
Abstract
Rationale
Ketamine, a non-competitive NMDA receptor antagonist, induces acute effects resembling the positive, negative and cognitive symptoms of schizophrenia. Chronic use has been suggested to lead to persistent schizophrenia-like neurobiological changes.
Objectives
This study aims to test the hypothesis that chronic ketamine users have changes in brain neurochemistry and increased subthreshold psychotic symptoms compared to matched poly-drug users.
Methods
Fifteen ketamine users and 13 poly-drug users were included in the study. Psychopathology was assessed using the Comprehensive Assessment of At-Risk Mental State. Creatine-scaled glutamate (Glu/Cr), glutamate + glutamine (Glu + Gln/Cr) and N-acetyl aspartate (NAA/Cr) were measured in three brain regions—anterior cingulate, left thalamus and left medial temporal cortex using proton magnetic resonance spectroscopy.
Results
Chronic ketamine users had higher levels of subthreshold psychotic symptoms (p < 0.005, Cohen’s d = 1.48) and lower thalamic NAA/Cr (p < 0.01, d = 1.17) compared to non-users. There were no differences in medial temporal cortex or anterior cingulate NAA/Cr or in Glu/Cr or Glu + Gln/Cr in any brain region between the two groups. In chronic ketamine users, CAARMS severity of abnormal perceptions was directly correlated with anterior cingulate Glu/Cr (p < 0.05, r = 0.61—uncorrected), but NAA/Cr was not related to any measures of psychopathology.
Conclusions
The finding of lower thalamic NAA/Cr in chronic ketamine users may be secondary to the effects of ketamine use compared to other drugs of abuse and resembles previous reports in individuals at genetic or clinical risk of schizophrenia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ketamine, a dissociative anaesthetic agent and non-competitive NMDA receptor antagonist, induces acute effects at subanaesthetic doses that resemble the positive, negative and cognitive symptoms of schizophrenia (Krystal et al. 1994). When administered to patients with stable schizophrenia, ketamine may lead to a relapse of symptoms indistinguishable from the idiopathic disease (Lahti et al. 1995).
Despite considerable interest in the NMDA receptor antagonist model of psychosis, acute ketamine administration has been suggested to be somewhat unlike idiopathic psychoses because of its relatively short-lived effects (Tsai and Coyle. 2002). In contrast, chronic ketamine administration may be more representative of the long-term NMDA receptor dysfunction hypothesised to occur in schizophrenia (Tsai and Coyle. 2002). In rats, repeated administration of ketamine has been reported to lead to neurodegenerative changes in brain regions resembling those affected in schizophrenia (Keilhoff et al. 2004). Reductions in brain N-acetyl aspartate (NAA), suggested to be a marker of neuronal integrity and, possibly, of glutamatergic neuronal function (Reynolds and Harte. 2007), have been reported in rats administered the NMDA receptor antagonist phencyclidine (Reynolds et al. 2005). NAA reductions measured using proton magnetic resonance spectroscopy (1H-MRS) are also a consistent finding in patients with schizophrenia, becoming more marked and affecting more regions with disease progression (Steen et al. 2005; Brugger et al. 2011). NAA reductions in thalamus have been reported in individuals at high clinical and genetic risk of psychosis, whilst more widespread reductions, including temporal cortex and frontal cortex, have been reported in patients with chronic schizophrenia (Brugger et al. 2011). These findings suggest that reduced thalamic NAA may be an indicator of early pathophysiological changes linked to the risk for psychosis.
In terms of brain glutamate levels, 1H-MRS findings have been mixed. In individuals at risk of psychosis, and in patients with first-episode psychosis, elevations in medial prefrontal glutamine or glutamate + glutamine levels have been reported (Bartha et al. 1997; Théberge et al. 2002; Tibbo et al. 2004; Stone et al. 2009; Marsman et al. 2013), but others have not found any evidence of difference in brain glutamate metabolites in these groups (Wood et al. 2007; Yoo et al. 2009). In contrast, in patients with chronic schizophrenia, brain glutamate levels have generally been reported as being normal or low (Szulc et al. 2013; Marsman et al. 2013), although this may be an effect of medication as a recent study demonstrated elevated medial prefrontal glutamate + glutamine levels in unmedicated patients with chronic schizophrenia (Kegeles et al. 2012).
To date there have been three reports of glutamate metabolite estimation with 1H-MRS following acute ketamine administration in humans. One reported increased anterior cingulate glutamine (Rowland et al. 2005), one reported increased anterior cingulate glutamate (Stone et al. 2012), and one reported no change in anterior cingulate glutamate + glutamine levels (Taylor et al. 2011), although the latter study was potentially underpowered, utilising a between-subject analysis with a sample size of nine subjects receiving placebo and eight receiving ketamine. No human studies of the effects of chronic ketamine use on brain glutamate have been reported, but in rats elevations in brain glutamate measured using 1H-MRS have been reported (Kim et al. 2011).
Although chronic NMDA receptor blockade is an established preclinical model of schizophrenia, it is not known if chronic NMDA receptor blockade in humans is associated with reductions in levels of brain NAA and increases in glutamate seen in preclinical models and in people with early symptoms of psychosis. It would not be ethical to administer daily ketamine to healthy volunteers over an extended period of months or years, but an alternative method to study chronic NMDA receptor hypofunction in humans is to study people who choose to take ketamine regularly. Ketamine users have been reported to have increased prefrontal dopamine D1 receptors (Narendran et al. 2005) as well as reduced gray matter volume in anterior cingulate cortex (Liao et al. 2011). In this study, we tested the hypothesis that compared to non-ketamine-using poly-drug users, individuals with chronic ketamine use have greater levels of subthreshold psychotic symptoms, elevations in creatine-scaled anterior cingulate glutamate (Glu/Cr) and glutamate + glutamine (Glu + Gln/Cr) and reductions in creatine-scaled thalamic, medial temporal cortex and anterior cingulate N-acetyl aspartate (NAA/Cr).
Methods
The study was approved by Imperial College London Research Ethics Committee, UK. Fifteen ketamine users (age range 20–29 years old, with a history of using ketamine at least three times per week for the last year) and 13 poly-drug-using controls (matched for age, sex and education level but who had no history of regular ketamine use) were included in the study. All participants completed information about their level and history of substance use including ketamine. Levels of subthreshold psychotic symptoms were assessed using the Comprehensive Assessment of At-Risk Mental State (CAARMS)—abnormalities of thought content (bizarre + non-bizarre), abnormalities of perceptions and abnormalities of speech production subscales (Yung et al. 2005), with participants describing their experiences when drug-free.
All participants then underwent MRI imaging on a Philips 3-T Intera magnetic resonance system, software release 2.1.3. An initial localizer scan was performed followed by acquisition of a whole-brain 3D-MPRAGE (TR = 9.6 ms, TE = 4.5 ms, flip angle 8°, slice thickness = 1.2 mm, 0.94 mm × 0.94 mm in plane resolution, 150 slices). PRESS (Point RESolved Spectroscopy) data were then acquired (TE = 35 ms; TR = 3,000 ms; 64 averages for anterior cingulate, 96 averages for medial temporal cortex and thalamus), utilising the default “Excite” water suppression routine. Auto-shimming was performed using second-order pencil beam shim. The 3D MPRAGE was used to place regions of interest for the PRESS analysis. The anterior cingulate region of interest (ROI) was prescribed from the midline sagittal slice, and the centre of the 20 mm × 20 mm × 20 mm ROI was placed 13 mm above the anterior section of the genu of corpus callosum at 90o to the AC-PC line (Fig. 1). The centre of the thalamus ROI (20 mm × 15 mm × 20 mm) was defined at the point in the axial slices where the thalamus was widest, using sagittal and axial planes, and rotating the ROI to ensure that it was clear of CSF (Fig. 2). The centre of the medial temporal cortex ROI (20 mm × 20 mm × 15 mm) was positioned superior to the anterior edge of the cerebellum and angled in line with the body of the hippocampus (Fig. 3).

Placement of anterior cingulate ROI

Placement of left thalamus ROI

Placement of left medial temporal cortex ROI
PRESS spectra (Fig. 4) were analyzed using LCModel version 6.3. The raw spectral data were read into LCMgui, the graphical user interface for LCModel. A standard basis set of 16 metabolites (l-alanine, aspartate, creatine, phosphocreatine, GABA, glucose, glutamine, glutamate, glycerophosphocholine, glycine, myo-inositol, l-lactate, N-acetyl aspartate, N-acetylaspartylglutamate, phosphocholine, taurine), included as part of LCModel and acquired with the same field strength (3 T), localization sequence (PRESS) and echo time (35 msec) as our study was used. Model metabolites and concentrations employed in the basis set are fully detailed in the LCModel manual (http://s-provencher.com/pages/lcm-manual.shtml). For all metabolites, we used the recommended cutoff of Cramer-Rao Lower Bounds (CRLB) of 20 % to exclude poorly fitted metabolite peaks. As absolute quantification is not possible at field strengths above 1.5 T due to the breakdown of the law of reciprocity (Jansen et al. 2006) and due to the fact that we did not have access to unsuppressed PRESS spectra for water scaling, we quantified all metabolites as a ratio to creatine.

Sample 1H-MRS spectra (LCModel output) from anterior cingulate, thalamus, and medial temporal cortex ROIs (control—left, ketamine—right)
Group differences in demographic, drug use, rating and metabolite data were assessed with Welch's two-sample t-test and the chi-square test was implemented in R version 2.14.1 (Ihaka and Gentleman. 1996). Correlations between metabolite levels and CAARMS severity of abnormal thought content, abnormal perceptions or speech production were investigated in ketamine users using Pearson's correlation.
Results
Ketamine users did not differ from controls in demographic or other substance use variables, except for amount of amphetamine used per month (Table 1). Ketamine users had significantly greater severity of CAARMS—abnormalities of thought content (mean score = 2.3 vs. 0.54; t = 3.36; p = 0.003, Cohen's d = 1.48), with six of the ketamine users and 0 controls reaching formal criteria for an at-risk mental state (Table 1) (Yung et al. 2005).
1H-MRS spectra were of reasonable quality in anterior cingulate and left thalamus, with a mean (SD) signal-to-noise ratio reported by LCModel of 16.9 (3.4) and 11.75 (3), respectively, and of passable quality in left medial temporal cortex with a mean (SD) signal-to-noise ratio of 8.1 (3.4). Line widths reported by LCModel followed a similar pattern with mean (SD) of 5.4 (0.97) Hz in anterior cingulate, 6.4 (1.4) Hz in left thalamus and 8.3 (3.3) Hz in left medial temporal cortex. Signal-to-noise ratio did not differ in any region between controls and ketamine users, but the line width in thalamus was wider in ketamine users (mean = 5.9 Hz, 7.0 Hz; p = 0.03). Glu + Gln/Cr was not measureable at CRLB < 20 % in thalamus in one ketamine user or in medial temporal cortex in two ketamine users and two controls. Medial temporal cortex NAA/Cr was not measureable at CRLB < 20 % in one ketamine user.
Ketamine users had significantly lower NAA/Cr in thalamus than controls (mean level = 1.17 vs 1.24; t = 2.84; p < 0.01, Cohen's d = 1.17). This did not appear to be due to differences in scan quality between ketamine users and controls as there was no relationship between LCModel-reported line width and NAA/Cr in thalamus (p = 0.2). There were no differences in medial temporal cortex or anterior cingulate NAA/Cr or in Glu + Gln/Cr or Glu/Cr in any brain region between the two groups (p > 0.05; Table 2).
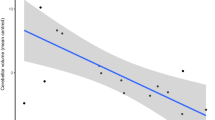
In ketamine users, there was a significant relationship between anterior cingulate Glu/Cr levels and CAARMS severity of abnormal perceptions (p = 0.02, r = 0.61; Fig. 5). A similar trend relationship was seen between anterior cingulate Glu + Gln/Cr and CAARMS severity of abnormal perceptions in ketamine users (p = 0.06, r = 0.48). However, these relationships were not corrected for multiple comparisons (18 tests in total) and were not present in the total sample (poly-drug controls plus ketamine users). There was no relationship between thalamic NAA levels and CAARMS severity of abnormal thought content, abnormal perceptions or speech production in ketamine users or controls.

Relationship between Glu/Cr in anterior cingulate and CAARMS severity of abnormal perceptions in ketamine users
Discussion
This is the first study, to our knowledge, to report brain 1H-MRS findings in chronic ketamine users. The finding that ketamine users have subthreshold psychotic symptoms as well as reduced levels of thalamic NAA/Cr reflects previous reports of reduced thalamic NAA in individals at clinical and generic risk of schizophrenia as well as in patients with first episode and chronic schizophrenia (Brugger et al. 2011). It suggests that adding chronic ketamine use to poly-drug use is associated with psychotic symptoms and brain alterations in the thalamus similar to those seen during the development of schizophrenia. This is of relevance in the treatment of people with chronic ketamine abuse as it suggests that they may be at increased risk of developing psychosis compared to poly-drug users.
The thalamus is thought to play a key role in the filtering of sensory information, integrating internal and external perceptions. Abnormalities of thalamic function have been hypothesised to be central to the development of psychotic symptoms (Carlsson and Carlsson. 1990; Vollenweider and Geyer. 2001; Clinton and Meador-Woodruff. 2004; Behrendt. 2006). Recent studies support this mechanism: the neurotoxic effects of ketamine in rodents appears to occur through action in the thalamus as opposed to direct effects on cortical brain regions (Sharp et al. 2001); reductions in thalamic blood flow have been shown to occur following administration of the serotonergic hallucinogen psilocybin (Carhart-Harris et al. 2012), and several case reports of patients with thalamic infarct describe the sudden onset of a schizophrenia-like psychosis (Arikan et al. 2009; Mittal and Khan. 2010; Crail-Melendez et al. 2012).
Current evidence suggests that alterations in thalamic structure and function may be one of the earliest detectable changes in the development of schizophrenia. Volumetric reductions in thalamus are a consistent finding in patients with schizophrenia, with a recent meta-analysis reporting that these reductions were more marked in unmedicated patients than those on antipsychotic treatment (Haijma et al. 2012). An MRI study of individuals at genetic high risk of developing schizophrenia has also reported reductions in thalamic volume (Lawrie et al. 1999).
Reductions in NAA, suggested to represent changes in neuronal integrity, are present at all stages of illness and in multiple brain regions, but the thalamus is the only region that has been reported to show NAA reductions in individuals with prodromal symptoms or at genetic risk of schizophrenia (Brugger et al. 2011). Thalamic NAA has been reported to be negatively correlated with duration of untreated psychosis (Jakary et al. 2005) and with length of prodrome (Theberge et al. 2004), while antipsychotic treatment is associated with an increase in thalamic NAA (Szulc et al. 2013). Reductions in thalamic NAA levels also appear to be related to other biological endophenotypes of schizhophrenia: we have reported that reductions in thalamic NAA in individuals with prodromal psychosis correlated with deficits in frontal P300 (Stone et al. 2010) as well as with reductions in cortical grey matter volume (Stone et al. 2009). The significance of the reductions in thalamic volume and NAA level in ARMS individuals and patients with psychosis is unclear, but in post-mortem studies on patients with chronic schizophrenia, reductions in thalamic neuron density have been found (Clinton and Meador-Woodruff. 2004), possibly due to loss of glutamatergic thalamocortical projection neurons (Danos et al. 1998).
The finding that chronic ketamine users did not differ from poly-drug-using controls in terms of brain Glu/Cr or Glu + Gln/Cr levels, although unexpected, is not altogether surprising. In patients with schizophrenia and first episode psychosis, there has been a great deal of heterogeneity in studies on brain glutamate levels. From a meta-analysis of the literature, we would predict that ketamine users, if they resemble patients with first episode psychosis, would have elevated glutamine levels in anterior cingulate (Marsman et al. 2013). Unfortunately, we were unable to reliably separate glutamine from glutamate in this study. Our finding of a relationship between the level of anterior cingulate Glu/Cr and perceptual distortions in ketamine users should be viewed with caution as it is uncorrected for multiple comparisons. We previously reported a relationship between anterior cingulate glutamate levels and positive psychotic symptoms following acute ketamine administration (Stone et al. 2012). Furthermore, we found elevated Glu/Cr in patients with first episode psychosis who remain symptomatic following antipsychotic treatment (Egerton et al. 2012). Thus, it is possible that ketamine users with more marked psychotic symptoms may have higher Glu/Cr levels in anterior cingulate, but the present study is not sufficiently powered to address this question.
The findings from this study are broadly in keeping with the literature on patients with schizophrenia and first episode psychosis in which NAA reductions are more robustly associated with illness trait but are not closely related to symptomatology (Brugger et al. 2011), whereas elevated anterior cingulate Glu levels, although less strongly associated with illness trait (Marsman et al. 2013), may be related to the severity of psychotic symptoms (Egerton et al. 2012; de la Fuente-Sandoval et al. 2013; Demjaha et al. 2013).
The study has a number of limitations:
-
(1)
Most notable is that ketamine users had taken a variety of other drugs. In particular, ketamine users had a significantly higher monthly intake of amphetamine than poly-drug-using controls. There was also a large amount of variance in level of use in the ketamine users group, and ketamine users generally had a higher mean level of all substances than controls. At present, although there is no evidence that other drugs affect thalamic NAA levels, cannabis, cocaine, methamphetamine and MDMA use has been associated with reduced NAA in frontal cortex (Chang et al. 1999; Reneman et al. 2002; Nordahl et al. 2005; Hermann et al. 2007; Salo et al. 2007; Cowan et al. 2009), and alcohol dependence has been reported to be associated with reduced thalamic grey matter volume and reduced temporal and parietal NAA levels (Gazdzinski et al. 2008). Another group reported that chronic cocaine use was associated with reduced anterior cingulate glutamate levels (Yang et al. 2009). It is thus not possible to be completely sure that the reported reductions in thalamic NAA/Cr were due to ketamine exposure rather than to amphetamine or other substances;
-
(2)
An associated limitation is due to the fact that the control group was poly-drug users. As drugs of abuse have been reported to have effects on reducing 1H-NAA levels (primarily in frontal cortex), as described earlier, the effect of ketamine may have been masked to a certain extent by these effects. The lack of a drug-free control group may make generalisation of the model to schizophrenia more problematic;
-
(3)
A third limitation is that the spectroscopy data were scaled to creatine rather than water. This approach has an advantage of not requiring correction for the amount of CSF in the ROI as brain metabolites such as Cr and NAA are present in brain tissue and not in CSF. On the other hand, scaling to creatine means that we cannot exclude the possibility that the differences in NAA/Cr were driven by group differences in creatine. The fact that there were no differences in other creatine-scaled metabolites between the two groups makes this unlikely, however;
-
(4)
Lastly, reverse causality is a possibility—individuals with an At-Risk Mental State, or with low thalamic NAA, may have been at a higher probability of becoming regular ketamine users. However, given that ketamine worsens psychotic symptoms (Lahti et al. 1995), it seems unlikely that individuals with subthreshold psychotic symptoms would recreationally abuse a drug that worsened their symptoms. Longitudinal studies are required to investigate this possibility.
In summary, this study suggests that chronic ketamine use, a naturalistic model of chronic NMDA receptor dysfunction, is associated with symptoms and neurochemical changes that have also been reported in individuals at high risk of developing schizophrenia. It supports the hypothesis that abnormalities of NMDA receptor function may drive symptoms and brain pathology in the early stages of the illness. Further work in our laboratory will seek to determine whether this is a unique effect of ketamine or whether other drugs of abuse, such as cannabis, are also associated with reductions in thalamic NAA.
References
Arikan MK, Kutukcu A, Karay A, Ozmen M (2009) A case of psychosis associated with left thalamic lacunar infarcts. Prog Neuropsychopharmacol Biol Psychiatry 33:729–730. doi:10.1016/j.pnpbp.2009.02.022
Bartha R, Williamson PC, Drost DJ, Malla A, Carr TJ, Cortese L, Canaran G, Rylett RJ, Neufeld RWJ (1997) Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch Gen Psychiatry 54:959–965. doi:10.1001/archpsyc.54.10.959
Behrendt RP (2006) Dysregulation of thalamic sensory “transmission” in schizophrenia: neurochemical vulnerability to hallucinations. J Psychopharmacol 20:356–372
Brugger S, Davis JM, Leucht S, Stone JM (2011) Proton magnetic resonance spectroscopy and illness stage in schizophrenia—a systematic review and meta-analysis. Biol Psychiatry 69:495–503. doi:10.1016/j.biopsych.2010.10.004
Carhart-Harris RL, Erritzoe D, Williams T, Stone JM, Reed LJ, Colasanti A, Tyacke RJ, Leech R, Malizia AL, Murphy K, Hobden P, Evans J, Feilding A, Wise RG, Nutt DJ (2012) Neural correlates of the psychedelic state as determined by fMRI studies with psilocybin. Proc Natl Acad Sci U S A 109:2138–2143. doi:10.1073/pnas.1119598109
Carlsson M, Carlsson A (1990) Schizophrenia: a subcortical neurotransmitter imbalance syndrome? Schizophr Bull 16:425–432
Chang L, Ernst T, Strickland T, Mehringer CM (1999) Gender effects on persistent cerebral metabolite changes in the frontal lobes of abstinent cocaine users. Am J Psychiatry 156:716–722
Clinton SM, Meador-Woodruff JH (2004) Thalamic dysfunction in schizophrenia: neurochemical, neuropathological, and in vivo imaging abnormalities. Schizophr Res 69:237–253
Cowan RL, Joers JM, Dietrich MS (2009) N-Acetylaspartate (NAA) correlates inversely with cannabis use in a frontal language processing region of neocortex in MDMA (Ecstasy) polydrug users: a 3 T magnetic resonance spectroscopy study. Pharmacol Biochem Behav 92:105–110. doi:10.1016/j.pbb.2008.10.022
Crail-Melendez D, Atriano-Mendieta C, Carrillo-Meza R, Ramirez-Bermudez J (2012) Schizophrenia-like psychosis associated with right lacunar thalamic infarct. Neurocase. doi:10.1080/13554794.2011.654211
Danos P, Baumann B, Bernstein HG, Franz M, Stauch R, Northoff G, Krell D, Falkai P, Bogerts B (1998) Schizophrenia and anteroventral thalamic nucleus: selective decrease of parvalbumin-immunoreactive thalamocortical projection neurons. Psychiatry Res 82:1–10
de la Fuente-Sandoval C, Leon-Ortiz P, Azcarraga M, Stephano S, Favila R, Diaz-Galvis L, Alvarado-Alanis P, Ramirez-Bermudez J, Graff-Guerrero A (2013) Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: a longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry 70:1057–1066. doi:10.1001/jamapsychiatry.2013.289
Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, McGuire PK (2013) Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry. doi:10.1016/j.biopsych.2013.06.011
Egerton A, Brugger S, Raffin M, Barker GJ, Lythgoe DJ, McGuire PK, Stone JM (2012) Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology. doi:10.1038/npp.2012.113
Gazdzinski S, Durazzo TC, Weiner MW, Meyerhoff DJ (2008) Are treated alcoholics representative of the entire population with alcohol use disorders? A magnetic resonance study of brain injury. Alcohol 42:67–76. doi:10.1016/j. alcohol .2008.01.002
Haijma SV, Van Haren N, Cahn W, Koolschijn PC, Hulshoff Pol HE, Kahn RS (2012) Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull. doi:10.1093/schbul/sbs118
Hermann D, Sartorius A, Welzel H, Walter S, Skopp G, Ende G, Mann K (2007) Dorsolateral prefrontal cortex N-acetylaspartate/total creatine (NAA/tCr) loss in male recreational cannabis users. Biol Psychiatry 61:1281–1289. doi:10.1016/j.biopsych.2006.08.027
Ihaka R, Gentleman R (1996) R: a language for data analysis and graphics. J Comput Graph Stat 5:299–314
Jakary A, Vinogradov S, Feiwell R, Deicken RF (2005) N-Acetylaspartate reductions in the mediodorsal and anterior thalamus in men with schizophrenia verified by tissue volume corrected proton MRSI. Schizophrenia Res 76:173–185
Jansen JF, Backes WH, Nicolay K, Kooi ME (2006) 1H MR spectroscopy of the brain: absolute quantification of metabolites. Radiology 240:318–332. doi:10.1148/radiol.2402050314
Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X, Gil R, Slifstein M, Abi-Dargham A, Lisanby SH, Shungu DC (2012) Elevated prefrontal cortex gamma-aminobutyric acid and glutamate–glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry 69:449–459. doi:10.1001/archgenpsychiatry.2011.1519
Keilhoff G, Becker A, Grecksch G, Wolf G, Bernstein HG (2004) Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience 126:591–598
Kim SY, Lee H, Kim HJ, Bang E, Lee SH, Lee DW, Woo DC, Choi CB, Hong KS, Lee C, Choe BY (2011) In vivo and ex vivo evidence for ketamine-induced hyperglutamatergic activity in the cerebral cortex of the rat: potential relevance to schizophrenia. NMR Biomed 24:1235–1242. doi:10.1002/nbm.1681
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB Jr, Charney DS (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214
Lahti AC, Holcomb HH, Medoff DR, Tamminga CA (1995) Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 6:869–872
Lawrie SM, Whalley H, Kestelman JN, Abukmeil SS, Byrne M, Hodges A, Rimmington JE, Best JJ, Owens DG, Johnstone EC (1999) Magnetic resonance imaging of brain in people at high risk of developing schizophrenia. Lancet 353:30–33
Liao Y, Tang J, Corlett PR, Wang X, Yang M, Chen H, Liu T, Chen X, Hao W, Fletcher PC (2011) Reduced dorsal prefrontal gray matter after chronic ketamine use. Biol Psychiatry 69:42–48. doi:10.1016/j.biopsych.2010.08.030
Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE (2013) Glutamate in schizophrenia: a focused review and meta-analysis of (1)H-MRS studies. Schizophr Bull 39:120–129. doi:10.1093/schbul/sbr069
Mittal M, Khan S (2010) Starvation causes acute psychosis due to anterior thalamic infarction. South Med J 103:701–703. doi:10.1097/SMJ.0b013e3181e1e2f3
Narendran R, Frankle WG, Keefe R, Gil R, Martinez D, Slifstein M, Kegeles LS, Talbot PS, Huang Y, Hwang DR, Khenissi L, Cooper TB, Laruelle M, Abi-Dargham A (2005) Altered prefrontal dopaminergic function in chronic recreational ketamine users. Am J Psychiatry 162:2352–2359
Nordahl TE, Salo R, Natsuaki Y, Galloway GP, Waters C, Moore CD, Kile S, Buonocore MH (2005) Methamphetamine users in sustained abstinence: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry 62:444–452. doi:10.1001/archpsyc.62.4.444
Reneman L, Majoie CB, Flick H, den Heeten GJ (2002) Reduced N-acetylaspartate levels in the frontal cortex of 3,4-methylenedioxymethamphetamine (Ecstasy) users: preliminary results. AJNR Am J Neuroradiol 23:231–237
Reynolds GP, Harte MK (2007) The neuronal pathology of schizophrenia: molecules and mechanisms. Biochem Soc Trans 35:433–436
Reynolds LM, Cochran SM, Morris BJ, Pratt JA, Reynolds GP (2005) Chronic phencyclidine administration induces schizophrenia-like changes in N-acetylaspartate and N-acetylaspartylglutamate in rat brain. Schizophr Res 73:147–152
Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, Barrow R, Yeo R, Lauriello J, Brooks WM (2005) Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry 162:394–396
Salo R, Nordahl TE, Natsuaki Y, Leamon MH, Galloway GP, Waters C, Moore CD, Buonocore MH (2007) Attentional control and brain metabolite levels in methamphetamine abusers. Biol Psychiatry 61:1272–1280. doi:10.1016/j.biopsych.2006.07.031
Sharp FR, Tomitaka M, Bernaudin M, Tomitaka S (2001) Psychosis: pathological activation of limbic thalamocortical circuits by psychomimetics and schizophrenia? Trends Neurosci 24:330–334
Steen RG, Hamer RM, Lieberman JA (2005) Measurement of brain metabolites by 1H magnetic resonance spectroscopy in patients with schizophrenia: a systematic review and meta-analysis. Neuropsychopharmacology 30:1949–1962
Stone JM, Day F, Tsagaraki H, Valli I, McLean MA, Lythgoe DJ, O'Gorman RL, Barker GJ, McGuire PK (2009) Glutamate dysfunction in people with prodromal symptoms of psychosis: relationship to gray matter volume. Biol Psychiatry 66:533–539. doi:10.1016/j.biopsych.2009.05.006
Stone JM, Bramon E, Pauls A, Sumich A, McGuire PK (2010) Thalamic neurochemical abnormalities in individuals with prodromal symptoms of schizophrenia—relationship to auditory event-related potentials. Psychiatry Res 183:174–176. doi:10.1016/j.pscychresns.2010.05.004
Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, Krystal JH, Nutt D, Barker GJ (2012) Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry PMID: 22212598; doi:10.1038/mp.2011.171
Szulc A, Waszkiewicz N, Bibulowicz D, Konarzewska B, Tarasow E (2013) Proton magnetic resonance spectroscopy changes after antipsychotic treatment. Curr Med Chem 20:414–427
Taylor MJ, Tiangga ER, Ni Mhuircheartaigh R, Cowen P (2011) Lack of effect of ketamine on cortical glutamate and glutamine in healthy volunteers: a proton magnetic resonance spectroscopy study. J Psychopharmacol. doi:10.1177/0269881111405359
Théberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J, Neufeld RW, Rogers J, Pavlosky W, Schaefer B, Densmore M, Al-Semaan Y, Williamson PC (2002) Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry 159:1944–1946. doi:10.1176/appi.ajp.159.11.1944
Theberge J, Al-Semaan Y, Drost DJ, Malla AK, Neufeld RW, Bartha R, Manchanda R, Menon R, Densmore M, Schaefer B, Williamson PC (2004) Duration of untreated psychosis vs. N-acetylaspartate and choline in first episode schizophrenia: a 1H magnetic resonance spectroscopy study at 4.0 Tesla. Psychiatry Res 131:107–114
Tibbo P, Hanstock C, Valiakalayil A, Allen P (2004) 3-T proton MRS investigation of glutamate and glutamine in adolescents at high genetic risk for schizophrenia. Am J Psychiatry 161:1116–1118
Tsai G, Coyle JT (2002) Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol 42:165–179. doi:10.1146/annurev.pharmtox.42.082701.160735
Vollenweider FX, Geyer MA (2001) A systems model of altered consciousness: integrating natural and drug-induced psychoses. Brain Res Bull 56:495–507
Wood SJ, Yücel M, Wellard RM, Harrison BJ, Clarke K, Fornito A, Velakoulis D, Pantelis C (2007) Evidence for neuronal dysfunction in the anterior cingulate of patients with schizophrenia: a proton magnetic resonance spectroscopy study at 3 T. Schizophr Res 94:328–331
Yang S, Salmeron BJ, Ross TJ, Xi ZX, Stein EA, Yang Y (2009) Lower glutamate levels in rostral anterior cingulate of chronic cocaine users—A (1)H-MRS study using TE-averaged PRESS at 3 T with an optimized quantification strategy. Psychiatry Res 174:171–176. doi:10.1016/j.pscychresns.2009.05.004
Yoo SY, Yeon S, Choi CH, Kang DH, Lee JM, Shin NY, Jung WH, Choi JS, Jang DP, Kwon JS (2009) Proton magnetic resonance spectroscopy in subjects with high genetic risk of schizophrenia: investigation of anterior cingulate, dorsolateral prefrontal cortex and thalamus. Schizophr Res 111:86–93
Yung AR, Yuen HP, McGorry PD, Phillips LJ, Kelly D, Dell'Olio M, Francey SM, Cosgrave EM, Killackey E, Stanford C, Godfrey K, Buckby J (2005) Mapping the onset of psychosis: the comprehensive assessment of at-risk mental states. Aust N Z J Psychiatry 39:964–971. doi:10.1111/j.1440-1614.2005.01714.x
Funding source
This work was supported by an MRC grant to Oliver Howes (grant code: MC_A656_5QD30).
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Celia Morgan and Oliver D. Howes contributed equally to this work.
Rights and permissions
About this article
Cite this article
Stone, J.M., Pepper, F., Fam, J. et al. Glutamate, N-acetyl aspartate and psychotic symptoms in chronic ketamine users. Psychopharmacology 231, 2107–2116 (2014). https://doi.org/10.1007/s00213-013-3354-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-013-3354-8