
Abstract
Rationale
Fragile X syndrome (FXS) is considered the leading inherited cause of intellectual disability and autism. In FXS, the fragile X mental retardation 1 (FMR1) gene is silenced and the fragile X mental retardation protein (FMRP) is not expressed, resulting in the characteristic features of the syndrome. Despite recent advances in understanding the pathophysiology of FXS, there is still no cure for this condition; current treatment is symptomatic. Preclinical research is essential in the development of potential therapeutic agents.
Objectives
This review provides an overview of the preclinical evidence supporting metabotropic glutamate receptor 5 (mGluR5) antagonists as therapeutic agents for FXS.
Results
According to the mGluR theory of FXS, the absence of FMRP leads to enhanced glutamatergic signaling via mGluR5, which leads to increased protein synthesis and defects in synaptic plasticity including enhanced long-term depression. As such, efforts to develop agents that target the underlying pathophysiology of FXS have focused on mGluR5 modulation. Animal models, particularly the Fmr1 knockout mouse model, have become invaluable in exploring therapeutic approaches on an electrophysiological, behavioral, biochemical, and neuroanatomical level. Two direct approaches are currently being investigated for FXS treatment: reactivating the FMR1 gene and compensating for the lack of FMRP. The latter approach has yielded promising results, with mGluR5 antagonists showing efficacy in clinical trials.
Conclusions
Targeting mGluR5 is a valid approach for the development of therapeutic agents that target the underlying pathophysiology of FXS. Several compounds are currently in development, with encouraging results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Martin–Bell syndrome, an X-linked intellectual disability, was first described in 1943 by James Purdon Martin and Julia Bell in multiple male members of a family (Martin and Bell 1943). Years later, in 1969, Herbert Lubs discovered the existence of a break on the X chromosome of affected males (Lubs 1969), which was termed “fragile site” by Frederick Hecht in 1970. This led to the name change from Martin–Bell syndrome to fragile X syndrome (FXS). It was only in 1991 that the gene responsible for FXS was identified on the X chromosome at position q27.3, and named fragile X mental retardation 1 gene (FMR1) (Verkerk et al. 1991). In FXS, the FMR1 gene is silenced, and consequently its gene product, fragile X mental retardation protein (FMRP), has reduced expression or is entirely absent. Lack of FMRP expression appears to be at the core of the intellectual disability and other features characteristic of FXS. The prevalence of FXS with the full mutation and intellectual disability is 1:4,000 in males and 1:6,000 in females (Sherman 2012; Turner et al. 1992), and it is considered to be the leading inherited single-gene cause of intellectual disability and autism.
The main clinical phenotype of FXS is intellectual disability. However, individuals typically present with features that are also common to autism spectrum disorder, including deficits in higher cognitive functions, such as delays in speech and language development, impaired theory of mind, and impaired social and emotional processing (Garber et al. 2008). Other characteristic features of FXS include anxiety, attention deficits, hyperactivity, irritability, and autistic-like behaviors including social deficits and hand-flapping, as well as physical characteristics including hypotonia, hypermobility of joints, and macroorchidism (Garber et al. 2008) (Table 1). Dendritic spine abnormalities have been reported in postmortem neuropathological studies in patients with FXS (Hinton et al. 1991; Irwin et al. 2000, 2001; Rudelli et al. 1985). Neuroimaging studies of patients with FXS revealed increased brain size, larger caudate nucleus, increased size of amygdala and hippocampus, cerebellar vermis hypoplasia (Reiss et al. 1995), and ventricular abnormalities (Schapiro et al. 1995), with the right side of the brain being apparently more affected.
The cognitive, physical, and behavioral phenotypes are relatively easy to observe and measure in patients with FXS. Conversely, the neuroanatomical phenotype is much more difficult to observe as it can only be studied in depth in postmortem brain material. Therefore, animal models that mimic the FXS phenotype have become critical in the search for suitable therapies.
The FMR1 gene and its product, FMRP
The FMR1 gene is located on the X chromosome at position q27.3. It has a length of 40 kb and contains 17 exons (Verkerk et al. 1991). Its 5′ untranslated region contains a CGG repeat with a length varying from 6 to 55 repeats in the general population. In some individuals, both males and females, this repeat can become unstable and can reach a length between 55 and 200 CGG repeats, leading to a so-called premutation (Fig. 1). These individuals are known as carriers with a premutation and have a high risk of developing fragile X-associated tremor/ataxia syndrome. Moreover, 20 % of the females carrying a premutation manifest premature ovarian insufficiency (Brouwer et al. 2009). In the case of individuals with FXS, the trinucleotide repeat length expands beyond 200 repeats (full mutation) (Oberle et al. 1991; Yu et al. 1991).

The unaffected, permutated, and mutated CCG repeat of the FMR1 gene
In affected individuals, cytosine residues in the CGG repeat sequence are methylated, with methylation extending to the 52 CpGs of the FMR1 promoter (Pieretti et al. 1991). Unmutated FMR1 alleles are also methylated, but in a region further upstream, separated from the FMR1 promoter by what appears to be a “boundary” that prevents methylation from spreading downstream (Naumann et al. 2009). This boundary is missing in full mutation alleles, and methylation occurs upstream of the CGG repeat region around the 13 weeks of embryonic development (Malter et al. 1997). As a consequence, FMR1 transcription is inhibited, leading to a reduction in or absence of FMRP from early on during development (Sutcliffe et al. 1992). There are very rare alleles that remain unmethylated despite containing >200 CGG repeats. These alleles maintain some transcriptional activity (Smeets et al. 1995; Tabolacci et al. 2008b; Tassone et al. 2000) and produce reduced levels of FMRP, compatible with “normal” intellectual development. In the case of premutation carriers, FMRP is produced but at a reduced level and, paradoxically, elevated levels of FMR1 messenger ribonucleic acid (mRNA) are produced (Tassone et al. 2007).
The epigenetic status of full mutation alleles is also characterized by deacetylation of histones H3 and H4, reduced methylation of lysine 4, and increased methylation of lysine 9 (H3K9) on histone H3 (Tabolacci et al. 2008b). These epigenetic changes promote a heterochromatic configuration that excludes the binding of specific transcription factors (Kumari and Usdin 2001), thus turning gene transcription off (Coffee et al. 1999). Notably, the rare unmethylated full mutation alleles maintain a normal epigenetic code, except for H3K9 status, which has methylation levels between normal and full mutation alleles, possibly explaining the reduced transcriptional level of unmethylated full mutations (Smeets et al. 1995; Tabolacci et al. 2008b).
The FMR1 gene has been highly conserved throughout evolution. Two autosomal paralogs have been identified, fragile X-related genes 1 and 2 (FXR1 and FXR2), located on chromosomes 3q28 and 17p13, respectively (Coy et al. 1995; Siomi et al. 1995; Zhang et al. 1995). Together the three genes form the FXR family. There is a high sequence similarity between FMR1 and FXR1/2, especially in their functional domains (Fig. 2) and overlap in tissue distribution. Despite this, FXR1P and FXR2P do not seem to be able to compensate for the lack of FMRP, suggesting that the FXR proteins may have different functions (Coffee et al. 2010).

FMR1P and its paralogs FXR1P and FXR2P. The KH domains and the C-terminal RGG box are the RNA-binding domains
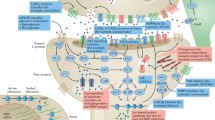
FMRP is a protein with four major isoforms between 70 and 80 kDa, expressed in many tissues, but predominantly in the brain. High levels are found in the hippocampus and cerebellum, and there is moderate expression in the cerebral cortex (Abitbol et al. 1993; Devys et al. 1993; Hinds et al. 1993; Khandjian et al. 1995). FMRP has also been shown to bind to nitric oxide synthase 1 transcripts during a specific period in human embryonic development (Kwan et al. 2012) that is important for the normal development and function of the nervous system, especially in processes like speech production, language recognition, attention, complex social behaviors, decision making, and emotional processing. In neurons, FMRP is localized mainly in the cell cytoplasm (Devys et al. 1993), where it binds to target mRNAs, including its own mRNA, and travels throughout the cell, and in and out of the nucleus (Devys et al. 1993; Feng et al. 1997; Ferrari et al. 2007; Willemsen et al. 1996). Importantly, FMRP travels into the dendrites via large RNA granules containing target mRNAs, motor proteins, other RNA binding proteins, and ribosomal subunits (de Diego et al. 2002). Target mRNAs of FMRP include: postsynaptic density (PSD)95 (Zalfa et al. 2007), SAPAP1-3 (Brown et al. 2001; Schutt et al. 2009), α-CaMKII, Arc/Ar3.1 (Kao et al. 2010; Zalfa et al. 2003), Shank1 (Schutt et al. 2009), and many more (Darnell and Richter 2012). FMRP regulates the local translation of these mRNAs into proteins at the synapse in the PSD. This process regulates the morphology of the spine and the functionality of the synapse (synaptic plasticity). FMRP also acts as a translational repressor of target mRNAs encoding proteins that regulate the internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) (Fig. 3a), which are essential in the proper function of the synapse.

Role of FMRP at the synapse—the mGluR theory: a unaffected—FMRP present and b FXS—absence of FMRP
The mGluR theory
Aberrant signaling via group 1 metabotropic glutamate receptors (mGluRs), is implicated in the pathophysiology of FXS (Bear et al. 2004). Group 1 mGluRs include mGluR1, expressed mainly in the cerebellum, thalamus, and CA3 hippocampal region and metabotropic glutamate receptor (mGluR)5), highly expressed in the CA1 and CA3 hippocampal regions, cortex and striatum (Dhami and Ferguson 2006; Lujan et al. 1996). FMRP regulates synaptic protein synthesis by binding to ribosomes and stalling translation of target mRNAs (Darnell et al. 2011). In 1997, the first connection between the FMRP and mGluR pathways was identified by Weiler et al. (1997) who observed that activation of group I mGluRs with 3,5-dihydroxyphenylglycine stimulated protein synthesis in synaptoneurosomes including the expression of FMRP (Weiler et al. 1997). In later studies in Fmr1 knockout (KO) mouse models, Huber et al. showed that the absence of FMRP leads to increased protein synthesis and altered synaptic plasticity, including enhanced long-term depression (LTD) (Huber et al. 2002). These observations led to the formulation of the mGluR theory (Bear et al. 2004), which states that the absence of FMRP in FXS results in excessive glutamatergic signaling via mGluR5. Consequently, this leads to increased local mRNA translation at the synapse, because FMRP is not present to regulate the process, and a high rate of AMPAR internalization and subsequent degradation, which in turn weakens the synapse (Fig. 3b). Increased internalization of AMPARs results in an increased number of longer immature dendritic spines, which could explain the intellectual disability found in patients with FXS. This immature spine morphology has been observed in both patients with FXS and in animal models mimicking FXS (Portera-Cailliau 2012).
Transgenic animal models for FXS
To date, preclinical FXS studies have been performed on fruit fly (Drosophila melanogaster) (Dhami and Ferguson 2006; Dockendorff et al. 2002; Gatto and Broadie 2008; Kanellopoulos et al. 2012; Pan et al. 2004; Zhang et al. 2001), zebrafish (Danio rerio) (den Broeder et al. 2009; Tucker et al. 2004; Tucker et al. 2006; van’t Padje et al. 2005), mouse (Mus musculus), and lately a rat model (Ratus norvegicus; SAGE Labs).
The most widely used animal model for FXS is the laboratory mouse (M. musculus). Several mouse models have been generated, such as Fmr1 KO, Fmr1 conditional KO, Fmr1 conditional restoration (Bakker et al. 1994; Mientjes et al. 2006), and recently a mouse model for the I304N mutation, Fmr1 I304N (Zang et al. 2009). All these lines are available in different strains, such as A/J, C57Bl/6, 129/Ola, FVB, Balb, DBA, and many more (Paradee et al. 1999; Pietropaolo et al. 2011; Spencer et al. 2011). The wide variety of mouse strains (Mouse Genome Database 2012) offers more possibilities for studying different phenotypic aspects of the syndrome, as each strain has different genetic characteristics. However, the interstrain differences sometimes lead to different results for the same aspect studied, and therefore, it is important to choose the correct strain for each study. Additional factors that may contribute to the differences in findings include the region studied, age of the mice and the method used. Despite these differences, it is generally concluded that the Fmr1 KO mice have abnormal dendritic spine morphology and increased spine density (for a recent review, see Portera-Cailliau 2012), similar to that found in patients with FXS (Hinton et al. 1991; Irwin et al. 2001; Rudelli et al. 1985).
Clinical evaluation scales used to assess cognitive and behavioral impairments in humans cannot be applied to mice. Consequently, several behavioral tests applicable to mice were developed to test processes like learning and memory, such as: T-maze, Morris water maze, fear conditioning test, object discrimination test and many more. Fmr1 KO mice have been found to exhibit decreased anxiety in open field; a higher latency to enter a dark box (Michalon et al. 2012); impairment in the acquisition of a visuospatial discrimination task (Krueger et al. 2011) and showed reduced freezing behavior to training context and sound (Guo et al. 2011). In addition, fear conditioning seems to be normal in Fmr1 KO on a C57BL/6 background, but impaired in KO mice with a C57xFVB background (Paradee et al. 1999), indicating that amygdala function could differ between strains. The same strain difference could account for the variation in results obtained with the Morris water maze, where some studies found impairment in the reversal trials in Fmr1 KO mice (Bakker et al. 1994; D’Hooge et al. 1997), while others have not observed any difference between KO and wild-type mice in learning and reversal tasks (Paradee et al. 1999). Other behavioral experiments showed that Fmr1 KO mice exhibited repetitive behavior (measured mainly by marble burying), abnormal social behavior (measured by the three chamber automatic test, or direct/indirect social interaction tests), and audiogenic seizures and anxiety deficiency (in open field or open-arms plus maze, dark–light box); however, results varied between studies (Bilousova et al. 2009; Chen and Toth 2001; de Vrij et al. 2008; Gantois et al. 2013; McNaughton et al. 2008; Michalon et al. 2012; Mineur et al. 2002; Moy et al. 2009; Nielsen et al. 2002; Peier et al. 2000; Pietropaolo et al. 2011; Restivo et al. 2005; Spencer et al. 2005; Spencer et al. 2011).
The abnormal neuroanatomical, cognitive, and behavioral phenotypes found in the Fmr1 KO mice have been investigated further at the molecular and functional level. Huber et al. showed that a form of LTD, dependent on mGluRs, is altered in the Fmr1 KO mouse model (Huber et al. 2002). This form of LTD is normally protein synthesis-dependent, but in the case of Fmr1 KO mice it occurs independently (Nosyreva and Huber 2006; Ronesi and Huber 2008). As mentioned above, the mGluR theory connects FMRP with long-term potentiation and LTD, mainly with increased internalization of AMPARs (Bear et al. 2004). Thus, several studies on the Fmr1 KO mice have looked at the levels of AMPAR subunits, N-methyl-D-aspartate receptor (NMDAR) subunits and mGluR5 and also at other postsynaptic proteins. Normal levels of AMPAR units GluA1 and GluA2/3 have been found in homogenate preparation from the cortex of 1-week-old Fmr1 KO mice, but reduced levels in the synaptoneurosome (SNS) fraction; whereas at 2 weeks of age, only the GluA1 subunit was reduced in SNS, while GluA2/3 and GluN2B levels were reduced in homogenates (Till et al. 2012). Giuffrida et al. reported normal levels of AMPA, NMDA and mGluR5 receptors in total protein homogenates and synaptic membrane preparations from the forebrain of Fmr1 KO mice (Giuffrida et al. 2005). However, homogenates and SNS fractions from the prefrontal cortex of KO mice had decreased levels of NR1, NR2A and NR2B subunits of NMDAR, SAPAP3, PSD-95, and Arc proteins (Krueger et al. 2011). FMRP loss of function has also been linked to GABAergic inhibition in FXS. A decreased level of mRNA for 8/18 GABAA receptor subunits have been found in the brains of Fmr1 KO mice (D’Hulst et al. 2006), and reduced levels of GABAA β subunit levels have been observed in the hippocampus and brainstem compared with control values (El et al. 2005). This may provide an explanation for amygdala dysfunction seen in Fmr1 KO mice.
FXS animal models, particularly the Fmr1 KO mouse model, have become invaluable in exploring therapeutic approaches in this field. However, there are some limitations of the KO mouse in modeling the human FXS. In the mouse model the Fmr1 gene is knocked out from conception, thus, FMRP is not expressed in any cell at any point during development (Oostra and Nelson 2006). Conversely, in humans, the FMR1 gene is methylated and silenced during embryonic development; therefore, some FMRP is expressed during the very early stages (Willemsen et al. 2002). Moreover, patients frequently present with mosaicism due to the presence of cells (neurons) containing a premutation (size mosaics; ∼50 % of patients with FXS) and because the FMR1 gene is not methylated in all cells (methylation mosaics) (Stöger et al. 2011). Finally, murine lines are inbred and genetically uniform, thus, they are a poor model for FXS in the human population and in particular for FXS treatment studies. Consequently, the genetic differences between humans with FXS and the corresponding murine KO model affect the extrapolation of the preclinical results found through mouse research to patients.
Considerations for drug development
There are several reasons that justify cautious optimism in finding an effective therapy that targets the underlying pathophysiology of FXS: the condition is a single gene disorder and genetically homogeneous, with very few exceptions; we have detailed knowledge of FMR1 gene structure; the open reading frame of the mutant gene remains intact, its transcription is stopped by reversible epigenetic changes; we have detailed knowledge of the consequences of the lack of FMRP at the level of dendritic post-synapses; and the clinical condition does not seem to entail irreversible damages to the CNS. For a recent review of potential therapeutic interventions, see Levenga et al. (2010) and Tranfaglia (2011).
The search for FXS targeted therapies was initiated following the identification and characterization of the genetic defect causing FXS (Verkerk et al. 1991). Two direct approaches are currently being investigated for FXS treatment: (1) reactivation of the affected gene and (2) compensating for the lack of FMRP. Restoring FMR1 gene activity is based on the concept that the epigenetic changes that block transcription are potentially reversible. The idea is to convert a nonfunctional methylated full mutation to a functional unmethylated full mutation. This approach has made significant contributions to the understanding of the genetic, epigenetic and protein translation mechanism in FXS. Two compounds, 5-Aza-deoxycytidine (Chiurazzi et al. 1999; Tabolacci et al. 2005) and valproic acid (Tabolacci et al. 2008a), have been shown to reactivate the FMR1 gene to some extent in fragile X cells. In a small, open-label trial of 10 boys with FXS, treatment with valproic acid resulted in a general improvement of hyperactivity and attention deficit, as measured by the Conners scale (Torrioli et al. 2010), although no increase in the mRNA levels of FMR1 could be measured.
The second approach is based on current knowledge of the signaling pathways impaired by the lack of FMRP, especially within the dendritic post-synaptic vesicles. Pharmacological and genetic rescue studies were mainly inspired by the mGluR theory of FXS. The rationale for the use of mGluR antagonists to treat FXS is strengthened by an elegant study in which mice heterozygous for the Grm5 gene (encoding mGluR5) were crossed with Fmr1 KO mice. The resulting 50 % reduction in mGluR5 protein levels led to the correction of some of the typical FXS phenotypic features, especially of the audiogenic seizures (Dölen et al. 2007).
The search for selective mGluR5 antagonists was initiated in 1992 following the cloning of the receptor by the team of S. Nakanishi (Abe et al. 1992). The aim was to identify agents which selectively inhibited mGluR5 and were tolerated in vivo. The first candidates identified were amino acid derivatives that did not distinguish between mGluR1 and mGluR5. The properties of these early agents did not allow them to be considered for further development or for use as pharmacological tools.
Significant progress in the understanding of the physiological role of mGluR5 and the potential therapeutic applications of mGluR5 ligands was made following the identification of the first potent and selective, noncompetitive antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) and its precursor molecules SIB1757 and SIB1893 (Gasparini et al. 1999; Varney et al. 1999). Following the discovery of MPEP and publication of its detailed mode of action (Pagano et al. 2000), drug discovery programs were initiated involving industry and academic research laboratories. These led to the identification of a number of candidate mGluR5 antagonists that are currently in preclinical and clinical development (Lindsley and Emmitte 2009; Rocher et al. 2011). Treatments with MPEP in Fmr1 KO mice resulted in suppression of the audiogenic seizure phenotype (Thomas et al. 2012; Yan et al. 2005), rescuing of the prepulse inhibition (PPI) (de Vrij et al. 2008) and a reduction in repetitive-like behavior (Burket et al. 2011; Thomas et al. 2012). In addition, following MPEP administration (2 weeks), the immature morphological phenotype of pyramidal neurons in the somatosensory cortex of Fmr1 KO mice was clearly rescued in neonate mice and less effective in 6 weeks old mice (Su et al. 2011). Very recently, Michalon et al. demonstrated in mice that chronic treatment with the novel long-acting mGluR5 antagonist, CTEP, starting at 4 weeks of age could restore cognitive deficits, auditory induced seizures, aberrant dendritic spine density, overactive ERK and mammalian target of rapamycin (mTOR) signaling, and partially corrects macroorchidism (Michalon et al. 2012). Clinically, fenobam, developed previously as an anxiolytic (Pecknold et al. 1982), was the first mGluR5 antagonist tested in FXS. Beneficial effects included reduced anxiety and hyperarousal, improved PPI, and better accuracy on a performance task (Berry-Kravis et al. 2009). Preclinical results of fenobam treatment showed improved motor learning deficiency on the Erasmus Ladder in mice (Vinueza Veloz et al. 2012), and rescuing of the dendritic spine abnormality of Fmr1 KO cultured neurons in vitro (de Vrij et al. 2008). In a recently completed study, Gantois et al. demonstrated that long-term treatment with AFQ056/mavoglurant, a selective mGluR5 antagonist, can rescue aberrant social behavior in the Fmr1 KO mice (Gantois et al. 2013). Furthermore, a recent clinical trial of mavoglurant identified a responder subgroup which reported significant improvements in Aberrant Behavior Checklist-Community Edition total score (−27.8 vs. placebo; P < 0.001), despite no significant improvements in the overall population (Jacquemont et al. 2011). The responder subgroup consisted of patients described as completely methylated according to a bisulfite-sequencing-based method; more sensitive than the widely used southern blot analysis. Patients who are partially methylated showed varying responses to treatment. An active effect on methylation was excluded, as treatment of FXS cell lines with mavoglurant was not found to result in either demethylation or transcriptional reactivation of the FMR1 gene (Tabolacci et al. 2012). Further clinical trials, currently underway, may provide a better understanding of the mode of action of mavoglurant and severity of the disease, especially with respect to the methylation pattern of patients.
Indirect approaches include targeting signaling pathways downstream or upstream of mGluRs, for example by decreasing the level of glycogen synthase kinase 3β, linked to group I mGluR signaling, which is upregulated in FXS (Min et al. 2009). This theory was supported by the use of lithium in a pilot study trial on 15 patients with FXS. Results showed that 2-month treatment with lithium had positive effects on behavioral adaptive skills (Berry-Kravis et al. 2008).
Currently, the most advanced investigational therapeutic interventions aim to modulate synaptic transmission, either through the reduction of synaptic excitability using selective mGluR5 inhibitors or through the reduction of neurotransmitter release via the activation of the presynaptic GABAB receptors. Such agents have been developed through large efforts in preclinical research and the use of model organisms such as the Fmr1 KO mouse model, as well as established and validated clinical evaluation scales (Sansone et al. 2012).
Additional approaches, currently being investigated preclinically in the Fmr1 KO mouse model, aim to modulate intracellular targets such as phosphoinositide 3-kinase (PI3K) (Gross et al. 2010), MTOR (Hoeffer et al. 2012), or MAP2K1 and MAP2K2 (MEK 1/2) (Wang et al. 2012). These agents are likely to be investigated in emerging cellular models involving the reprogramming of patient tissue samples in inducible pluripotent cells with a subsequent differentiation in neuronal cells (Sheridan et al. 2011). This novel approach has the potential to improve the validation of biological hypotheses as well as to investigate the effects of new agents without compromising patient safety.
Conclusions
The monogenic cause of FXS leads to a relatively genetically homogeneous patient population, and offers a unique and favorable situation for research towards developing effective therapies. It also facilitates the use of a variety of transgenic animal models mimicking the FXS phenotype. Although these models do not completely reflect the true human FXS phenotype, they are invaluable for research, understanding the pathophysiology of FXS, and particularly for assessing novel therapeutic approaches.
Despite a genetically homogeneous population, individuals with FXS display significant heterogeneity in clinical phenotype and drug response. A possible explanation might be variance in the epigenetic regulation of the FMRI gene and differences in the residual levels of FMRP. However, it is not completely clear how these differences at the molecular level reflect in the overall clinical phenotype.
Research on pharmacological therapies for FXS has been mainly focused on mGluR5 antagonists. Preclinical data from animal research on these agents are encouraging, and there are positive signs from clinical trials in patients with FXS. The results from phase III mavoglurant trials are eagerly awaited and, if positive, could quickly lead to the registration of the first therapy to specifically target the underlying pathophysiology in an intellectual disability syndrome.
References
Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S (1992) Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem 267:13361–13368
Abitbol M, Menini C, Delezoide AL, Rhyner T, Vekemans M, Mallet J (1993) Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet 4:147–153
Bakker CE, Verheij C, Willemsen R, van der Helm R, Erlemans F, Vermey M, Ygrave A, Oogeveen AT, Oostra BA (1994) Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch–Belgian Fragile X Consortium. Cell 78:23–33
Bear MF, Huber KM, Warren ST (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci 27:370–377
Berry-Kravis E, Sumis A, Hervey C, Nelson M, Porges SW, Weng N, Weiler IJ, Greenough WT (2008) Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr 29:293–302
Berry-Kravis E, Hessl D, Coffey S et al (2009) A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet 46:266–271
Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM (2009) Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet 46:94–102
Brouwer JR, Willemsen R, Oostra BA (2009) The FMR1 gene and fragile X-associated tremor/ataxia syndrome. Am J Med Genet B Neuropsychiatr Genet 150B:782–798
Brown V, Jin P, Ceman S et al (2001) Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107:477–487
Burket JA, Herndon AL, Winebarger EE, Jacome LF, Deutsch SI (2011) Complex effects of mGluR5 antagonism on sociability and stereotypic behaviors in mice: possible implications for the pharmacotherapy of autism spectrum disorders. Brain Res Bull 86:152–158
Chen L, Toth M (2001) Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103:1043–1050
Chiurazzi P, Pomponi MG, Pietrobono R, Bakker CE, Neri G, Oostra BA (1999) Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum Mol Genet 8:2317–2323
Coffee B, Zhang F, Warren ST, Reines D (1999) Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 22:98–101
Coffee RL Jr, Tessier CR, Woodruff EA 3rd, Broadie K (2010) Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis Model Mech 3:471–485
Coy JF, Sedlacek Z, Bachner D, Hameister H, Joos S, Lichter P, Delius H, Poustka A (1995) Highly conserved 3′ UTR and expression pattern of FXR1 points to a divergent gene regulation of FXR1 and FMR1. Hum Mol Genet 4:2209–2218
Darnell JC, Richter JD (2012) Cytoplasmic RNA-binding proteins and the control of complex brain function. Cold Spring Harb Perspect Biol 4:a012344
Darnell JC, Van Driesche SJ, Zhang C et al (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146:247–261
de Diego OY, Severijnen LA, van Cappellen G, Schrier M, Oostra B, Willemsen R (2002) Transport of fragile X mental retardation protein via granules in neurites of PC12 cells. Mol Cell Biol 22:8332–8341
de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R (2008) Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis 31:127–132
den Broeder MJ, van der Linde H, Brouwer JR, Oostra BA, Willemsen R, Ketting RF (2009) Generation and characterization of FMR1 knockout zebrafish. PLoS One 4:e7910
Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL (1993) The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet 4:335–340
Dhami GK, Ferguson SS (2006) Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacol Ther 111:260–271
D’Hooge R, Nagels G, Franck F, Bakker CE, Reyniers E, Storm K, Kooy RF, Oostra BA, Willems PJ, De Deyn PP (1997) Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76:367–376
D’Hulst C, De GN, Reeve SP, Van DD, De Deyn PP, Hassan BA, Kooy RF (2006) Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res 1121:238–245
Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA (2002) Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34:973–984
Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF (2007) Correction of fragile X syndrome in mice. Neuron 56:955–962
El IA, Ding XH, Scalia J, Trenkner E, Brown WT, Dobkin C (2005) Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci Lett 377:141–146
Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM (1997) Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci 17:1539–1547
Ferrari F, Mercaldo V, Piccoli G, Sala C, Cannata S, Achsel T, Bagni C (2007) The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol Cell Neurosci 34:343–354
Gantois I, Pop AS, de Esch CE, Buijsen RA, Pooters T, Gomez-Mancilla B, Gasparini F, Oostra BA, D’Hooge R, Willemsen R (2013) Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav Brain Res 239:72–79
Garber KB, Visootsak J, Warren ST (2008) Fragile X syndrome. Eur J Hum Genet 16:666–672
Gasparini F, Lingenhohl K, Stoehr N et al (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38:1493–1503
Gatto CL, Broadie K (2008) Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development 135:2637–2648
Giuffrida R, Musumeci S, D’Antoni S, Bonaccorso CM, Giuffrida-Stella AM, Oostra BA, Catania MV (2005) A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J Neurosci 25:8908–8916
Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K, Warren ST, Bassell GJ (2010) Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci 30:10624–10638
Guo W, Allan AM, Zong R et al (2011) Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med 17:559–565
Hinds HL, Ashley CT, Sutcliffe JS, Nelson DL, Warren ST, Housman DE, Schalling M (1993) Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome. Nat Genet 3:36–43
Hinton VJ, Brown WT, Wisniewski K, Rudelli RD (1991) Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet 41:289–294
Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, Whelan AM, Zukin RS, Klann E, Tassone F (2012) Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav 11:332–341
Huber KM, Gallagher SM, Warren ST, Bear MF (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99:7746–7750
Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10:1038–1044
Irwin SA, Patel B, Idupulapati M et al (2001) Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 98:161–167
Jacquemont S, Curie A, des Portes V et al (2011) Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med 3:64ra1
Kanellopoulos AK, Semelidou O, Kotini AG, Anezaki M, Skoulakis EM (2012) Learning and memory deficits consequent to reduction of the fragile X mental retardation protein result from metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J Neurosci 32:13111–13124
Kao DI, Aldridge GM, Weiler IJ, Greenough WT (2010) Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc Natl Acad Sci U S A 107:15601–15606
Khandjian EW, Fortin A, Thibodeau A, Tremblay S, Cote F, Devys D, Mandel JL, Rousseau F (1995) A heterogeneous set of FMR1 proteins is widely distributed in mouse tissues and is modulated in cell culture. Hum Mol Genet 4:783–789
Krueger DD, Osterweil EK, Chen SP, Tye LD, Bear MF (2011) Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A 108:2587–2592
Kumari D, Usdin K (2001) Interaction of the transcription factors USF1, USF2, and alpha –Pal/Nrf-1 with the FMR1 promoter. Implications for fragile X mental retardation syndrome. J Biol Chem 276:4357–4364
Kwan KY, Lam MM, Johnson MB et al (2012) Species-dependent posttranscriptional regulation of NOS1 by FMRP in the developing cerebral cortex. Cell 149:899–911
Levenga J, de Vrij FM, Oostra BA, Willemsen R (2010) Potential therapeutic interventions for fragile X syndrome. Trends Mol Med 16:516–527
Lindsley CW, Emmitte KA (2009) Recent progress in the discovery and development of negative allosteric modulators of mGluR5. Curr Opin Drug Discov Devel 12:446–457
Lubs HA (1969) A marker X chromosome. Am J Hum Genet 21:231–244
Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P (1996) Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci 8:1488–1500
Malter HE, Iber JC, Willemsen R, de Graaff E, Tarleton JC, Leisti J, Warren ST, Oostra BA (1997) Characterization of the full fragile X syndrome mutation in fetal gametes. Nat Genet 15:165–169
Martin JP, Bell J (1943) A pedigree of mental defect showing sex-linkage. J Neurol Psychiatry 6:154–157
McNaughton CH, Moon J, Strawderman MS, Maclean KN, Evans J, Strupp BJ (2008) Evidence for social anxiety and impaired social cognition in a mouse model of fragile X syndrome. Behav Neurosci 122:293–300
Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L (2012) Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74:49–56
Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, Zu T, Hoogeveen-Westerveld M, Severijnen L, Rife M, Willemsen R, Nelson DL, Oostra BA (2006) The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis 21:549–555
Min WW, Yuskaitis CJ, Yan Q, Sikorski C, Chen S, Jope RS, Bauchwitz RP (2009) Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 56:463–472
Mineur YS, Sluyter F, de Wit S, Oostra BA, Crusio WE (2002) Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus 12:39–46
Mouse Genome Database (2012) Mouse Genome Informatics Website. The Jackson Laboratory, Bar Harbor, Maine
Moy SS, Nadler JJ, Young NB, Nonneman RJ, Grossman AW, Murphy DL, D’Ercole AJ, Crawley JN, Magnuson TR, Lauder JM (2009) Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav 8:129–142
Naumann A, Hochstein N, Weber S, Fanning E, Doerfler W (2009) A distinct DNA-methylation boundary in the 5′- upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome. Am J Hum Genet 85:606–616
Nielsen DM, Derber WJ, McClellan DA, Crnic LS (2002) Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res 927:8–17
Nosyreva ED, Huber KM (2006) Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol 95:3291–3295
Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL (1991) Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252:1097–1102
Oostra BA, Nelson DL (2006) Animal models of fragile X syndrome: mice and flies. In: Wells BD, Ashizawa T (eds) Genetic instabilities and neurological diseases. Elsevier, Amsterdam, pp 175–193
Pagano A, Ruegg D, Litschig S et al (2000) The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem 275:33750–33758
Pan L, Zhang YQ, Woodruff E, Broadie K (2004) The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol 14:1863–1870
Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST (1999) Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94:185–192
Pecknold JC, McClure DJ, Appeltauer L, Wrzesinski L, Allan T (1982) Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J Clin Psychopharmacol 2:129–133
Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, Nelson DL (2000) (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum Mol Genet 9:1145–1159
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66:817–822
Pietropaolo S, Guilleminot A, Martin B, D’Amato FR, Crusio WE (2011) Genetic-background modulation of core and variable autistic-like symptoms in Fmr1 knock-out mice. PLoS One 6:e17073
Portera-Cailliau C (2012) Which comes first in fragile X syndrome, dendritic spine dysgenesis or defects in circuit plasticity? Neuroscientist 18:28–44
Reiss AL, Abrams MT, Greenlaw R, Freund L, Denckla MB (1995) Neurodevelopmental effects of the FMR-1 full mutation in humans. Nat Med 1:159–167
Restivo L, Ferrari F, Passino E, Sgobio C, Bock J, Oostra BA, Bagni C, Ammassari-Teule M (2005) Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc Natl Acad Sci U S A 102:11557–11562
Rocher JP, Bonnet B, Bolea C, Lutjens R, Le PE, Poli S, Epping-Jordan M, Bessis AS, Ludwig B, Mutel V (2011) mGluR5 negative allosteric modulators overview: a medicinal chemistry approach towards a series of novel therapeutic agents. Curr Top Med Chem 11:680–695
Ronesi JA, Huber KM (2008) Metabotropic glutamate receptors and fragile x mental retardation protein: partners in translational regulation at the synapse. Sci Signal 1:e6
Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM (1985) Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol 67:289–295
Sansone SM, Widaman KF, Hall SS, Reiss AL, Lightbody A, Kaufmann WE, Berry-Kravis E, Lachiewicz A, Brown EC, Hessl D (2012) Psychometric study of the aberrant behavior checklist in fragile X syndrome and implications for targeted treatment. J Autism Dev Disord 42:1377–1392
Schapiro MB, Murphy DG, Hagerman RJ et al (1995) Adult fragile X syndrome: neuropsychology, brain anatomy, and metabolism. Am J Med Genet 60:480–493
Schutt J, Falley K, Richter D, Kreienkamp HJ, Kindler S (2009) Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J Biol Chem 284:25479–25487
Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ (2011) Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 6:e26203
Sherman S (2012) Epidemiology. In: Hagerman RJ, Hagerman PJ (eds) Fragile X syndrome: diagnosis, treatment and research. The Johns Hopkins University Press, Balitmore, pp 136–168
Siomi MC, Siomi H, Sauer WH, Srinivasan S, Nussbaum RL, Dreyfuss G (1995) FXR1, an autosomal homolog of the fragile X mental retardation gene. EMBO J 14:2401–2408
Smeets HJ, Smits AP, Verheij CE, Theelen JP, Willemsen R, van de Burgt I, Hoogeveen AT, Oosterwijk JC, Oostra BA (1995) Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet 4:2103–2108
Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R (2005) Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav 4:420–430
Spencer CM, Alekseyenko O, Hamilton SM, Thomas AM, Serysheva E, Yuva-Paylor LA, Paylor R (2011) Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res 4:40–56
Stöger R, Genereux DP, Hagerman RJ, Hagerman PJ, Tassone F, Laird CD (2011) Testing the FMR1 promoter for mosaicism in DNA methylation among CpG sites, strands, and cells in FMR1-expressing males with fragile X syndrome. PLoS One 6:e23648
Su T, Fan HX, Jiang T, Sun WW, Den WY, Gao MM, Chen SQ, Zhao QH, Yi YH (2011) Early continuous inhibition of group 1 mGlu signaling partially rescues dendritic spine abnormalities in the Fmr1 knockout mouse model for fragile X syndrome. Psychopharmacology (Berlin) 215:291–300
Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST (1992) DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1:397–400
Tabolacci E, Pietrobono R, Moscato U, Oostra BA, Chiurazzi P, Neri G (2005) Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur J Hum Genet 13:641–648
Tabolacci E, De Pascalis I, Accadia M, Terracciano A, Moscato U, Chiurazzi P, Neri G (2008a) Modest reactivation of the mutant FMR1 gene by valproic acid is accompanied by histone modifications but not DNA demethylation. Pharmacogenet Genomics 18:738–741
Tabolacci E, Moscato U, Zalfa F, Bagni C, Chiurazzi P, Neri G (2008b) Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur J Hum Genet 16:1487–1498
Tabolacci E, Pirozzi F, Gomez-Mancilla B, Gasparini F, Neri G (2012) The mGluR5 antagonist AFQ056 does not affect methylation and transcription of the mutant FMR1 gene in vitro. BMC Med Genet 13:13
Tassone F, Hagerman RJ, Loesch DZ, Lachiewicz A, Taylor AK, Hagerman PJ (2000) Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am J Med Genet 94:232–236
Tassone F, Beilina A, Carosi C, Albertosi S, Bagni C, Li L, Glover K, Bentley D, Hagerman PJ (2007) Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA 13:555–562
Thomas AM, Bui N, Perkins JR, Yuva-Paylor LA, Paylor R (2012) Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology (Berlin) 219:47–58
Till SM, Wijetunge LS, Seidel VG, Harlow E, Wright AK, Bagni C, Contractor A, Gillingwater TH, Kind PC (2012) Altered maturation of the primary somatosensory cortex in a mouse model of fragile X syndrome. Hum Mol Genet 21:2143–2156
Torrioli M, Vernacotola S, Setini C, Bevilacqua F, Martinelli D, Snape M, Hutchison JA, Di Raimo FR, Tabolacci E, Neri G (2010) Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am J Med Genet A 152A:1420–1427
Tranfaglia MR (2011) The psychiatric presentation of fragile x: evolution of the diagnosis and treatment of the psychiatric comorbidities of fragile X syndrome. Dev Neurosci 33:337–348
Tucker B, Richards R, Lardelli M (2004) Expression of three zebrafish orthologs of human FMR1-related genes and their phylogenetic relationships. Dev Genes Evol 214:567–574
Tucker B, Richards RI, Lardelli M (2006) Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum Mol Genet 15:3446–3458
Turner G, Robinson H, Laing S, van den Berk M, Colley A, Goddard A, Sherman S, Partington M (1992) Population screening for fragile X. Lancet 339:1210–1213
van’t Padje S, Engels B, Blonden L, Severijnen LA, Verheijen F, Oostra BA, Willemsen R (2005) Characterisation of Fmrp in zebrafish: evolutionary dynamics of the fmr1 gene. Dev Genes Evol 215:198–206
Varney MA, Cosford ND, Jachec C et al (1999) SIB-1757 and SIB-1893: selective, noncompetitive antagonists of metabotropic glutamate receptor type 5. J Pharmacol Exp Ther 290:170–181
Verkerk AJ, Pieretti M, Sutcliffe JS et al (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914
Vinueza Veloz MF, Buijsen RA, Willemsen R, Cupido A, Bosman LW, Koekkoek SK, Potters JW, Oostra BA, De Zeeuw CI (2012) The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav 11:325–331
Wang X, Snape M, Klann E, Stone JG, Singh A, Petersen RB, Castellani RJ, Casadesus G, Smith MA, Zhu X (2012) Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J Neurochem 121:672–679
Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT (1997) Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A 94:5395–5400
Willemsen R, Bontekoe C, Tamanini F, Galjaard H, Hoogeveen A, Oostra B (1996) Association of FMRP with ribosomal precursor particles in the nucleolus. Biochem Biophys Res Commun 225:27–33
Willemsen R, Bontekoe CJ, Severijnen LA, Oostra BA (2002) Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet 110:601–605
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP (2005) Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49:1053–1066
Yu S, Pritchard M, Kremer E et al (1991) Fragile X genotype characterized by an unstable region of DNA. Science 252:1179–1181
Zalfa F, Giorgi M, Primerano B, Moro A, di Penta A, Reis S, Oostra B, Bagni C (2003) The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112:317–327
Zalfa F, Eleuteri B, Dickson KS et al (2007) A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci 10:578–587
Zang JB, Nosyreva ED, Spencer CM et al (2009) A mouse model of the human fragile X syndrome I304N mutation. PLoS Genet 5:e1000758
Zhang Y, O’Connor JP, Siomi MC, Srinivasan S, Dutra A, Nussbaum RL, Dreyfuss G (1995) The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J 14:5358–5366
Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K (2001) Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107:591–603
Financial disclosures and acknowledgments
FG and BGM are employees of Novartis Pharma AG and hold shares with Novartis Pharma AG. FG and BGM have also received reimbursement from Novartis Pharma AG for travel expenses. GN received honoraria and compensation from Novartis for providing biological samples as part of a collaborative project. This work was supported by The Netherlands Organisation for Health Research and Development (ZonMw) 912-07-022 (AP and RW), E-Rare program entitled “Cure FXS” (no. EU/FIS PS09102673) (RW), FRAXA Research Foundation (RW and GN), Telethon (GGP10150) (GN) and Associazione Italiana Sindrome X Fragile (GN). Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. We thank Emma Burke, of iMed Comms, who provided medical writing assistance with this review. The authors have full control of all primary data and agree to allow Psychopharmacology to review the data if requested.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Pop, A.S., Gomez-Mancilla, B., Neri, G. et al. Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology 231, 1217–1226 (2014). https://doi.org/10.1007/s00213-013-3330-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-013-3330-3