
Abstract
The neutral Si n K (n = 2–8) clusters and their anions have been systematically studied by means of the higher level of Gaussian-3 schemes. Equilibrium geometries and electron affinities have been calculated and are discussed for each considered size. For neutral Si n K clusters, the ground state structure is found to be “attaching structure”, in which the K atom is bound to Si n clusters. The most stable isomer for their anions, however, is found to be “substitutional structures”, which is derived from Si(n+1) by replacing the Si atom with a K. The dissociation energies of K atom from the lowest energy structures of Si n K have also been estimated to examine relative stabilities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Clusters of silicon and metal have received considerable attention in the past decade. Specially, alkali–silicon clusters possess scientific value because it has been known that they serve as promoters in catalysts, and can be used as power source material for spaceflight aero-crafts, emitters, and many other products [1–8]. Extensive experimental [9–13] and theoretical [14–22] studies on alkali metal–silicon clusters have recently been reported. For example, the electron affinities and ionization potentials of Si n Na m have been measured by means of photoelectron spectra [9–13]. Rabilloud et al. [14, 15] have performed investigation of structures and properties of neutral and positively charged Si n Li + p and Si n Na + p (n ≤ 6, p ≤ 2) clusters by means of B3LYP/6-31+G(d) method and concluded that the structures of the neutral (Si n Li p and Si n Na p ) and their cations clusters keep the frame of the corresponding Si n , Li, and Na species being adsorbed at the surface. We [7] have carried out G3 calculations for Si n Li (n = 2–8) and their anions, and found that the ground state structures of neutral Si n Li clusters are “attaching structure” and the most stable structures of Si n Li− anions are “substitutional structures”.
The potassium–silicon clusters have been more sparsely studied than lithium– and sodium–silicon clusters. For neutrals of potassium–silicon clusters, the geometries, population analysis, and electric dipole moments of Si n K p (n ≤ 6, p ≤ 2) have only been reported by Rabilloud et al. [8, 16]. Compared with Si n Li p and Si n Na p , the most stable structures of Si n A p are found to be similar for A = Li, Na, and K [8, 16]. For anions of potassium–silicon clusters, Li et al. [21] have explored the ground state structures of Si n K− with n ≤ 10. In this study, we have investigated the reliable electronic structures, electron affinities, and dissociation energies of small silicon–potassium clusters with the aim of understanding how their properties differ from those of bare silicon clusters. We found that the most stable isomer for negatively charged ion Si n K− is “substitutional structures”. In addition, we found that the ground state structures of Si n K− with n = 6, 7, and 8 are different from those reported previously [21]. The ground state structures of Si n K− with n = 6, 7, and 8 presented in this paper are more stable than those reported previously [21].
2 Computational methods
All of calculations have been performed using the Gaussian-3 (G3) method [23, 24] and the Gaussian 03 package [25]. The G3 theory is a composite technique in which the geometry optimization is carried out at the MP2(full)/6-31G(d) level. The energy, a series of single-point energy calculation at the levels of QCISD(T)/6-31G(d), MP4/6-31G(d), MP4/6-31+G(d), MP4/6-31G(2df,p), and MP2(full)/G3large with the MP2(full)/6-31G(d) geometry is carried out. Then, this energy is modified by a series of corrections. Finally, the HF/6-31G(d) vibrational frequencies, scaled by 0.893, are applied for the zero-point vibrational energy (ZPVE) correction at 0 K. The combined G3 methods are the higher level of ab initio calculations of molecular energies of compounds containing first-, second-, and third-row atoms. The average absolute deviation from experiment for the electron affinities are only 0.99 kcal/mol for a set of 63 molecules [23, 24].
Two types of initial geometries for both neutral Si n K (n = 2–8) and their anions are taken into account. One is the “substitutional structure”, which can be regarded as being derived from Si(n+1) by replacing a Si atom with a K atom. In addition, the other is the “attaching structure” in which the K atom is bound to Si n geometry.Footnote 1 For the “attaching structure”, two types of structures are also taken into account. One is the bridge-site type and the other is the apex-site type, in which the potassium atom is bound to one of the silicon atoms. Nevertheless, the apex-site type is found to be either a saddle point or a local minimal point on the potential surface. An important fact is that the ground state structure of neutral Si n K is “attaching structure” and the ground state structure of anion Si n K− is “substitutional structures”.
3 Result and discussion
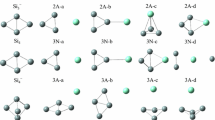
The geometries optimized with MP2(full)/6-31G(d) level for Si n K (n = 2–8) clusters and their anions are displayed in Fig. 1. Frequencies are calculated to verify that the structures are local minima on the potential energy surface and not transition states at MP2(full)/6-31G(d) level.

The optimized geometries for neutral Si n K (n = 2–8) and their anions in which only silicon atoms are numbered. The MP2(full)/6-31G(d) bond lengths are shown in Å
3.1 Si2K and Si2K−
For neutral Si2K, Rabilloud et al. [8, 16] have presented that its ground state structure is C 2v symmetry. Similarly to Si2H [26], Si2Li [6, 7], and Si2Na [10, 22], neutral Si2K has a 2A1 ground state with a low-lying 2B1 excited state. The latter state is only 0.05 eV higher in energy at the G3 level of theory. The bond distance of K–Si is predicted to be 3.325 Å at MP2(full)/6-31G(d) level, which is in excellent agreement with the value of 3.33 Å predicted by the B3LYP/6-31+G(d) scheme [8, 16].
For negatively charged ion Si2K−, the ground state structure displays C 2v symmetry with 1A1 state. This result agrees with those reported by Li’s report [21]. The bond length of K–Si is predicted to be 3.118 Å at MP2(full)/6-31G(d) level, which agrees excellently with the value of 3.120 Å predicted by the MP2(full)/6-311+G(d) method. As can be seen from Fig. 1, the K–Si bond distance of anion Si2K− is shorter than that of neutral by 0.207 Å. The reason, similarly to Si n Na system described by Kishi et al. [11], is that the additional electron going into the SOMO of the neutral Si n K becomes doubly occupied in the anion, which localizes mainly on the Si n framework. However, the electron back-donation from the Si n framework to the K atom is induced and makes the bond between the Si n and K atom strong.
3.2 Si3K and Si3K−
The results of G3 calculation show that the geometries of the ground state structure of Si3K display C 2v symmetry, which are the same as those reported by Rabilloud et al. [8, 16]. Similarly to Si3H [27, 28], Si3Li [6, 7], and Si3Na [10], Si3K has a 2A1 ground state with a low-lying 2B2 excited state. The latter state is only higher in energy by 0.16 eV at the G3 level. The K–Si bond lengths are calculated to be 3.209 and 3.240 Å for 2A1 and 2B2 electronic state, respectively. Rabilloud et al. [8, 16] presented a K–Si bond distances of 3.26 Å at B3LYP/6-31+G(d) level of theory.
For negatively charged ion Si3K−, the ground state structures possess C 2v symmetry with 1A1 state. The K–Si bond lengths are calculated to be 3.162 Å, which are shorter than that of neutral with 2A1 state by 0.047 Å. The reason is described earlier. At MP2(full)/6-311+G(d) level, the K–Si bond length of Si3K− is predicted to be 3.142 Å [21].
3.3 Si4K and Si4K−
Three minima for Si4K are shown in Fig. 1. Both Si4K−I and Si4K−II are “attaching structure” and display C s symmetry, whereas the former is 2A′ state, and the latter is 2A″ state. The 2A″ state is higher in energy than the 2A′ state by 0.13 eV at the G3 level. This result is different from those reported previously [8, 16]. We have also performed DFT calculations. At the B3LYP level with G3Large basis set [23, 24], the planar isomer of Si4K-II is also higher in energy than that of the Si4K-I structure by 0.06 eV. This case is different from Si4Li. For Si4Li, the planar isomer of the 2A″ state is slightly lower in energy than that of the 2A′ state at the B3LYP/G3Large level [7]. The ground state of Si4A (A = Li, Na, K) is different from Si4H. In fact, the H–Si bonds of ground state structures of Si n H with the exception of Si2H and Si3H are stretched bounds [29]. However, the A–Si bonds for ground state geometries of Si n A are bridged [6–16]. The C 2v symmetry of the 2B2 Si4K-III isomer is “substitutional structure”. That is, it can be regarded as being derived from Si5 by replacing a Si atom with a K atom. Energetically, it is higher in energy than the Si4K-I by 0.47 eV. As can be seen from Fig. 1, the bond distances for the two equivalent K–Si bonds of Si4K-I structure are predicted to be 3.389 Å at MP2(full)/6-31G(d) level.
For negatively charged ion Si4K−, two isomers are shown in Fig. 1. The Si4K−-I, “substitutional structure”, with C 2v symmetry and 1A1 state is predicted to be the most stable. The Si4K−-II displays C s symmetry with 1A′ state and belongs to “attaching structure”. That is, it can be regarded as being derived from Si4 by attaching to a K atom. Energetically, it is higher in energy than the Si4K−-I by 0.13 eV at the G3 level of theory. The geometries of Si4K−-I are similar to that of structure predicted at MP2(full)/6-311+G(d) level of theory [21]. The shorter K–Si bond lengths for Si4K−-I are calculated to be 3.068 Å.
3.4 Si5K and Si5K−
Two minima for Si5K are shown in Fig. 1. The Si5K-I structure derived from Si5 by edge capping with a K atom displays C 2v symmetry with 2B 2 state. The Si5K-II isomer derived from Si5 by face capping with a K atom displays C s symmetry with 2A′ state. At B3LYP/6-31+G(d) level of theory, Rabilloud et al. reported that the Si5K-II isomer was the ground state structure for Si5K [8, 16]. However, the Si5K-I structure is more stable than that of Si5K-II isomer by 0.32 eV in energy at the G3 level of theory. The two equivalent K–Si bond lengths for Si5K-I are calculated to be 3.230 Å.
For negatively charged ion Si5K−, the ground state structure is C s symmetry with 1A′ state as can be seen from Fig. 1. This result is the same as the previous result predicted at MP2(full)/6-311+G(d) level of theory [21]. The geometry of ground state of Si5K− can be regarded as being derived from not only Si5 by face capping with a K atom (“attaching structure”) but also Si6 by replacing a Si atom with a K atom (“substitutional structure”). The bond lengths of Si5K− are calculated to be 3.146 Å for the two equivalent K–Si bonds and 3.324 Å for K−Si3 bond. All of these K–Si bond lengths are shorter than those of corresponding neutral (Si5K-II) by 0.145 and 0.018 Å, respectively. The reason is described earlier.
3.5 Si6K and Si6K−
The geometry of the ground state of neutral Si6K, “attaching structure”, displays C 2v symmetry with 2B2 state (see Fig. 1). This result agrees with the result predicted by B3LYP/6-31+G(d) scheme [8, 16]. The bond lengths are predicted to be 3.295 Å for the two equivalent K−Si5 and K−Si6 bonds and 3.450 Å for the another two equivalent K−Si3 and K−Si4 bonds, all of which are shorter than the B3LYP/6-31+G(d) K–Si bond lengths by 0.065 and 0.020 Å, respectively [8, 16].
For anion Si6K−, the lowest-energy structure, Si6K−-I shown in Fig. 1, displays C 2v symmetry with 1A1 state. This result is different from those reported previously [21]. Li et al. [21] presented that the ground state structure of Si6K− displayed C 3v symmetry as Si6K−-II shown in Fig. 1. At the G3 level of theory, the Si6K−-I structure is more stable than the Si6K−-II by 0.07 eV in energy. In fact, the geometry of ground state of Si6K−-I belongs to not only “attaching structure”, but also “substitutional structure”. As can be seen from Fig. 1, the K–Si bond distances of anion Si6K−-I are again shorter than that of neutral Si6K.
3.6 Si7K and Si7K−
The geometry of the ground state of neutral Si7K, “attaching structure”, possesses C s symmetry with 2A′ state (see Fig. 1). This result is different from the ground state structure of Si7Li and Si7Na [7, 10, 18], which are C 2v symmetry with 2B1 state. For Si7K, the geometry of C 2v symmetry with 2B1 state is higher in energy than that of C s symmetry with 2A′ state by 0.05 eV. The bond lengths of the ground state structure are predicted to be 3.315 Å for the two equivalent K−Si3 and K−Si4 bonds and 3.289 Å for K−Si2 bonds.
Two minima for anion Si7K− are shown in Fig. 1. The Si7K−-I is “substitutional structure” and displays C 1 symmetry. The Si7K−-II is “attaching structure” and displays C s symmetry with 1A′ state. Li et al. [21] presented that the ground state structure of Si7K− was Si7K−-II isomer at MP2(full)/6-311+G* level. We also performed the MP2(full)/6-311+G* calculation. The results show that the Si7K−-I structures are more stable than the Si7K−-II isomers by 0.22 eV in energy at MP2(full)/6-311+G*. At the G3 level, both the Si7K−-I and the Si7K−-II isomers are nearly identical in energy (the Si7K−-I structures are slightly lower than the Si7K−-II isomers by −0.007 eV at the G3 level with ZPVE correction). Compared to the single-point energy at the levels of QCISD(T)/6-31G(d), MP4/6-31G(d), MP4/6-31+G(d), MP4/6-31G(2df,p), and MP2(full)/G3large with MP2(full)/6-31G(d) geometry, the results show that Si7K−-I structures are more stable than the Si7K−-II isomers by 0.12, 0.27, 0.33, 0.28, and 0.19 eV in energy, respectively. In this case, we assign the Si7K−-I geometry to the ground state structure of anion Si7K−.
3.7 Si8K and Si8K−
The geometry of the ground state of neutral Si8K, “attaching structure”, has C 2v symmetry with 2B1 state (shown in Fig. 1). The bond lengths are predicted to be 3.327 Å for the two equivalent K−Si4 and K−Si6 bonds, 3.298 Å for the equivalent K−Si8 and K−Si3 bonds.
For anion Si8K−, two isomers are shown in Fig. 1. The Si8K−-I is not only “attaching structure”, but also “substitutional structure”, and displays C 2v symmetry with 1A1 state. Compared with the neutral Si8K, the K–Si bond distances of anion Si8K−-I are again shorter by 0.130 and 0.123 Å, respectively. The Si8K−-II isomer with C s symmetry and 1A′ state is similar to the C 1 structure reported by Li et al. [21]. Energetically, the Si8K−-II isomer is less stable than the Si8K−-I structure by 0.74 eV in energy at the G3 level. At MP2(full)/6-311+G* level, the Si8K−-II isomer is less stable than the Si8K−-I structure by 0.63 eV.
It is interesting to note that the ground state structures of anion Si n K− can be regarded as being derived from Si(n+1) by replacing a Si atom with a K atom, that is, “substitutional structure”. Although the ground state structures of Si n K− (n = 2, 3, 5, 6, 8) can also be regarded as being derived from Si n by attaching to a K atom, the ground state structures of Si4K− and Si7K− are conclusive evidence that the lowest-energy structures are “substitutional structures”. For neutral Si n K, the most stable structure is “attaching structure” in which the K atom is bound to at least two silicon atoms. This result is the same as that of previous studies [8, 16].
3.8 Electron affinities
The adiabatic electron affinities (EA) (defined as the difference of total energies in the manner EA = E (optimized neutral) – E (optimized anion) of Si n K clusters are calculated at the G3 level. The ZPVE corrected electron affinities of Si n K are predicted to be 1.48 (1.48) eV for Si2K, 1.49 (1.49) eV for Si3K, 1.36 (1.36) eV for Si4K, 2.19 (2.18) eV for Si5K, 1.75 (1.76) eV for Si6K, 1.62 (1.61) eV for Si7K, and 2.69 (2.71) eV for Si8K (presented in parentheses without ZPVE correction). Compared to Si n Li and Si n (see Fig. 2), it can be found that (1) the variation trend of the calculated EAs of Si n K and Si n Li clusters vary in parallel curves with local maxima around n = 5 and 8 and local minima at n = 4 and 7. As expected the EAs of Si n K is always lower than that of Si n Li cluster; and (2) the EA of Si n K cluster is lower than that of the corresponding Si n at the cluster size n ≤ 7, whereas it is higher at n ≥ 8. For Si n Li clusters, the EA is lower than that of the corresponding Si n at the cluster size n ≤ 4, and it is higher at n ≥ 5 [7]. The reason can be explained as follows. (1) When a alkali metal atom is adsorbed on Si n clusters, the charge transfer from alkali metal atom to silicon clusters (in fact, alkali metal atom is positive charge and silicon cluster is negative charge in Si n A) results in decrease of the electron affinities of Si n cluster. However, with the increase of silicon cluster size, the average charge obtained by each silicon atom became less and less. That is, the effect of decrease of electron affinities becomes weak with the increase of silicon cluster size. (2) The change from closed-shell to open-shell (that is, the Si n cluster is generally closed-shell and Si n A cluster is open-shell) results in increase of the electron affinities. (3) The negative charge of Si n clusters in Si n K is more negative than that in Si n Li because the ionization potential of lithium atom is larger than that of potassium (that is, the ability of capture electron for Si n K is weaker than for Si n Li). All of these lead to the results described above.

The ZPVE corrected adiabatic electron affinities (EA) in eV for Si n , Si n Li, and Si n K (n = 2–8) clusters versus the clusters size n. The EAs of Si n and Si n Li clusters are taken from Ref. [7]
3.9 Dissociation energies
The dissociation energy (D e) (defined as the energy required in the reaction Si n K → Si n +K) of Si n K clusters are predicted with the G3 scheme to be 2.43 (2.45) eV for Si2K, 2.20 (2.23) eV for Si3K, 1.93 (1.94) eV for Si4K, 2.27 (2.31) eV for Si5K, 1.88 (1.91) eV for Si6K, 1.36 (1.38) eV for Si7K, and 2.09 (2.12) eV for Si8K (presented in parentheses without ZPVE correction). From the D e, the stability of bonding a K atom to silicon clusters can be found. The higher the values of these dissociation energies are, the more stable the clusters bonding of a K atom are. A better way of comparing the local relative stabilities of different size clusters is by means of the incremental binding energies [30]. The D e of the Si n K and Si n Li [7] clusters as a function of the size of the clusters is shown in Fig. 3. As can be seen from Fig. 3, the two parallel oscillating curves, the top curve for Si n Li and the lower curve for Si n K, show that (1) the Si n K (and Si n Li) for n = 4 and 7 are less stable than for n = 2, 5, and 8 because the dissociation energies are local minima for n = 4 and 7 and local maxima for n = 2, 5, and 8. These also indicate that Si n for n = 4 and 7 are more stable and for n = 2, 5, and 8 are less stable. This result is consistent with previous result with respect to Si n [30–32]. And (2) as expected the dissociation energies of lithium atom are larger than that of potassium atom since the small size of Li atom results in a higher stability. That is, Li adsorption on surface of silicon clusters is more stable than K.

The ZPVE corrected dissociation energies (D e) in eV for Si n Li and Si n K (n = 2–8) clusters versus the clusters size n. The D e of Si n Li clusters are taken from Ref. [7]
4 Conclusions
The equilibrium geometries and electronic structures of small Si n K clusters (n = 2–8) and their anions have been systematically investigated by means of the higher level of the G3 scheme. Similarly, to Si n Li, the ground-state structures of neutral Si n K are predicted to be “attaching structure” in which the potassium atom is bound to Si n clusters. The ground-state structures of anion Si n K−, however, are “substitutional structures” which is derived from Si(n+1) by replacing a Si atom with a K−. The adiabatic electron affinities of Si n K have been calculated. The reliable adiabatic electron affinities are predicted to be 2.43 (2.45) eV for Si2K, 2.20 (2.23) eV for Si3K, 1.93 (1.94) eV for Si4K, 2.27 (2.31) eV for Si5K, 1.88 (1.91) eV for Si6K, 1.36 (1.38) eV for Si7K, and 2.09 (2.12) eV for Si8K (presented in parentheses without ZPVE correction). The dissociation energies of K from the lowest energy structure of Si n K clusters have been calculated and used to reveal relative stability. The dissociation energies are predicted to be 2.43 (2.45) eV for Si2K, 2.20 (2.23) eV for Si3K, 1.93 (1.94) eV for Si4K, 2.27 (2.31) eV for Si5K, 1.88 (1.91) eV for Si6K, 1.36 (1.38) eV for Si7K, and 2.09 (2.12) eV for Si8K (presented in parentheses without ZPVE correction). As expected both the electron affinities and the dissociation energies of Si n K are smaller than that of Si n Li. To the best of our knowledge, there are no experimental data regarding the electron affinity and dissociation energy for Si n K systems. Our results may thus provide a reference for further investigations.
References
Beck SM (1989) J Chem Phys 90:6306
Ohara M, Koyasu K, Nakajima A, Kaya K (2003) Chem Phys Lett 371:490
Binning RC Jr, Bacelo DE (2005) J Phys Chem A 109:754
Koyasu K, Akutsu M, Mitsui M, Nakajima A (2005) J Am Chem Soc 127:4998
Jaeger JB, Jaeger TD, Duncan MA (2006) J Phys Chem A 110:9310
Yang JC, Lin LH, Zhang YS, Jalbout AF (2008) Theor Chem Account 121:83
Hao DS, Liu JR, Yang JC (2008) J Phys Chem A 112:10113
Rabilloud F, Sporea CJ (2007) Comput Meth Sci Eng 7:273
Kaya K, Sugioka T, Taguwa T, Hoshino K, Nakajima A (1993) Z Phys D 26:S201
Kishi R, Iwata S, Nakajima A, Kaya K (1997) J Chem Phys 107:3056
Kishi R, Kawamata H, Negishi Y, Iwata S, Nakajima A, Kaya K (1997) J Chem Phys 107:10029
Zubarev DY, Boldyrev AI, Li X, Cui LF, Wang LS (2005) J Phys Chem A 109:11385
Zubarev DY, Alexandrova AN, Boldyrev AI, Cui LF, Li X, Wang LS (2006) J Chem Phys 124:124305
Sporea C, Rabilloud F, Cosson X, Allouche AR, Aubert-Frécon M (2006) J Phys Chem A 110:6032
Sporea C, Rabilloud F, Allouche AR, Frécon M (2006) J Phys Chem A110:1046
Sporea C, Rabilloud F, Aubert-Frécon M (2007) J Mol Struct Theochem 802:85
Sporea C, Rabilloud F (2007) J Chem Phys 127:164306
Wei S, Barnett RN, Landman U (1997) Phys Rev B 55:7953
Zhao GF, Sun JM, Liu X, Guo LJ, Luo YH (2008) J Mol Struct Theochem 851:348
Wang H, Lu WC, Li ZS, Sun CC (2005) J Mol Struct Theochem 730:263
Li SD, Ren GM, Jin ZH (2003) J Chem Phys 119:10063
Lin LH, Yang JC, Ning HM, Hao DS, Fan HW (2008) J Mol Struct Theochem 851:197
Curtiss LA, Raghavachari K, Redfern PC, Rassolov V, Pople JA (1998) J Chem Phys 109:7764
Curtiss LA, Redfern PC, Rassolov V, Kedziora G, Pople JA (2001) J Chem Phys 114:9287
Gaussian 03, Revision C.02, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian Inc., Wallingford
Pak C, Rienstra-Kiracofe JC, Schaefer HF (2000) J Phys Chem A 104:11232
Xu C, Taylor TR, Burton GR, Neumark DM (1998) J Chem Phys 108:7645
Xu WG, Yang JC, Xiao WS (2004) J Phys Chem A 108:11345
Yang JC, Bai X, Li CP, Xu WG (2005) J Phys Chem A 109:5717
Raghavachari K (1986) J Chem Phys 84:5672
Raghavachari K, Rohlfing CM (1988) J Chem Phys 89:2219
Yang JC, Xu WG, Xiao WS (2005) J Mol Struct Theochem 719:89
Acknowledgments
This work has been financially supported by a research grant (Grant No. NJ05052) administered by the Science and Research Foundation of Higher Education of Inner Mongolia and by the NCET Grant (Grant No. NCET-06-0267) from the Ministry of Education of the People’s Republic of China.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hao, DS., Liu, JR., Wu, WG. et al. Study on structures and electron affinities of small potassium–silicon clusters Si n K (n = 2–8) and their anions with Gaussian-3 theory. Theor Chem Acc 124, 431–437 (2009). https://doi.org/10.1007/s00214-009-0635-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00214-009-0635-8