energy gap indicates high chemical stability. Figure 6 displays the energy gap for Na+Nen clusters as a function of the number of neon atoms.

Variation of HOMO–LUMO energy gap of Na+Nen clusters as a function of the cluster size
The energy gap curve of Na+Nen presents a maximum at n = 8 and 12, implying the high stability of these clusters. However, smaller values of HOMO–LUMO gap mean the high chemical reactivity of their corresponding clusters. These facts are similar to those obtained by using binding energy, first-order energy difference, the second-order difference in energy and the fragmentation energy discussed above.
The dipolar properties are of great importance to understand the interatomic interactions and to study electrostatic and optical properties of the clusters. The DFT approach is considered as an interesting method to determine these properties with a good accuracy, contrary to other quantum methods which find many challenges in this determination [58, 59]. The dipole moment as a function of clusters size for the Na+Nen complexes is computed by DFT method and is depicted in Fig. 7. As clearly seen, all clusters present a polar character except for Na+Nen (n = 8, 9, 10 and 12), which possesses a very weak value of dipole moment, near to zero. The Na+Ne2 cluster presents the highest dipole moment (5.86 D), and then the dipole moment fluctuates as a function of the number of neon atoms and presents an irregular variation. The clusters presenting a large dipole moment have a high charge separation [60]. According to Fig. 7, the Na+Ne8 and Na+Ne12 clusters have small values of dipole moment, i.e., a low charge separation due to their high stabilities.

Dipole moment of Na+Nen as a function of cluster size
3.3 Topological analysis
The Na–Ne interactions within Na+Nen clusters are analyzed through a topological atom-in-molecules (AIM) analysis [61]. According to AIM approach, the interaction between two neighboring atoms is characterized by the presence of a bond critical point (BCP). In fact, the presence of the BCP point (3, − 1) is indispensable to bind between the two atoms. The topological parameters found at the BCP allow us to conclude about the nature and the strength of such interaction. The electron density ρBCP and its Laplacian ∇2ρBCP at a given BCP are the chief parameters. In the AIM approach, the interactions are classified into two types: the closed shell interaction (non-covalent interaction), such as hydrogen bond, ionic and van der Waals interactions, and the shared shell interaction (covalent interaction). The sign of the Laplacian ∇2ρBCP at the BCP (3, − 1) is an important indicator which informs about the type of binding. Non-covalent interactions are generally defined by ρ(r) < 0.1 a.u. and ∇2ρ(r) > 0 a.u. [62]. For van der Waals interaction, in requirement to the Pauli exclusion principle the electron density is contracting away from the interatomic surface that allows to low values of electron density of about 10−3 a.u. [62]. In Fig. 8, we display the electron density and its Laplacian, respectively, as a function of cluster size. Accordingly, all electron density values computed at Na–Ne BCPs are smaller, of order of 10−3 a.u., and their Laplacian are positive, which confirms the van der Waals interactions within the investigated clusters. The electron density and its Laplacian curves present remarkable peaks for n = 4, n = 8 and 12. Upper to the last size, these topological parameters appear closely constant. One may conclude that high stabilities of Na+Ne8 and Na+Ne12 have been proved by the large values of the electron density and its Laplacian compared to their neighboring sizes. The average topological energy ENa–Ne of the Na–Ne interactions is plotted in Fig. 9. For all studied sizes the values of ENa–Ne are found in the range of 5 and 10 kJ/mol which confirm the van der Waals interactions between Na and Ne atoms. The topological energy curve shows pronounced peaks for the sizes n = 4, 8 and 12 which explain the high stabilities of these clusters. Additionally, the energy density H(r) is a complement indicator to distinguish the type of interactions. The positive values H(r) indicate the non-covalent interactions, and the negative values correspond to covalent interaction. As shown in Fig. 10, our results are typical for non-covalent interactions. It is remarkable that the values of H(r) obtained for Na+Ne4, Na+Ne8 and Na+Ne12 clusters are greater than those for the neighboring clusters, confirming the previous topological results. Further, another indicator used in this work to confirm the type of interactions is the ratio G(r)/ρ (G(r) is the Lagrangian kinetic energy at the BCP). The values of ρ(r), G(r) and G(r)/ρ are regrouped in Table 3. As reported in this table, G(r)/ρ values are greater than unity (G(r)/ρ > 1) defining the non-covalent binding into the Na+Nen clusters.

Size dependence of the electron density and its Laplacian of the Na+Nen clusters

Variation of the average interaction energy at Na–Ne BCPs as a function of the cluster size

Variation of the average energy density H(r) as a function of the cluster size
3.4 Reduced gradient density
The topological RDG analysis is generally used to identify the nature of intermolecular interactions (such as hydrogen bonding, van der Waals and repulsive steric interactions) [63]. The VMD code provides a visualization of the RDG isosurface in which the type of interactions is determined by color coding: The blue color shows the attractive interactions, the red color is used for the repulsive interactions, and the green color defines the van der Waals interactions. Figure 11 presents the RDG function vs the electron density ρ multiplied by the sign of the second largest value of Hessian matrix of electron density, λ2, for the Na+Nen clusters. The position and the width of the observed spikes allow us to conclude about the interatomic interactions within the cluster. Here, the RDG analysis is performed at isosurface equal to 0.5. Obviously, the peaks take place in the range of [− 0.01; 0.005] and exhibit well the van der Waals interactions in the Na+Nen clusters. This fact is confirmed by the green color in the RDG isosurface. As seen in Fig. 11, at 0.5 isosurface value, the spikes width varies from one cluster to another. The smaller clusters until n = 12 possess only one peak localized in the repulsive region (λ2 > 0) and two peaks in the attractive region (λ2 < 0) closely between − 0.002 and − 0.01 which are much wider than the repulsive peaks. The attractive effect increases by going to n = 10 and then decreases for n = 11. At this size (n = 11), we remark that the attraction effect is slightly more important than the repulsive one. For n = 12, equal contributions between attraction and repulsive interactions explain probably the stability of the Na+Ne12 cluster. Above this size, the spikes in the attractive region increase again and are larger than the repulsive peaks. These observations are in good agreement with the topological analysis.



Reduced density gradient (RDG) function versus the sign of second eigenvalue λ2 and VMD determination of the RDG isosurfaces (inside plot) for the Na+Nen clusters
4 Conclusions
In this work, we have presented a structural study of the Na+Nen clusters. The geometrical optimization has been carried out by using the pairwise approach and DFT calculation. For the pairwise method, the core–core Na+–Ne potential is fitted by the use of the Tang and Toennies analytical form, and for the Ne–Ne interaction, we used the Lennard-Jones potential. The DFT calculation was performed at M06-2X/aug-cc-pVTZ level of theory. For small clusters (n = 1–12), the comparison between the two methods displays systematically the same structures with a small difference in their geometrical parameters except for n = 6; the geometry obtained by the DFT is found to be more stable than that in the pairwise method. The first solvation layer is closed at n = 12, and it corresponds to the icosahedral structure (C5h). The stability of Na+Nen clusters is discussed in terms of binding energy, first- and second-order derivative energy, fragmentation energy and HOMO–LUMO energy gap. Accordingly, the icosahedral structure Na+Ne12 is found to be the most stable cluster. Also, the results show that all Na+Nen clusters present a polar character. The Na+Ne8 and of Na+Ne12 clusters possess a low value of dipole moment and thus a low charge separation related to the high stability of these clusters. Topological analysis has been performed using the AIM approach, and it gives a positive Laplacian of electron density revealing the van der Waals interaction within the clusters. Reduced density gradient analysis (RDG) was also performed. The RDG plots show the dominance of the attractive effect regarding the repulsive one. Exceptionally for n = 12, we note that equal contribution between attractive and repulsive interactions within the Na+Ne12 cluster may be responsible to its high stability.
References
Prekas D, Luder C, Velegrakis M (1997) J Chem Phys 108:4450
Froudakisa GE, Farantosa SC, Velegrakis M (2000) Chem Phys 258:13
Ryan M, Collier M, de Pujo P, Crépin C, McCaffrey JG (2010) J Phys Chem A 114:3011
Fanourgakis GS, Farantos SC, Lüder C, Velegrakis M, Xantheas SS (1998) J Chem Phys 109:108
Velegrakis M, Luder C (1994) Chem Phys Lett 223:139
Luder C, Prekasf D, Velegrakis M (1997) Laser Chem 17:109
Wright TG, Breckenridge WH (2010) J Phys Chem A 114:3182
Whetten RL, Schriver KE, Persson JL, Hahn MY (1990) J Chem Soc Faraday Trans 86:2375
Farges J, De Feraudy MF, Raoult B, Torchet G (1983) J Chem Phys 78:5067
Farges J, De Feraudy MF, Raoult B, Torchet G (1986) J Chem Phys 84:3491
Gaied W, Ben El Hadj Rhouma M (2011) Int J Quantum Chem 111:652
Issa K, Issaoui N, Ghalla H, Yaghmour SJ, Mahros AM, Oujia B (2016) J Mol Phys 114:118
Bilalbegović G (2003) Phys Lett A 308:61
Di Paola C, Sebastianelli F, Bodo E, Baccarelli I, Gianturco FA (2005) J Chem Theory Comput 1:1045
Sebastianelli F, Bodo E, Baccarelli I, Di Paola C, Gianturco FA, Yurtsever M (2006) J Comput Mater 35:261
Sapse AM, Dumitra A, Jain DCA (2003) J Clust Sci 14:21
Gardner AM, Withers CD, Graneek JB, Wright TG (2010) J Phys Chem A 114:7631
Bu X, Zhong C, Jalbout AF (2004) Chem Phys Lett 387:410
Xinping B, Chongli Z (2004) Chem Phys Lett 392:181
Abdessalem K, Habli H, Ghalla H, Yaghmour SJ, Calvo F, Oujia B (2014) J Chem Phys 141:154308
Hernández-Rojas J, Wales DJ (2003) J Chem Phys 119:7800
Fujisaki A (2002) J Chem Phys 117:5271
Nagata T, Aoyagi M, Lwata S (2004) J Phys Chem A 108:683
Giju KT, Roszack S, Gora RW, Leszczynski J (2004) Chem Phys 391:112
Fujisaki A (1995) J Chem Phys 102:8485
Breckenridge WH, Ayles VL, Wright TG (2007) Chem Phys 333:77
Lan YZ, Feng YL (2009) Phys Rev A 79:033201
He J, Wu K, Sa R, Li Q, Wei Y (2010) Chem Phys Lett 490:132
He J, Wu K, Liu C, Sa R (2009) Chem Phys Lett 483:30
Issaoui N, Abdessalem K, Ghalla H, Yaghmour SJ, Calvo F, Oujia B (2014) J Chem Phys 141:174316
Gueorguiev GK, Pacheco JM, Stafstrom S, Hultman L (2006) Thin Solid Films 515:1192
Shi S, Liu Y, Zhang C, Denga B, Jiang GA (2015) Comput Theor Chem 1054:8
Bhattacharjee D, Mishra BK, Chakrabartty AK, Deka RC (2014) Comput Theor Chem 1034:61
Liu Y, Tiana Z, Cheng L (2016) RSC Adv 6:4705
Chuang FC, Hsu CC, Hsieh YY, Albao MA (2010) Chin J Phys 48:82
Li Z, Zhao Z (2017) Mater Chem Phys 187:54
Li GL, Ma WL, Gao AM, Chen HY, Finlow D, Li QS (2012) J Theor Comput Chem 11:185
Maa QM, Xie Z, Wang BR, Liu Y, Li YC (2011) Solid State Commun 151:806–810
Lin L, Yang J (2015) J Mol Model 21:155
Wales DJ, Doye JKP (1997) J Phys Chem A 101:5111
Naumkin FY, Wales DJ (1998) Chem Phys Lett 290:164
Doye JPK, Wales DJ (1998) Phys Rev Lett 80:1357
Leary RH, Doye JPK (1999) Phys Rev E 60:R6320
Naumkin FY, Wales DJ (2000) Mol Phys 98:219
Wolf MD, Landman U (1998) J Phys Chem A 102:6129
Xiao Y, Williams DE (1993) Chem Phys Lett 215:17
Zeiri Y (1995) Phys Rev E 51:R2769
Deaven DM, Tit N, Morris JR, Ho KM (1996) Chem Phys Lett 256:195
Ahlrichs R, Böhm HJ, Brode S, Tang KT, Toennies JP (1988) J Chem Phys 88:6290
Tang KT, Toennies JP (1984) J Chem Phys 80:3726
Frisch MJ, Trucks GW, Schlegel HB et al (2013) Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford, CT
Zhang J, Dolg M (2015) ABCluster: the artificial bee colony algorithm for cluster global optimization Phys. Chem Chem Phys 17:24173–24181
Zhang J, Dolg M (2016) Global optimization of clusters of rigid molecules using the artificial bee colony algorithm. Phys Chem Chem Phys 18:3003e10
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215
Zhao Y, Truhlar DG (2006) J Phys Chem A 110:13126
Bililign S, Gutowski M, Simons J, Breckenridge WH (1994) J Chem Phys 100:8212
Aziz RA, Slaman MJ (1989) Chem Phys 130:187
Reis H, Papadopoulos MG, Boustani I (2000) Int J Quantum Chem 78:131
Hickey AL, Rowley CN (2014) J Phys Chem A 118:3678
Dutta A, Mondal P (2017) Comput Theor Chem 1115:284
Bader RFW (1990) Atoms in molecules: a quantum theory. Oxford University Press, Oxford
Pakiari AH, Eskandari KJ (2006) J Mol Struct THEOCHEM 759:51
Johnson ER, Keinan S, Paula MS, Julia CG, Cohen AJ, Yang WT (2010) J Am Chem Soc 132:6498
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ben Hadj Ayed, M., Osmani, T., Issaoui, N. et al. Structures and relative stabilities of Na+Nen (n = 1–16) clusters via pairwise and DFT calculations. Theor Chem Acc 138, 84 (2019). https://doi.org/10.1007/s00214-019-2476-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-019-2476-4