
Abstract
Eight density functionals, CAM-B3LYP, \(\hbox {LC-}\omega \hbox {PBE}\), M11, MN12SX, N12SX, \(\omega \hbox {B97}\), \(\omega \hbox {B97X}\), and \(\omega \hbox {B97XD}\), related to the Def2TZVP basis sets, were assessed together with the SMD solvation model for the calculation of the molecular properties and structure of the therapeutic peptide of marine origin Parasin I. All the chemical reactivity descriptors for the system are calculated via conceptual density functional theory (CDFT). The active sites suitable for nucleophilic, electrophilic, and radical attacks are selected by linking them with the Fukui function indices, nucleophilic and electrophilic Parr functions, and condensed dual descriptor \(\Delta {f(r)}\), respectively. The study reveals that the MN12SX and N12SX density functionals are the most appropriate ones for predicting the chemical reactivity of the molecule under study. Additionally, the pKa value for the peptide is predicted with great accuracy based on our previously published methodology. Moreover, the ability of the studied molecule in acting as an efficient inhibitor of the formation of advanced glycation endproducts (AGEs), which constitutes a useful knowledge for the development of drugs for fighting diabetes, Alzheimer and Parkinson diseases is also presented. Finally, the bioactivity scores for Parasin I are predicted through different methodologies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
For the consideration of therapeutic peptides from the point of view of medicine, it is necessary to know their molecular properties and their bioactivity. It is our belief that the bioactivity of these peptides is intimately related to their chemical reactivity from a molecular perspective. For this reason, we consider it essential to study the chemical reactivity of natural products that have the potential to become medicines through the tools provided by computational chemistry and molecular modeling. Probably, the most powerful tool currently available to study the chemical reactivity of molecular systems from the point of view of computational chemistry and molecular modeling is conceptual DFT [1, 2], also called chemical reactivity theory, which using a series of global and local descriptors allow to predict the interactions between the molecules and understand the way in that chemical reactions proceed.
Considering that the knowledge of the chemical reactivity is essential for the development of new medicines, we have decided to study in this work Parasin I which is an antimicrobial peptide derived from histone H2A in the catfish [3] and that could be the basis for the design of new therapeutic peptides.
Thus, the objective of this work is to study the chemical reactivity of the Parasin I antimicrobial peptide of marine origin using the techniques of the conceptual DFT, determining its global properties (that is, of the molecule as a whole) as well as the local properties that allow to understand and predict the active reaction sites, both electrophilic and nucleophilic. Likewise, the pKa values of the peptide will be predicted based on a methodology previously developed by us [4], the ability of this potentially therapeutic peptide to act as inhibitor of the formation of advanced glycation endproducts (AGEs) will be established according to our previous ideas [5], and the descriptors of bioavailability and bioactivity (bioactivity scores) will be calculated through different procedures described in the literature [6,7,8,9,10,11,12,13,14,15].
2 Theoretical background
The Kohn–Sham theory involves calculating the molecular density, the energy of the system, and the molecular orbital energies, particularly those associated with the frontier orbitals including the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) [16,17,18,19]. This theory is necessary for establishing the quantitative values of the various conceptual DFT descriptors. Recently, there has been an increased interest in using range-separated (RS) exchange correlation functionals in Kohn–Sham DFT [20,21,22,23]. These functionals tend to partition the \(\hbox {r}_{12}^{-1}\) operator and exchange the parts into long- and short-ranged parts, whose range-separation parameter, \(\omega \), controls the rate of attaining the long-range behavior. It is possible to fix the value of \(\omega \) or “tune” it by a system-by-system mechanism that minimizes a tuning norm. The basis of the optimal tuning approach is the fact that the energy of the HOMO, \(\epsilon _H(N)\), in the case of the exact Kohn–Sham (KS) theory as well as the generalized KS theory for an N electron system should be \(-I(N)\). Here, I represents the vertical ionization potential, which is calculated as the energy difference, \(E(N-1) - E(N)\), by considering a particular density functional. If approximate density functionals are used, it would possibly lead to considerable differences between \(\epsilon _H{(N)}\) and \(-I(N)\). Optimal tuning involves determining the system-specific range-separation parameter \(\omega \) nonempirically with an RSE functional. Alternatively, it also implies that several other parameters including \(\epsilon _H{(N)} = -{I(N)}\) are optimally satisfied [24,25,26,27,28,29,30,31]. Even though there is no equivalent form to match this prescription for deriving the electron affinity A together with the LUMO energy for the case of neutral species, it is possible to say that \(\epsilon _H{(N+1)} = {-A(N)}\), which facilitates obtaining the optimized value of \(\omega \), which is then optimized to establish both properties. This would make it easy to predict the Conceptual DFT descriptors. In the past, a simultaneous prescription referred to as the “KID procedure” (Koopmans in DFT), owing to its correspondences with the Koopmans’ theorem, was proposed by the authors [32,33,34,35,36,37,38,39].
3 Settings and computational methods
This study obtained the molecular structure of the Parasin I peptide of marine origin from PubChem (https://pubchem.ncbi.nlm.nih.gov), a website that serves as the public repository for information pertaining chemical substances along with their associated biological activities. The pre-optimization of the resultant system involved selecting the five most stable conformers as it is customary for these kind of studies. The selection was done using random sampling that involved molecular mechanics techniques and inclusion of the various torsional angles via the general MMFF94 force field [40,41,42,43,44] involving the Marvin View 17.15 program, which constitutes as an advanced chemical viewer suited to multiple and single chemical queries, structures, and reactions (https://www.chemaxon.com). After that, the chemistry of the structures was checked and the 3D structures of the stereoisomers was generated using the same MarvinView 17.15 program. The chirality at the stereogenic centers was verified in accordance to the Cahn–Ingold–Prelog priority rules. The resulting geometries were further refined as it was explained before and the lowest energy conformation for each system was chosen to calculate the electronic energy and the HOMO and LUMO orbitals at the DFT functional level as mentioned in the next paragraph, choosing in the end the lowest energy conformation between the five conformers to proceed to the next step. It must be stressed that although, in general the properties of the molecular systems are strongly dependent on their conformations, from our experience working with small and large peptides, the global reactivity descriptors will be almost the same irrespective of the conformations (at least for the five lowest energy conformers). Indeed, this is observed in the five conformations considered in this work as we shall see in the Results and Discussion section, although in the manuscript we have presented the results only for the conformation considered as the global energy minimum.
Consistent with our previous work [32,33,34,35,36,37,38,39], the computational studies were performed with the Gaussian 09 [45] series of programs that implement density functional methods. The basis set Def2SVP was used in this work for the geometry optimization and frequency determination, while the Def2TZVP basis set was used for calculating the electronic properties [46, 47]. All calculations were performed in the presence of water as solvent under the solvation model density (SMD) parameterization of the integral equation formalism-polarized continuum model (IEF-PCM) [48].
To calculate the molecular structure and properties of the studied system, we have chosen eight density functionals which are known to consistently provide satisfactory results for several structural and thermodynamic properties: CAM-B3LYP [22], \(\hbox {LC-}\omega \hbox {PBE}\) [49], M11 [50], MN12SX [51], N12SX [51], \(\omega \hbox {B97}\) [52], \(\omega \hbox {B97X}\) [52], and \(\omega \hbox {B97XD}\) [21].
The SMILES notation of the studied compound was fed into the online Molinspiration software from Molinspiration Cheminformatics (www.molinspi-ration.com) for the calculation of the molecular properties (Log P, total polar surface area, number of hydrogen bond donors and acceptors, molecular weight, number of atoms, number of rotatable bonds, etc.) and for the prediction of the bioactivity scores (GPCR ligands, kinase inhibitors, ion channel modulators, enzymes and nuclear receptors). The bioactivity scores were compared with those obtained through the use of other software like MolSoft from Molsoft L.L.C. (http://molsoft.com/mprop/) and ChemDoodle Version 9.02 from iChemLabs L.L.C. (www.chemdoodle.com).
4 Results and discussion
4.1 Geometry optimization and global reactivity descriptors calculation
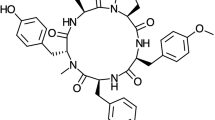
The molecular structure of Parasin I, which graphical sketch is shown in Fig. 1, was preoptimized in the gas phase by considering the DFTBA model available in Gaussian 09 and then reoptimized using the eight density functionals mentioned in the previous section together with the Def2SVP basis set and the SMD solvent model using water as the solvent. After verifying that each of the structures corresponded to the minimum energy configurations through a frequency calculation analysis, the electronic properties were determined by using the same model chemistry but with the Def2TZVP basis set instead of that used for the geometry optimization. The optimized molecular structure of Parasin I is also displayed in the Supplementary Information in PDF format.

Graphical sketch of the Parasin I molecule
The analysis of the results obtained in the study aimed at verifying that the KID procedure was fulfilled. On doing it previously, several descriptors associated with the results that the HOMO and LUMO calculations obtained are related with results obtained using the vertical I and A following the \(\Delta \)SCF procedure. A link exists between the three main descriptors and the simplest conformity to the Koopmans’ theorem by linking \(\epsilon _H\) with –I, \(\epsilon _L\) with –A, and their behavior in describing the HOMO-LUMO gap as \(J_{\text {I}} = |\epsilon _{\text {H}}+E_{{\text {gs}}}(N-1)-E_{{\text {gs}}}(N)|\), \(J_{\text {A}} = |\epsilon _{\text {L}}+E_{{\text {gs}}}(N-)-E_{{\text {gs}}}(N+1)|\), and \(J_{\text {HL}} = \sqrt{J_I^2+J_A^2}\). Notably, the \(J_{\text {A}}\) descriptor consists of an approximation that remains valid only when the HOMO that a radical anion has (the SOMO) shares similarity with the LUMO of the neutral system. Consequently, we decided to design another descriptor \(\Delta \)SL as the difference between the energies of the SOMO and the LUMO to guide in verifying the accuracy of the approximation [32,33,34,35,36,37,38,39]. The results of this analysis are presented in Table 1.
As can be seen in Table 1, the results for the descriptors show values that are consistent with our previous findings for the case of the melanoidins [32,33,34,35,36,37,38,39]; that is, only the MN12SX and N12SX density functionals are capable of giving HOMO and LUMO energies that allow to verify the agreement with the approximate Koopmans’ theorem. This is not only true because the JHL values are almost zero, but due to the fact that the \(\Delta \hbox {SL}\) descriptor, which relates to the difference between the LUMO of the neutral and the HOMO of the anion, is also close to zero. Indeed, these values cannot be exactly equal to zero, but the small differences mean that errors in the prediction of the global reactivity descriptors will be negligible. Moreover, it can be seen in Table 1 that the MN12SX and N12SX density functionals are the only ones that predict negative values for the LUMO energies which will represent positive values of the electron affinity A. An opposite and incorrect (unphysical) behavior is observed in Table 1 for the other density functionals considered in this work.
By taking into account the KID procedure presented in our previous works together with the finite difference approximation, the global reactivity descriptors can be expressed as:
Electronegativity | \(\chi = -\frac{1}{2} (I + A) \approx \frac{1}{2} (\epsilon _{\text {L}} + \epsilon _{\text {H}})\) | |
Global Hardness | \(\eta = (I - A) \approx (\epsilon _{\text {L}} - \epsilon _{\text {H}})\) | |
Electrophilicity | \( \omega = \frac{\mu ^2}{2 \eta } = \frac{(I + A)^2}{4 (I - A)} \approx \frac{(\epsilon _{\text {L}} + \epsilon _{\text {H}})^2}{4 (\epsilon _{\text {L}} - \epsilon _{\text {H}})}\) | [53] |
Electrodonating Power | \(\omega ^{-} = \frac{(3 I + A)^{2}}{16(I - A)} \approx \frac{(3 \epsilon _{\text {H}} + \epsilon _{\text {L}})^{2}}{16 \eta }\) | [54] |
Electroaccepting Power | \(\omega ^{+} = \frac{(I + 3 A)^{2}}{16(I - A)} \approx \frac{(\epsilon _{\text {H}} + 3 \epsilon _{\text {L}})^{2}}{16 \eta }\) | [54] |
Net Electrophilicity | \(\Delta \omega ^{\pm } = \omega ^{+} - (-\omega ^{-}) = \omega ^{+} + \omega ^{-}\) | [55] |
where \(\epsilon _{\text {H}}\) and \(\epsilon _{\text {L}}\) are the energies of the HOMO and LUMO, respectively.
In order to provide evidence about our assertion mentioned in the Settings and Computational Methods section stating that there is only an almost negligible change of the global reactivity descriptors with the molecular structure of the five lowest energy conformers, we are presenting in Fig. 2 a graphical sketch of these values for the case of the MN12SX density functional. The results presented in Fig. 2 represent a confirmation of the validity of our assertion.

Graphical sketches showing the evolution of the global reactivity descriptors with the change in the molecular structures of the five lowest energy conformers, labeled as C1 to C5: a electronegativity, global hardness, and electrophilicity; b electrodonating power, electroaccepting power, and net electrophilicity
According to our previous discussion and the information given in Table 1, the results for the global reactivity descriptors based on the values of the HOMO and LUMO energies will be significative only for the MN12SX and N12SX density functionals. Thus, these results are presented in Table 2.
As expected from the molecular structure of this species, its electrodonating ability is more important than its electroaccepting character. There are no significative differences between the values obtained by using either of the density functionals for the calculation of the global reactivity descriptors. Notwithstanding, after an inspection of Table 1, it can be said that the MN12SX density functional is a somewhat better than the N12SX density functional in verifying the approximate Koopmans behavior. Thus, only the MN12SX density functional will be considered for the remaining of this work.
4.2 Local reactivity descriptors calculation
Applying the same ideas as before, the definitions for the local reactivity descriptors will be:
Nucleophilic Fukui Function | \(f^{+}(\mathbf {r})=\rho _{N+1}(\mathbf {r})-\rho _{N}(\mathbf {r})\) | |
Electrophilic Fukui Function | \(f^{-}(\mathbf {r})=\rho _{N}(\mathbf {r})-\rho _{N-1}(\mathbf {r})\) | |
Dual Descriptor | \({\Delta }f(\mathbf {r}) = \left( \frac{\partial \,f(\mathbf {r})}{\partial \,N}\right) _{\upsilon (\mathbf {r})}\) | |
Nucleophilic Parr Function | \(P{^-}(\mathbf {r}) = \rho _{s}^{rc} (\mathbf r)\) | |
Electrophilic Parr Function | \(P{^+}(\mathbf {r}) = \rho _{s}^{ra} (\mathbf r)\) |
where \(\rho _{N+1}(\mathbf {r})\), \(\rho _{N}(\mathbf {r})\), and \(\rho _{N-1}(\mathbf {r})\) are the electronic densities at point \(\mathbf {r}\) for a system with \(N+1\), N, and \(N-1\) electrons, respectively, and \(\rho _{s}^{rc} (\mathbf r)\) and \(\rho _{s}^{ra} (\mathbf r\)) are related to the atomic spin density (ASD) at the r atom of the radical cation or anion of a given molecule, respectively [64].
Starting from the total electronic densities arising from calculations for the systems with \(N+1\), N, and \(N-1\) electrons, the electrophilic Fukui function \(f^{-}(\mathbf {r})\) and nucleophilic Fukui function \(f^{+}(\mathbf {r})\) for the Parasin I molecule estimated according to the above difference formulas are shown in Figs. 2a, b, respectively.

a Electrophilic Fukui function \(f^-\)(r) and b nucleophilic Fukui function \(f^{+}(r)\) for the Parasin I molecule where reddish zones indicate positive values and greenish zones denote negative values
As it has been stated by Martínez-Araya in a recent work [61], while the Fukui function is a nice descriptor to understand the local reactivity of the molecules, it can be demonstrated that the dual descriptor in its condensed form \(\Delta f_k\) will perform better for the prediction of the preferred sites for the electrophilic and nucleophilic attacks. For this reason, we have decided to present the results for the condensed dual descriptor \(\Delta f_k\) as calculated from either Mulliken population analysis in comparison with the nucleophilic Parr function \(P^+_k\) and electrophilic Parr function \(P^-_k\) proposed by Domingo et al [62, 63] considering atomic spin densities coming from the Hirshfeld population analysis (Fig.3).
The results for the calculation of these local reactivity descriptors for the Parasin I molecule are presented in Table 3. It must be noted that we are presenting only the results for those atomic sites where the \(\Delta f_k\) are greater than 1. The values for \(\Delta f_k\), \(P^+_k,\) and \(P^-_k\) are multiplied by 100 for easier comparison. Also, the H atoms are not shown. As can be seen in Table 3, the local reactivity descriptors calculated from the different formulations are able to recognize the nucleophilic and electrophilic sites for chemical reactivity with great accuracy. Moreover, there is an impressive agreement between the results coming from the condensed dual descriptor \(\Delta f_k\) and the nucleophilic and electrophilic Parr functions \(P^+_k\) and \(P^-_k\) which means that their use in this and future works related to the study of therapeutic peptides will be a warranty of success.
4.3 Determination of pKa value of the peptide
We have recently presented a study of the computational prediction of the pKas of small peptides through conceptual DFT descriptors [4]. In that work, we concluded that the relationship \(\hbox {pKa} = 16.3088 - 0.8268 \eta \) could be a valuable starting point for the prediction of the pKa of larger peptides of interest for the development of AGE inhibitors.
Thus, we have now applied the mentioned relationship to the calculation of the pKa of the Parasin I molecule giving a result of 12.421. This result could be of interest when designing pharmaceutical drugs starting from these peptide allowing to explain the mechanisms of action and the drug delivery procedures. For example, starting from this result, it can predict a solubility of approximately 20 mg L\(^{-1}\) at the physiological pH of 7.4. This is very important because only dissolved drugs can be absorbed. Therefore, the solubility of a drug is an extremely important parameter in planning the development of tablets.
Additionally, the solubility can be also inferred from the value of the \(\Delta {G}\) of solvation which can be estimated from the results of our calculations. It must be remarked that we have chosen to perform them by considering the SMD approximation for modeling the solvent. This is the recommended choice for computing the \(\Delta {G}\) of solvation, which accomplished by performing gas phase and SCRF=SMD calculations for the system of interest and taking the difference the resulting energies with the Gaussian 09 program [45]. A rapid estimation of this magnitude gives a value of − 179.91 kcal mol\(^{-1}\) for the Parasin I molecule which is an identification that this peptide is fully soluble in water, a result in agreement with the experimental findings.
These results help to explain why Parasin I shows a strong antimicrobial activity toward gram-negative bacteria, gram-positive bacteria, and fungi without any hemolytic activity. The minimum inhibitory concentration (MIC) of Parasin I is in the range of 1–4 mg/ml [3]. The most potent antimicrobial peptides have been reported to kill susceptible bacteria in the range of 0.25–4 mg/ml, which indicates that Parasin I is one of the most potent antimicrobial peptides found so far.
4.4 Quantification of the AGEs inhibition ability
The Maillard reaction between a reducing carbonyl and the amino group of a peptide or protein leads to the formation of a Schiff base which through a series of steps renders different molecules known as advanced glycation endproducts or AGEs. It is believed that the presence of these AGEs is one of the main reasons for the developing of some diseases like diabetes, Alzheimer, and Parkinson.
Among several strategies that have been considered for the prevention of the formation of AGEs, it is worth to mention the use of compounds presenting amino groups in their structure capable of interacting with the reducing carbonyl group of carbohydrates and being competitive with the amino acids, peptides, and proteins present in our body. Many compounds have been devised as drugs to achieve this goal and to name a few, we can include pyridoxamine, aminoguanidine, carnosine, metformin, pioglitazone, and tenilsetam.
It can be proposed that peptides having amino and amido groups could be thought as potential therapeutic drugs for preventing the formation of AGEs. In a previous work, we have studied the ability of a group of proposed molecules to act as inhibitors of the formation of AGEs by quantifying their behavior in terms of conceptual DFT reactivity descriptors [5]. It was concluded that the key factor in the study of the chemical reactivity of the potential AGEs inhibitors was on their nucleophilic character and although there are several definitions of nucleophilicity [65], our results suggested that the inverse of the net electrophilicity \(\Delta \omega ^{\pm }\) could be a good definition for the nucleophilicity N. On the basis of the mentioned analysis, we were able to find some qualitative trends for the studied molecular systems.
In this work, we will extend this correlation to the Parasin I peptide in order to see if it can be considered as a precursor of therapeutic drugs for the inhibition of the formation of AGEs. As the model chemistry employed in both works is the same, the comparison is straightforward:
This qualitative trend is representative of the known pharmacological properties of the studied AGEs inhibitors [66, 67], and it can be seen that Parasin I possesses AGEs inhibition ability similar to that of pyridoxamine, being larger than the value for pioglitazone.
4.5 Bioactivity scores
When considering a given molecular system as a potential therapeutic drug, it is customary to check if the considered species follows the Lipinsky rule of five which is used to predict whether a compound has or not has a drug-like character [68]. The molecular properties related to the drug-like character were calculated with the aid of the MolSoft and Molinspiration software and are presented in Table S1 of the Supplementary Information, where miLogP represents the octanol/water partition coefficient, TPSA is the molecular polar surface area (in Å\(^2\)), natoms is the number of atoms of the molecule, nON and nOHNH are the number of hydrogen bond acceptors and hydrogen bond donors, respectively, nviol is the number of violations of the Lipinsky rule of five, nrotb is the number of rotatable bonds, volume is the molecular volume (in Å\(^3\)), and MW is the molecular weight of the studied system (in g mol\(^{-1}\)).
However, what the Lipinsky rule of five really measures is the oral bioavailability of a potential drug because this is desired property for a molecule having drug-like character [69]. Indeed, this criteria cannot be applied to peptides, even when they are small, as we can see in Table S1 of the Supplementary Information, due to the inherent molecular weight and number of hydrogen bonds.
In a more recent work, Martin [70] has developed what she called “A Bioavailability Score” (ABS) for avoiding these problems. The rule for the ABS established that the bioavailability score for neutral organic molecules must be 0.55 if they pass the Lipinsky rule of five and 0.170 if they fail. The ABS value for all the Parasin I peptide considered in this work has been calculated by using the ChemDoodle software, and the result was equal to 0.170.
Then, a different approach was followed by considering similarity searches in the chemical space of compounds with structures that can be compared to those that are being studied and with known pharmacological properties.
As has been mentioned in the Settings and Computational Methods section, this task can be accomplished using the online Molinspiration software for the prediction of the bioactivity scores (GPCR ligands, kinase inhibitors, ion channel modulators, enzymes, and nuclear receptors). Rather than relying in a universal drug-likeness score, the methodology focuses on those particular dug classes through the development of specific activity scores for each of these classes. The determination is done by applying Bayesian statistics to compare structures of representative ligands active on particular target with structures of inactive molecules and to identify substructure features (which in turn determine physicochemical properties) typical for active molecules. The results are named bioactivity scores, and the values for the Parasin I are presented in Table 4.
These bioactivity scores for organic molecules can be interpreted as active (when the bioactivity score > 0), moderately active (when the bioactivity score lies between − 5.0 and 0.0), and inactive (when the bioactivity score < − 5.0). Thus, Parasin I was found to be moderately bioactive in all cases.
5 Conclusions
In this paper, we have presented the results of a study of the chemical reactivity of the Parasin I antimicrobial peptide of marine origin based on the conceptual DFT as a tool to explain the molecular interactions.
The knowledge of the values of the global and local descriptors of the molecular reactivity of the Parasin I peptide studied could be useful in the development of new drugs based on this compound or some analogs.
In a similar manner, the pKa values for the potentially therapeutic peptide have been predicted by resorting to the value of the chemical hardness \(\eta \) following a previously proposed methodology and the information that resulted would be helpful in understanding not only the chemical reactivity but other important properties like the water solubility.
A point of special interest has been the quantification of the ability of the peptide to act as an inhibitor in the formation of AGEs, and this could be of importance for the design of medicines for fighting diseases like diabetes, Alzheimer, or Parkinson.
Finally, the molecular properties related to bioavailability have been predicted using different methodologies already described in the literature, and the descriptors used for the quantification of the bioactivity allowed to characterize the studied peptide as being moderately bioactive in all cases considered in the study.
References
Parr R, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, New York
Geerlings P, De Proft F, Langenaeker W (2003) Chem Rev 103:1793
Park IY, Park CB, Kim MS, Kim SC (1998) FEBS Lett 437(3):258
Frau J, Hernández-Haro N, Glossman-Mitnik D (2017) Chem Phys Lett 671:138
Frau J, Glossman-Mitnik D (2017) Molecules 22(1):226
Chattaraj P (ed) (2009) Chemical reactivity theory—a density functional view. CRC Press, Boca Raton
Bultinck P, De Winter H, Langenaeker W, Tollenaere JP (eds) (2004) Computational medicinal chemistry for drug discovery. Taylor & Francis, New York
Gore M, Jagtap UB (2018) Computational drug discovery and design. Springer, New York
Gupta GK, Kumar V (2016) Chemical drug design. Walter de Gruyter GmbH, Berlin
Dunn BM (ed) (2015) Peptide chemistry and drug design. Wiley, Hoboken
Castanho M, Santos NC (eds) (2011) Peptide drug discovery and development. Wiley, Weinheim
Otvos L (ed) (2008) Peptide-based drug design—methods and protocols. Humana Press, Totowa
Zhou P, Huang J (eds) (2015) Computational peptidology. Humana Press, New York
Tamamura H, Kobayakawa T, Ohashi N (eds) (2018) Mid-size drugs based on peptides and peptidomimetics—a new drug category. Springer, Singapore
sunna A, Care A, Bergquist PL (eds) (2017) Peptides and peptide-based biomaterials and their biomedical applications. Springer, Cham
Lewars E (2003) Computational chemistry—introduction to the theory and applications of molecular and quantum mechanics. Kluwer Academic Publishers, Dordrecht
Young D (2001) Computational chemistry—a practical guide for applying techniques to real-world problems. Wiley, New York
Jensen F (2007) Introduction to computational chemistry, 2nd edn. Wiley, Chichester
Cramer C (2004) Essentials of computational chemistry—theories and models, 2nd edn. Wiley, Chichester
Iikura H, Tsuneda T, Yanai T, Hirao K (2001) J Chem Phys 115(8):3540
Chai J, Head-Gordon M (2008) Phys Chem Chem Phys 10:6615
Yanai T, Tew DP, Handy NC (2004) Chem Phys Lett 393(1–3):51
Heyd J, Scuseria G (2004) J Chem Phys 121(3):1187
Stein T, Kronik L, Baer R (2009) J Chem Phys 131(24):244119
Stein T, Kronik L, Baer R (2009) J Am Chem Soc 131(8):2818
Stein T, Eisenberg H, Kronik L, Baer R (2010) Phys Rev Lett 105(26):266802
Karolewski A, Stein T, Baer R, Kümmel S (2011) J Chem Phys 134(15):151101
Kuritz N, Stein T, Baer R, Kronik L (2011) J Chem Theory Comput 7(8):2408
Ansbacher T, Srivastava HK, Stein T, Baer R, Merkx M, Shurki A (2012) Phys Chem Chem Phys 14:4109
Kronik L, Stein T, Refaely-Abramson S, Baer R (2012) J Chem Theory Comput 8(5):1515
Stein T, Autschbach J, Govind N, Kronik L, Baer R (2012) J Phys Chem Lett 3(24):3740
Frau J, Glossman-Mitnik D (2018) Molecules 23(3):559
Frau J, Glossman-Mitnik D (2018) Theor Chem Acc 137(5):1210
Frau J, Glossman-Mitnik D (2018) Front Chem 6(136):1
Frau J, Glossman-Mitnik D (2018) Contemp Chem 1(1):1
Frau J, Glossman-Mitnik D (2018) Comput Theor Chem 1134:22
Frau J, Glossman-Mitnik D (2018) Chem Sci Int J 22(4):1
Frau J, Glossman-Mitnik D (2018) J Mol Model 24(138):1
Frau J, Flores-Holguín N, Glossman-Mitnik D (2018) Mar Drugs 16(9):302
Halgren TA (1996) J Comput Chem 17(5–6):490
Halgren TA (1996) J Comput Chem 17(5–6):520
Halgren TA (1999) J Comput Chem 20(7):720
Halgren TA, Nachbar RB (1996) J Comput Chem 17(5–6):587
Halgren TA (1996) J Comput Chem 17(5–6):616
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2016) Gaussian 09 Revision E.01. Gaussian Inc., Wallingford
Weigend F, Ahlrichs R (2005) Phys Chem Chem Phys 7:3297
Weigend F (2006) Phys Chem Chem Phys 8:1057
Marenich A, Cramer C, Truhlar D (2009) J Phys Chem B 113:6378
Henderson TM, Izmaylov AF, Scalmani G, Scuseria GE (2009) J Chem Phys 131(4):044108
Peverati R, Truhlar DG (2011) J Phys Chem Lett 2(21):2810
Peverati R, Truhlar DG (2012) Phys Chem Chem Phys 14(47):16187
Chai J, Head-Gordon M (2008) J Chem Phys 128:084106
Parr R, Szentpaly L, Liu S (1999) J Am Chem Soc 121:1922
Gázquez J, Cedillo A, Vela A (2007) J Phys Chem A 111(10):1966
Chattaraj P, Chakraborty A, Giri S (2009) J Phys Chem A 113(37):10068
Toro-Labbé A (ed) (2007) Theoretical aspects of chemical reactivity. Elsevier, Amsterdam
Morell C, Grand A, Toro-Labbé A (2005) J Phys Chem A 109:205
Morell C, Grand A, Toro-Labbé A (2006) Chem Phys Lett 425:342
Martínez-Araya JI (2012) J Mol Model 18:4299
Martínez-Araya JI (2012) J Mol Model 19(7):2715
Martínez-Araya JI (2015) J Math Chem 53(2):451
Domingo LR, Pérez P, Sáez J (2013) RSC Adv 3:1486
Chamorro E, Pérez P, Domingo LR (2013) Chem Phys Lett 582:141
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Molecules 21:748
Domingo LR, Perez P (2011) Org Biomol Chem 9:7168
Rahbar S, Figarola JL (2003) Arch Biochem Biophys 419:63
Peyroux J, Sternberg M (2006) Pathol Biol 54:405
Leeson P (2012) Nature 481(7382):455
Santos GB, Ganesan A, Emery FS (2016) ChemMedChem 11(20):2245
Martin YC (2005) J Med Chem 48(9):3164
Acknowledgements
This work has been partially supported by CIMAV, SC, and Consejo Nacional de Ciencia y Tecnología (CONACYT, Mexico) through Grant 219566-2014 for Basic Science Research. Daniel Glossman-Mitnik conducted this work, while a Visiting Lecturer at the University of the Balearic Islands from which support is gratefully acknowledged. Norma Flores-Holguín and Daniel Glossman-Mitnik are researchers of CIMAV and CONACYT. This work was cofunded by the Ministerio de Economía y Competitividad (MINECO) and the European Fund for Regional Development (FEDER) (CTQ2014-55835-R).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest regarding the publication of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published as part of the special collection of articles derived from the 11th Congress on Electronic Structure: Principles and Applications (ESPA-2018).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Flores-Holguín, N., Frau, J. & Glossman-Mitnik, D. Computational prediction of bioactivity scores and chemical reactivity properties of the Parasin I therapeutic peptide of marine origin through the calculation of global and local conceptual DFT descriptors. Theor Chem Acc 138, 78 (2019). https://doi.org/10.1007/s00214-019-2469-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-019-2469-3