
Abstract
Systematic studies of the excited-state tautomerization in the 7-azaindole–(EtOH) n (n = 1, 2) complexes in the gas phase were theoretically investigated. Structures and energies for reactant, transition state and product were computed at the CASSCF levels with the 6-31G(d,p) and 6-311G(d,p) basis sets. The barrier heights and reaction energies were corrected by the second-order multireference perturbation theory (MRPT2) to consider the dynamic electron correlation. The excited-state double-proton transfer in 7AI–EtOH occurs in a concerted but asynchronous mechanism. Similarly, such paths are also found in the two transition states during the excited-state triple-proton transfer of 7AI–(EtOH)2 complex. One path is that the proton from the pyrrole ring moved first to ethanol, and the other path is that the ethanol proton moved first to pyridine ring. The CASSCF level with the MRPT2 correction clearly showed that the former path was much preferable to the latter. The preferable barrier height for the 7-azaindole–(EtOH)2 complex was 4.3 kcal/mol with the zero-point energy correction. Additionally, the effect on the substitution of the ethyl group for the methyl group in the 7AI–(MeOH) n (n = 1, 2) was discussed. The replacement of the methyl group by the ethyl group obviously increased the barrier height and the asynchronousity of proton transfer in the 7AI–(EtOH)2 complex and had little effect on the 7AI–(EtOH) complex.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Excited-state proton/hydrogen-atom transfer (ESPT/HAT) reaction has received considerable attention in recent years since it exists in a wide variety of biological and chemical processes [1–8]. In biological systems, proton/hydrogen-atom transfer usually takes place over a long distance across hydrogen-bonded chains between donors and acceptors which can provide an effective pathway for the rapid migration protons/hydrogen-atoms. The mechanism of ESPT, which relates to the strength of a hydrogen bond and dynamics of proton motion, provides much valuable information on the basic of chemical reaction in biological systems. Thus, it is fundamentally and practically important to understand the details of such long-range proton transfer processes. However, it is extremely hard to observe this long-range proton transfer process along a proton wire experimentally at the atomic level due to the structural complexity and massiveness of protein [4–7]. Therefore, it is necessary to establish a simplified model in order to simulate the proton transfer process.
Among numerous ESPT molecules, 7-azaindole (7AI) molecule is a very important model system to study the proton transfer process since it is similar to the molecule with the DNA base pair [9]. 7AI contains one proton-donor nitrogen atom in the five-member ring and one proton-acceptor nitrogen atom in the six-member ring and therefore shows simple hydrogen-bonding structures upon dimerization and complexation with polar solvents. Then, the excited-state proton transfer takes place through forming a cyclic hydrogen-bonding complex. Compared to the isolated 7AI, the energy barrier of tautomerization in 7AI was dramatically reduced by the complexation with one water molecule when the dynamic electron correlation was considered [10]. Detailed studies of intermolecular excited-state proton transfer (ESPT) in given media have increased dramatically for understanding the basic proton transfer dynamics [10–14]. One fundamental interest regarding the proton transfer is in whether the mechanism incorporates the concerted or stepwise pattern. In the concerted mechanism, multiple protons transferred simultaneously in a single step, while in the stepwise mechanism, multiple protons transferred sequentially with more than one step, where a stable intermediate compound was formed on the way. Another basic interest on the proton transfer process is the order of migration proton, namely which proton moves first to trigger the proton transfer process. If the direct proton relay from an acid to a base is not considered, there are two possible reaction paths in the solvent-mediated proton transfer process named protolysis path and solvolysis path [2, 15], respectively. This two reaction paths are corresponding to the different order of migration proton. The solvolysis path is the path that proton transfers from a solvent molecule to a base followed by deprotonation of an acid by the solvent molecule. On the contrary, if the proton transfers from an acid to a solvent molecule with subsequent proton scavenging by a base, this path is the protolysis path.
Recently, Sakota et al. [16] investigated the excited-state multiple-proton/hydrogen-atom transfer reactions in the 7-azaindole ethanol clusters, 7-azaindole–(EtOH) n (n = 1–3), in the gas phase by combining electronic spectroscopy and quantum chemical calculations. They found that the geometry of 7AI–(EtOH)2 is very similar to the 7AI–(CH3OH)2 geometry; the solvent molecules bridge the heteroaromatic N atom and the NH group in the five-member ring by intermolecular hydrogen bonds, forming a cyclic structure. The multiple-proton transfer is solvent and cluster-size selective and the excited-state tautomerization was observed only when n = 2 in the gas phase, and no clear evidence for the existence of excited-state double-proton transfer (ESDPT) and the excited-state quadruple-proton transfer (ESQPT) has been obtained. However, the detailed ESPT mechanism has not been clarified for 7AI–(EtOH) n clusters.
Kwon et al. [17] found that the rate constants of 7AI–alcohol complexes were dependent on the acidity of the alcohol in heptane; thus, they proposed that the proton transfer of 7AI was triggered by initial transfer of a proton from the alcohol to the pyridine nitrogen of 7AI, forming a cationic 7AI intermediate species, and was completed by rapid proton transfer from the pyrrole nitrogen of the intermediate to the transient alkoxide. We reported high-level calculations of potential energy surfaces for the ESPT processes in the 7AI–(CH3OH) n (n = 1, 2) cluster successfully using complete active space self-consistent field (CASSCF) method with the second-order multireference perturbation theory (MRPT2) as well as the time-dependent density functional theory (TDDFT) with long-range correction [18–20]. The theoretical results made us conclude that the concerted mechanism is more favorable in these clusters and that the two/three protons transfer asynchronously. The proton moved first from the N–H group in pyrrole ring of 7AI to methanol with subsequent proton scavenging by the N atom in pyridine ring, which indicates the opposite proton transfer pathway compared to the Kwon’s observations [17].
Either excited-state proton transfer or hydrogen-atom transfer process occurs depending on the energy order of the S ππ* and S πσ* in the target systems. These states are very important to confirm the nature of the excited-state reactions. Proton transfer is along the S ππ* state, while hydrogen-atom transfer is along the S πσ* state [21–24]. Particularly, it has been reported by investigating the energy paths that the S πσ* state lies well above the S ππ* state without intersections [25, 26].
Until now, the multiple-proton transfer reactions are reported for very limited molecular systems in the gas phase, and further information is necessary to investigate the proton transfer mechanism. Thus, the investigation of the multiple-proton transfer in 7AI–(EtOH) n clusters in the gas phase will throw light on obtaining detailed information about the solvent-assisted multiple-proton transfer, which is useful to reveal the proton transfer dynamics in complicated molecular systems such as enzymes and proteins. The purpose of this work is to perform a systematic study of tautomerization in the biologically interesting complexes: 7AI–EtOH and 7AI–(EtOH)2. One goal is to illustrate the excited-state multiple-proton transfer mechanisms. The other interest is to prove whether the substitution of the ethyl group for the methyl group in the 7AI–(MeOH) n (n = 1, 2) complex has any effect on the geometric structures and the ESPT dynamics, i.e., barrier height, proton transfer mechanism (concerted or stepwise, synchronous or asynchronous).
2 Computational details
The geometries of the reactant, product and transition state (TS) of the excited-state tautomerization in the 7AI–(EtOH) n (n = 1, 2) complexes were fully optimized at the CASSCF level with 6-31G(d,p) and 6-311G(d,p) basis sets using the Gaussian 09 program [27] in the gas phase. To carry out the CASSCF calculation, the crucial step is to select the proper active space. For the ESPT process in the 7AI–(EtOH) n (n = 1, 2) complexes, the single electronic state S ππ* was considered. Therefore, the obvious choice for an active space in 7AI complexes would include four π bonds, four corresponding antibonding orbitals, and one nitrogen π lone pair. This results in the active space of 10 electrons in 9 orbitals and is denoted CASSCF(10, 9). The active orbitals of TS in the 7AI–(EtOH) n (n = 1, 2) complexes are shown in Figure SI–SIII in the Supporting Information. Optimized structures were verified by calculating the vibrational frequencies and establishing that there are no imaginary frequency for the reactant and product and only one imaginary frequency for the TS. Single-point energy calculations were also performed using the second-order multireference perturbation theory (MRPT2) [28–33] for stationary points. The general theoretical background [29] is as follows.
The state-specific Rayleigh–Schrödinger perturbation theory based on the unperturbed eigenvalue equation
leads to the first few \(E_{I}^{(k)}\) as
V is a perturbation, R is the resolvent operator
where Q = 1 − P, P is defined as an active space, and the remaining part of Hilbert space is called the orthogonal space Q.
\(E_{I}^{(0)}\) is given in terms of orbital energies as
and the orbital energies are defined as
with
where \(D_{ij}\) is the one-electron density matrix. The MCSCF orbitals are resolved to make the \(F_{ij}\) matrix as diagonal as possible. The definition of an active space, the choices of active orbitals and the specification of the zeroth-order Hamiltonian completely determine the perturbation approximation. When a CASSCF wave function is used as the reference, the zeroth- plus first-order energy is equal to the CASSCF energy. The lowest non-trivial order is therefore the second order. Let the reference function \(\left| {\Psi _{\alpha }^{(0)} } \right\rangle\) be a CASSCF wave function,
The energy up to the second order is given by
where \(\left\{ {\left| I \right\rangle } \right\}\) is the set of all singly and doubly excited configurations from the reference configurations in CASSCF. All MRPT2 calculations were performed using the GAMESS program [34].
3 Results and discussion
3.1 7AI–EtOH complex
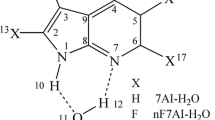
The structures of the reactant, TS and product in 1:1 7AI:EtOH complex were obtained by means of CASSCF methods with 6-31G(d,p) and 6-311G(d,p) basis sets. The optimized structures of reactant and product were confirmed by the vibrational frequency calculations. Structures of the stationary points in the 7AI–EtOH complex optimized at the CASSCF(10,9)/6-311G(d,p) level are shown in Fig. 1. For the 7AI–EtOH complex, the H-bond distances, H10–O16, H17–N6 in the reactant and N1–H10, O16–H17 in the product at the CASSCF level using 6-31G(d,p) basis set were 2.147, 2.117, 2.189 and 2.176 Å, respectively (Table 1). When the larger 6-311G(d,p) basis set was used, all H-bond distances became larger. The shorter the H-bond length, the higher the H-bond energy; therefore, the 6-31G(d,p) basis set predicted strong H-bonds compared to the 6-311G(d,p) basis set.

Reactant, product and the transition state of the ESDPT in the 7AI–C2H5OH complex at the CASSCF(10,9)/6-311G(d,p) level
TS structures for the excited-state proton transfer reaction in 7AI–EtOH were fully optimized and confirmed by frequency calculations and the intrinsic reaction coordinate (IRC) calculations, and some of the geometric parameters are listed in Table 1. In the TS at the CASSCF/6-31G(d,p) level, the H10 atom moved more than halfway along the reaction coordinate toward O16 (Fig. 1), whereas the H17 atom rarely moved. In this double-proton transfer, the H10 atom moved first followed by the H17 atom. This double-proton transfer process has occurred in a typical concerted but asynchronous protolysis path. When the 6-311G(d,p) basis set was applied, CASSCF method predicted a similar TS structure, in which the H10 atom moved about 0.30 Å from N1 to O16 along the reaction coordinate, whereas the H17 atom rarely moved.
A correlation plot between the hydrogen bond length and the proton transfer coordinate is shown in Fig. 2. Hydrogen bond coordinates q 1 = (1/2)(r AH − r BH) and q 2 = r AH + r BH were used to represent the correlation between r AH and r BH in many hydrogen-bonded complexes (A–H…B) [35, 36]. In the A–H…B complexes, the r AH and r BH distances depend on each other, leading to allowing r AH and r BH values based on the following Pauling equations under the assumption that the sum of two bond orders is conserved, n AH + n BH = 1:
where r 0AH and r 0BH are the equilibrium lengths of the free AH and BH bonds, and b AH and b BH are the parameters describing the decrease in the AH and the HB unit bond valences with the corresponding distances. When the symmetric hydrogen bonds are OHO and NHN, b are 0.37 and 0.404 Å, respectively [35, 36]. For a linear H-bond, q 1 represents the distance of H from the H-bond center and q 2 represents the distance between atoms A and B. A strong H-bond results in short r BH and slightly elongated r AH distances. Bond distance depends on bond energy and bond order. This type of correlation, i.e., the “bond energy bond order method,” has been used for many years to study hydrogen-atom transfer. When H is transferred from A to B in the A–H…B complex, q 1 increases from negative to positive values and q 2 goes through a minimum, which is located at q 1 = 0. Limbach et al. [35, 36] suggested that both proton transfer and hydrogen-bonding coordinates could be combined into the same correlation. It is a good choice to use this correlation to study the characteristics of transition state, such as earliness or lateness, bond order and synchronicity. The q 1 value of TS is negative or positive when the TS is either early or late, respectively. In addition, the two q 1 values for the double-proton transfer in TS should be very similar and different in the synchronous and asynchronous mechanism, respectively.

Correlation of the H-bond distances, q 2 = r 1 + r 2, with the proton transfer coordinate, q 1 = (1/2)(r 1 − r 2), for the 7AI–C2H5OH and 7AI–CH3OH complexes in the gas phase. All points are for the transition states in S1 optimized at the CASSCF/6-311G(d,p) level. The solid lines designate the correlation that satisfies conservation of the bond order. The parameters for Pauling equations were from the literature [35]. The structural parameters of the 7AI–CH3OH complex were from Ref. [18]. The region above and below the black line is where the sum of bond order is smaller and larger than unity, respectively
The correlations between N1–H10 and H10–O16 distances (H10 transfer), and N6–H17 and H17–O16 distances (H17 transfer) for the 7AI–EtOH complex at the CASSCF/6-311G(d,p) level are shown in Fig. 2. It is interesting to note that all points for the reactant, product and TS were very close to the black line, which suggests that the sum of bond order at all stationary points was approximately conserved. In the proton transfer process, two q 1 values for two proton transfers in the TS should be approximately the same. These q 1 values of H10 and H17 transfer at the TS were a little positive and very negative, respectively, which resulted in a highly asynchronous TS (slightly late and very early TS in terms of H10 and H17 transfers, respectively).
3.2 7AI–(EtOH)2 complex
The excited-state tautomerization of 7AI in alcohols [17, 37, 38] has been discussed as a two-step process. The first step is the solvent reorganization to form cyclic hydrogen-bonded 7AI–alcohol complexes, and the second step is an intrinsic proton transfer. If the solvent motion was rate limiting, no significant kinetic isotope effect (KIE) would be expected. However, KIEs for excited-state tautomerization have been observed in 7AI complexes with various alcohols, and Moogs and Maroncelli [37] suggested that both solvent reorganization and the intrinsic proton transfer step could determine the reaction rate. Since the ESPT is very fast, the hydrogen-bonded complexes, either cyclic or non-cyclic, should be present before excitation. Therefore, it is necessary to investigate what kind of hydrogen-bonded complex is most likely to be formed. Sakota et al. [16] reported that four structural isomers of 7AI–(EtOH)2 complex have been identified by RE2PI spectra. DFT calculations have predicted four different conformations for 7AI–(EtOH)2 in the ground state, in good agreement with the observation of the four structural isomers in the RE2PI spectra. The differences in the binding energies between the four isomers are very small. The most stable cyclic hydrogen-bonded structure of 7AI–(EtOH)2 complex [16] is energetically more favorable in the gas phase and has been chosen as the target structure to study the ESTPT process. In the 7AI–(EtOH)2 complex, one C–C bond of ethanol is almost parallel and the other C–C bond of ethanol is nearly perpendicular to the long axis of 7AI.
Optimized structural parameters of the reactant, product and TS for the 7AI–(EtOH)2 complexes are listed in Table 2, and structures at the CASSCF(10,9)/6-311G(d,p) level are depicted in Fig. 3. All H-bond distances in the reactant and product in the 7AI–(EtOH)2 complexes were smaller than those in 7AI–EtOH complex. The H-bonds in 7AI–(EtOH)2 complexes were more linear and shorter than those of the 7AI–EtOH complexes. Geometry of the TS was fully optimized at the CASSCF(10,9) level and was confirmed by frequency calculations and the intrinsic reaction coordinate (IRC) calculations. It was interesting that two TS structures were found (see Fig. 3). In the first TS (denoted as TS1), the H10 moved more than halfway from N1 toward the O16 atom with the H17 and H23 rarely moving, which generated a EtOH2 +-like moiety in a portion of the TS (at O16). However, in the second TS (denoted to TS2), the H23 moved more than halfway from the O22 to N6 atom, but the H10 and H17 rarely moved, resulting in a EtO−-like moiety in a portion of the TS (at O22). This two proton transfer paths found in TS1 and TS2 are exactly corresponding to the protolysis and solvolysis path, respectively. Because of the fact that only one proton moved substantially, while the other two protons moved, a stepwise mechanism with a possible intermediate was predicted. However, all calculations to find the intermediate end up either the reactant or the product. These results show that concerted but asynchronous processes exist in the ESTPT.

Reactant, product and two transition states (TS1 and TS2) of the ESTPT in the 7AI–(C2H5OH)2 complex at the CASSCF(10,9)/6-311G(d,p) level
The correlation between q 1 and q 2 for the ESTPT in the 7AI–(EtOH)2 complex at the CASSCF/6-311G(d,p) level is depicted in Fig. 4. For the TS1 in the 7AI–(EtOH)2 complex, the q 1 values of H10 transfer and H23 transfer were very positive and negative, respectively, whereas those for the TS2 were opposite. These results indicate that the asynchronousity of the two processes is opposite in terms of the order of H10 and H23 transfers. It is interesting to note that the correlation points for both TSs were under the solid line, which suggests that the total bond order at TS is increased. The formation of EtOH2 +-like moieties at TS and EtO−-like moiety at TS2 might induce coulomb interactions to increase the bond order.

Correlation of the H-bond distances, q 2 = r 1 + r 2, with the proton transfer coordinate, q 1 = (1/2)(r 1 − r 2), for the 7AI–(C2H5OH)2 and 7AI–(CH3OH)2 complexes in the gas phase. All points are for the transition states in S1 optimized at the CASSCF/6-311G(d,p) level. The solid lines designate the correlation that satisfies conservation of the bond order. The parameters for Pauling equations were from the literature [35]. The structure parameters of the 7AI–(CH3OH)2 complex were from Ref. [18]. The region above and below the black line is where the sum of bond order is smaller and larger than unity, respectively
3.3 The energetics of ESPT
Barrier heights (ΔV) and excited-state tautomerization energies (ΔE) for the 7AI–EtOH and 7AI–(EtOH)2 complexes are listed in Table 3. We used the CASSCF(10,9) method with 6-31G(d,p) and 6-311G(d,p) basis set followed by a single-point MRPT2 correction to calculate the energetics of the ESPT in the 7AI–EtOH and 7AI–(EtOH)2 complexes. Tautomerization energies and barrier heights depend much on the dynamic electron correlation and basis set. The tautomerization energies of the 7AI–EtOH complex at the MRPT2/6-311G(d,p) level were −18.7 and −17.0 kcal/mol with and without the ZPE corrections, respectively. The ΔE values of the 7AI–(EtOH)2 complex were −17.6 and −17.0 kcal/mol using the 6-31G(d,p) and 6-311G(d,p) basis sets, respectively, with the ZPE corrections. The tautomerization energies predicted for the 7AI–EtOH and 7AI–(EtOH)2 complexes were within 2 kcal/mol.
The MRPT2 barrier height of the ESDPT in the 7AI–EtOH complex was 8.8 and 8.1 kcal/mol using the 6-31G(d,p) and 6-311G(d,p) basis sets without ZPE correction, respectively. For the triple-proton transfer in the 7AI–(EtOH)2 complex, two transition states were predicted at the CASSCF level, and the barrier heights of TS1 and TS2 with the MRPT2 correction were 7.4 and 9.3 kcal/mol with the 6-31G(d,p) basis set. TS2 was 1.9 kcal/mol and higher than TS1 in barrier height. These results suggest that the triple-proton transfer in the excited state occurred preferably via TS1. When the ZPE correction was included using the frequencies calculated at the CASSCF/6-311G(d,p) level, the MRPT2 barrier height of 7AI–EtOH complex was 5.3 kcal/mol. According to the transition-state theory, the rate constant of ESPT in the 7AI–EtOH complex was calculated. Kwon et al. [17] reported that the rate constant k HH in heptane was 1.05 × 1010 s−1. The rate constant obtained in this work in the gas phase was 10 times smaller than the experimental value due to overestimated barrier height by about 1.5 kcal/mol. Considering the lack of solvent effect and tunneling effect in this work, the agreement between our calculations and experiment is very good. At the same time, the ZPE-corrected MRPT2 barrier height of TS1 and TS2 in the 7AI–(EtOH)2 complex were 5.4 and 5.5 kcal/mol with the 6-31G(d,p) basis set. No obvious difference was found between the ΔV values of TS1 and TS2 with the 6-31G(d,p) basis set. When using 6-311G(d,p) basis set, the ZPE-corrected ΔV values of TS1 and TS2 with the MRPT2 correction were 4.3 and 8.2 kcal/mol, respectively. The TS2 barrier height was 3.9 kcal/mol and higher in energy than TS1. The difference in the ZPE-corrected barrier height between the 7AI–EtOH and 7AI–(EtOH)2 complexes was 1.0 kcal/mol, using the 6-311G(d,p) basis set at the MRPT2 level.
Sakota et al. [16] reported the vibrational-mode specific nature of the ESTPT; the excitation of 154, 166, 167 and 170 cm−1 vibrational modes, which belong to four structural isomers, accelerated the reaction rate. We have found a similar vibrational mode to this specific vibrational mode, 116 cm−1 at the CASSCF(10,9)/6-311G(d,p) level. This mode is a heavy-atom breathing motion without hydrogenic motions, which brings two oxygen atoms and nitrogen atoms of 7AI closer, and shortens the reaction path to reach the transition state and hence to speed up the reaction. These results are consistent with the vibrational-mode specific nature of the ESTPT. The normal mode of vibration with 116 cm−1 of frequency in 7AI–(EtOH)2 was shown in Figure SIV in the Supporting Information.
The excited-state protropic tautomerization for 7AI in bulk solvents [9, 37, 39–41] implies that solvation plays a key role in proton transfer dynamics. According to our previous results, the solvent effects reduced barrier height of 7AI–(Solvent) n (solvent: H2O, MeOH, n = 1, 2) by 3–5 kcal/mol. Whereas the mechanism of proton transfer process in solution was same as in gas phase. However, the simulation discussed here for the 7AI–(EtOH)2 complex is limited in the gas phase due to the expensive costs (more than 5 days for the frequency calculations on a 8 cores).
3.4 The effect of ethyl group substitution
By comparing the results on 7AI–(EtOH) n (n = 1, 2) with those on 7AI–(MeOH) n (n = 1, 2) reported preciously [18], some similarities of excited-state multiple-proton transfer (ESMPT) processes in these complexes can be found. The multiple-proton transfer reaction has been observed in both the 7AI–alcohol 1:1 and 1:2 clusters. For the 1:1 cluster, a concerted but asynchronous protolysis path has been found. Such concerted reaction paths are also found in the two transition states during the excited-state triple-proton transfer (ESTPT) in the 7AI–alcohol 1:2 clusters. The protolysis and solvolysis ESTPT paths are by means of TS1 and TS2, respectively. The last feature is the specific vibrational-mode of the ESTPT obtained in the 1:2 7AI–alcohol clusters. This mode shows that the excitation of this vibrational mode shortens the reaction path to reach the transition state and hence to speed up the reaction. Additionally, some differences of ESMPT processes in the 7AI–alcohol 1:n (n = 1, 2) clusters existed and discussed as follows.
At first, the geometric structures in the 7AI–(EtOH) n (n = 1, 2) and 7AI–(MeOH) n (n = 1, 2) complexes [18] are compared at the CASSCF/6-311G(d,p) level. When the methyl group in the 7AI–CH3OH complex has been replaced by ethyl group, the structural parameters changed little. The H-bond distances, H10–O16, H17–N6 in the reactant and N1–H10, O16–H17 in the product in the 7AI–EtOH complex were only 0.007, 0.002, 0.003 and 0.003 Å longer than those in the 7AI–MeOH complex, respectively. For the 7AI–ROH (R: – CH3, – C2H5) complex, one TS has been found, in which the proton from the NH group in the pyrrole ring of 7AI moved first to alcohol. The alcohol molecule accepts proton first from the 7AI molecule, which means that the basicity of mediating ROH has the effect on the geometry and energy of the 7AI–ROH (R: – CH3, – C2H5) complex. Compared to the corresponding geometric parameters of TS in the 7AI–MeOH complex, the N1–H10 and N6–H17 distances of TS in the 7AI–EtOH complex were increased 0.002 and 0.016 Å, respectively, whereas the H10–O16 and O16–H17 distances decreased 0.001 and 0.005 Å, respectively. These geometrical changes in TS were probably attributed to the electron-donating ethyl group that made the basicity (gas-phase proton affinity) of ethanol bigger than that of methanol [42, 43]. These small changes on the structural parameters resulted in few differences which can be seen in the correlation plot. As shown in Fig. 2, q 2 and q 1 values of H10 and H17 transfer at the reactant and product in the 7AI–MeOH and 7AI–EtOH complexes were almost identical. In the TS, the H17 correlation point in the 7AI–EtOH complex moved a little to the upper-left side along the black line, while the H10 correlation point was moved little. Positions of the TS on the H17 transfer reaction coordinates became a bit late.
For the 7AI–(EtOH)2 complex, H-bond H10–O16, H17–O20 and H19–N6 distances in the reactant are 0.194, 0.085 and 0.125 Å longer than the corresponding values in the 7AI–(MeOH)2 complex, respectively. In the product, the H-bond distances N1–H10, O16–H17 and O20–H19 in the 7AI–(EtOH)2 complexes are only 0.003, 0.007 and 0.035 Å longer than the corresponding values in the 7AI–(MeOH)2 complex, respectively. For the 7AI–(ROH)2 (R: – CH3, – C2H5) complex, two TSs have been obtained at the CASSCF level. ESTPT in TS1 is the path that the proton from the pyrrole ring of 7AI moved first to alcohol, while ESTPT in TS2 is the path that the proton from the alcohol moved first to pyridine ring of 7AI. Therefore, the basicity of mediating alcohol will affect the structural parameters and the barrier height in TS1, and the acidity of alcohol will affect the structural parameters and the barrier height in TS2. When the TS1 structural parameters in 7AI–(EtOH)2 were compared to those in 7AI–(MeOH)2, the N1–H10 and O16–H10 distances were increased by 0.039 Å and decreased by 0.011 Å, respectively. These results are consistent with larger proton affinity (larger basicity) of EtOH than MeOH. And the protolytic pathway in ESPT would be favored if methyl is substituted by ethyl in the ROH molecule. When the TS2 structural parameters in 7AI–(EtOH)2 were compared to those in 7AI–(MeOH)2, the O22–H23 and N6–H23 distances were almost the same although ethanol has a little larger gas-phase acidity than the corresponding value in methanol. The structural changes in the 7AI–(EtOH)2 complex led to the changes in the correlation plot (Fig. 4). Comparing the correlation H10 and H23 points in the 7AI–(EtOH)2 complex to those points in the 7AI–(MeOH)2 complex, the q1 values of H10 transfer in TS1 and TS2 were more positive and negative, respectively. However, the q 1 values of H23 transfer in TS1 and TS2 were unchanged. These results mean that the asynchronousity of proton transfer was increased because of the substitution the methyl group with the ethyl group.
Otherwise, the barrier height was affected since the methyl group was replaced by ethyl group. We found that the ZPE-corrected barrier height of the ESDPT in the 7AI–EtOH has slightly larger tautomerization barrier than that in the 7AI–MeOH complex at the MRPT2/6-311G(d,p) level. For the 7AI–(EtOH)2 complex, the TS1 and TS2 barrier heights with ZPE correction at the MRPT2/6-311G(d,p) level are 2.5 and 1.6 kcal/mol higher than those values in the 7AI–(MeOH)2 complex.
4 Conclusions
In the present work, systematic studies of the excited-state proton transfer reactions in the gas phase were performed on 7AI–(EtOH) n (n = 1, 2) complexes using CASSCF method. The energetics of the excited-state tautomerization depends on the dynamic electron correction. For the 7AI–(EtOH)2 complex, CASSCF levels predicted two concerted but asynchronous paths of ESTPT: one where the proton moved first from the pyrrole ring of 7AI to ethanol and the other where the ethanol proton moved first to the pyridine ring. No obvious difference was found between the barrier heights of the two paths without considering the dynamic electron correlation. However, the MRPT2 correction clearly showed that the former path was much referable to the latter. The tautomerization barrier of the 7AI–(EtOH)2 complex was 4.3 kcal/mol at the MRPT2/CASSCF(10,9)/6-311G(d,p) level, which is lower than that of the 7AI–EtOH complex.
The effect of the substitution of ethyl group for the methyl group in the bridge methanol molecule in the 7AI–MeOH complex has been discussed. Some similarities and differences of excited-state multiple-proton transfer (ESMPT) processes in these complexes were found. For the one ethanol complex, the ethyl group in the complex had little effect on the structures and the dynamic mechanism. However, for the two ethanol complex, the substitution of ethyl group for the methyl group led to the higher barrier height, increased the asynchronousity of proton transfer and changed the geometric parameters obviously.
References
Tuckerman ME, Marx D, Parrinello M (2002) Nature 417:925–929
Mohammed OF, Pines D, Nibbering ETJ, Pines E (2007) Angew Chem Int Ed 46:1458–1461
Lill MA, Helms V (2002) Proc Natl Acad Sci 99:2778–2781
Mathias G, Marx D (2007) Proc Natl Acad Sci 104:6980–6985
Faxen K, Gilderson G, Adelroth P, Brzezinski P (2005) Nature 437:286–289
Lu D, Voth GA (1998) J Am Chem Soc 120:4006–4014
Kohen A, Cannio R, Bartolucci S, Klinman JP (1999) Nature 399:496–499
Hay S, Pudney CR, McGrory TA, Pang J, Sutcliffe MJ, Scrutton NS (2009) Angew Chem Int Ed 48:1452–1454
Taylor CA, El-Bayoumi MA, Kasha M (1969) Proc Natl Acad Sci 63:253
Chaban MG, Gordon MS (1999) J Phys Chem A 103:185
Nakano H (1993) J Chem Phys 99:7983
Casadesús R, Moreno M, Lluch JM (2003) Chem Phys 290:319
Fernandez-Ramos A, Smedarchina Z, Siebrand W, Zgierski MZ (2001) J Chem Phys 114:7518
Kina D, Nakayama A, Noro T, Taketsugu T, Gordon MS (2008) J Phys Chem A 112:9675–9683
Park SY, Jang DJ (2010) J Am Chem Soc 132:297–302
Sakota K, Komure N, Ishikawa W, Sekiya H (2009) J Chem Phys 130:224307
Kwon OH, Lee YS, Park HJ, Kim YH, Jang DJ (2004) Angew Chem Int Ed 43:5792–5796
Fang H, Kim YH (2011) J Phys Chem A 115:13743
Fang H, Kim YH (2011) J Phys Chem B 115:15048
Fang H, Kim YH (2015) Photochem Photobiol 91(2):306
Guqlielmi M, Tavernelli I, Rothlisberger U (2009) Phys Chem Chem Phys 11:4549–4555
Tanner C, Manca C, Leutwyler S (2003) Science 302:1736–1739
Tanner C, Manca C, Leutwyler S (2005) J Chem Phys 122:204326/1
Ashfold MNR, Cronin B, Devine AL, Dixon RN, Nix MGD (2006) Science 312:1637–1640
Sakota K, Jouver C, Dedonder C, Fujii M, Sekiya H (2010) J Phys Chem A 114(42):11161–11166
Kungwan N, Plasser F, Aquion AJA, Barbatti M, Wolschann P, Lischka H (2012) Phys Chem Chem Phys 14:9016–9025
Frisch MJ, Truck GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian Inc, Wallingford
Nakano H, Nakayama K, Hirao K, Dupuis M (1997) J Chem Phys 106:4912–4917
Hirao K (1992) Chem Phys Lett 190:374–380
Hirao K (1992) Chem Phys Lett 196:397–403
Hirao K (1992) Int J Quantum Chem S26:517–526
Hirao K (1993) Chem Phys Lett 201:59–66
Hashimoto T, Nakano H, Hirao K (1998) J Mol Struc (THEOCHEM) 451:25–33
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993) J Comput Chem 14:1347
Limbach HH, Pietrzak M, Benedict H, Tolstoy PM, Golubev NS, Denisov GS (2004) J Mol Struct 706:115–119
Limbach HH, Lopez JM, Kohen A (2006) Philos Trans R Soc B 361:1399
Moog RS, Maroncelli M (1991) J Phys Chem 95:10359
Waluk J (2003) Acc Chem Res 36:832
McMorrow D, Aartsma T (1986) Chem Phys Lett 125:581
Koijnenberg J, Huizer AH, Varma CAGO (1988) J Chem Soc Faraday Trans 2(84):1163
Smirnov AV, English DS, Rich RL, Lane J, Teyton L, Schwabacher AW, Luo S, Thornburg RW, Petrich JW (1997) J Phys Chem B 101:2758
Hunter EP, Lias SG (1998) J Phys Chem Ref Data 27:413–656
Bartmess JE, Scott JA, Mclver RT Jr (1979) J Am Chem Soc 101(20):6046–6056
Acknowledgments
This work was supported from the National Nature Science Foundation of China (No. 011101009), the Nature Science Foundation of Jiangsu province (No. 164101920) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (No. 011101011). The author thanks Professor Kim Yongho, working in Department of Applied Chemistry, Kyung Hee University, for providing computing resources.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fang, H. Theoretical study on excited-state proton transfer via hydrogen-bonded ethanol (EtOH) wire for 7AI in the gas phase. Theor Chem Acc 134, 142 (2015). https://doi.org/10.1007/s00214-015-1723-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-015-1723-6