
Abstract
Rationale
Oxytocin (OXT) has been proposed as a potential therapeutic agent for post-traumatic stress disorder (PTSD).
Objectives
We aimed to verify whether pharmacological manipulation of the brain OXT system affects cued fear conditioning and fear extinction.
Methods
Male rats and mice were intracerebroventricularly administered synthetic OXT (rats, 0.1 or 1.0 μg/5 μl; mice, 0.1 or 0.5 μg/2 μl) and/or an OXT receptor antagonist (OXTR-A; rats, 0.75 μg/5 μl) either prior to fear conditioning or extinction training.
Results
Preconditioning administration of OXT did not affect fear conditioning in rats, but decreased fear expression and facilitated fear extinction. In contrast, preconditioning blockade of OXT neurotransmission by OXTR-A did not affect fear conditioning or fear expression, but impaired fear extinction. When administered before extinction training, OXT impaired fear extinction in both rats and mice, indicating that the effects of OXT on fear extinction are conserved across species. This impairment was OXTR-mediated, as the inhibitory effect of OXT on fear extinction was abolished by prior treatment with OXTR-A. The impaired fear extinction was not a result of reduced locomotion in rats, whereas an apparent decrease in fear expression and facilitation of fear extinction with the higher OXT dose in mice was the result of behavioral hyperactivity.
Conclusions
These results suggest that increasing OXT neurotransmission during traumatic events is likely to prevent the formation of aversive memories. In contrast, OXT treatment before fear extinction training, which would be the comparable timepoint for psychotherapy in PTSD patients, rather delays fear extinction and, therefore, caution is needed before recommending OXT for the treatment of PTSD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pavlovian fear conditioning is a form of learning in which an association between a stimulus and its aversive consequences is made. Cued fear conditioning has been used in laboratory animals as a model of post-traumatic stress disorder (PTSD) and involves the presentation of a neutral stimulus, such as a tone or light (conditioned stimulus [CS]) in association with an aversive stimulus, such as a mild footshock (unconditioned stimulus [US]). Through repeated CS–US associations, animals learn that the CS predicts the US, and a conditioned response, such as freezing (Fanselow 1980), is elicited in the absence of the US. Fear extinction is regarded as a form of new learning (for reviews, see Cammarota et al. 2007; Quirk et al. 2010) and is defined as the attenuation of the conditioned response by repeated exposure to the CS without the US. Inability to extinguish fear memories was shown to involve hyperactivity of the amygdala (Rauch et al. 2000; Stein et al. 2002; Dilger et al. 2003) and is a core symptom in several psychiatric disorders, such as specific phobias, generalized and social anxiety disorder, panic disorder, and PTSD. The current treatment for PTSD consists of psychotherapy, often combined with antidepressant, benzodiazepine, and antipsychotic treatment, with selective serotonin reuptake inhibitors providing the best response rates (Marshall and Pierce 2000; Stein et al. 2006, 2009). However, treatment-resistant PTSD patients achieve only partial symptom remission or show a high rate of relapse after treatment discontinuation (Davidson et al. 2004; Bisson et al. 2007; Brunello et al. 2001; Ipser et al. 2006). Therefore, the development of approaches that combine psychotherapy with novel pharmacotherapy is still needed.
Neuropeptides, which have discrete synthesis, release, and receptor sites in the brain (Landgraf and Neumann 2004; Wotjak et al. 2008), have emerged as viable research candidates with respect to both pathophysiology and treatment of PTSD (Gülpinar and Yegen 2004; Viero et al. 2010). One such neuropeptide, oxytocin (OXT), which is synthesized in the paraventricular and supraoptic nuclei of the hypothalamus and centrally released within these hypothalamic and other limbic regions, including the septum, hippocampus, and central amygdala (CeA) in response to various stressful stimuli (Ebner et al. 2005; Neumann 2007), has been recently proposed as a potential therapeutic agent for PTSD (Olff et al. 2010). Both synthetic and endogenous OXT exert anxiolytic properties in rodents (McCarthy et al. 1996; Ring et al. 2006; Waldherr and Neumann 2007; Blume et al. 2008) and inhibit the activity of the hypothalamic–pituitary–adrenal (HPA) axis (Windle et al. 1997; Neumann et al. 2000). Comparable effects were also found in humans (Heinrichs et al. 2003), as OXT was shown to reduce the activation of the amygdala to threatening faces, thereby reducing the autonomic and behavioral manifestation of fear in healthy volunteers and social anxiety disorder patients (Kirsch et al. 2005; Labuschagne et al. 2010). More indirect evidence for the anxiolytic and antistress effects of OXT in humans comes from nursing mothers who are calmer and less anxious during stressful situations, possibly due to high brain OXT activity (Heinrichs et al. 2001; Carter et al. 2001; Slattery and Neumann 2008). Given that PTSD is marked by deficits in anxiety/stress regulation and hyperactivity of the amygdala (Rauch et al. 2000; Shin et al. 2004), OXT might be a good candidate for the treatment of PTSD (Olff et al. 2010; Viviani et al. 2011). Therefore, we aimed to study in detail whether OXT affects fear conditioning and fear extinction and whether such effects depend on the timing of administration. The classical fear conditioning paradigm involves acquisition, expression, and extinction of fear memories, and drugs can differentially affect these processes (Lattal and Abel 2001; Makkar et al. 2010). Therefore, we manipulated the OXT system by intracerebroventricular (icv) administration of synthetic OXT and/or an OXT receptor antagonist (OXTR-A) either prior to conditioning (also referred to as acquisition) or extinction training. In order to be able to draw more general conclusions, we performed the experiments in both rats and mice and hypothesized that OXT would facilitate fear extinction in both species.
Materials and methods
Animals
Male Wistar rats (280–300 g) and male CD1 mice (35–40 g) were purchased from Charles River, Sulzfeld, Germany. Animals were group-housed in polycarbonate cages (rats, 55 × 22 × 18 cm; mice, 42 × 26 × 15 cm) for 1 week before surgery and kept under standard laboratory conditions (12:12 light/dark cycle, lights on at 6 am, 22 °C, 60 % humidity, food and water ad libitum). After surgery, animals were single-housed in observation cages (rats, 40 × 24 × 36 cm; mice, 30 × 23 × 36 cm). All behavioral procedures took place during the light phase and were conducted in accordance with the local government of the Oberpfalz (Bavaria, Germany) and the guidelines of the National Institute of Health.
Cannula implantation
Guide cannula implantation was performed under isoflurane anesthesia (Forene®, Abbott GmbH & Co. KG, Wiesbaden, Germany). To avoid post-surgical infections, all animals received antibiotics (s.c.; 3 mg/30 μl Baytril®, Bayer Vital GmbH, Leverkusen, Germany). Animals were mounted on a stereotaxic frame, and a guide cannula (21 G; rats, 12 mm length; mice, 8 mm length; Injecta GmbH, Klingenthal, Germany) was implanted above the right lateral ventricle (rats: AP +1.0 mm from bregma, ML +1.6 mm, V +1.8 mm; mice: AP +0.2 mm, ML +1.0 mm, V +1.4 mm). The guide cannula was fixed with two stainless steel screws using dental cement (Kallocryl, Speiko-Dr. Speier GmbH, Münster, Germany) and closed by a stainless steel dummy cannula. After surgery, animals were handled daily (stroking, holding, and cleaning of dummy cannulas) for 5 days to minimize nonspecific stress responses during the experiment.
Intracerebral infusions
Animals received icv infusions of either vehicle (sterile Ringer solution, pH 7.4; rats, 5 μl; mice, 2 μl), synthetic OXT (Sigma-Aldrich, Munich, Germany; rats, 0.1 or 1.0 μg/5 μl; mice, 0.1 or 0.5 μg/2 μl—from this point onward referred to as lower and higher OXT doses for rats and mice, respectively), or a selective OXTR-A (desGly-NH2,d(CH2)5[Tyr(Me)2,Thr4]OVT; rats, 0.75 μg/5 μl) (Manning et al. 2008) via an infusion cannula (25 G, extended 2 mm beyond the guide cannula) connected via polyethylene tubing to a Hamilton syringe. The infusion system was left in place for 30 s following the infusion to allow diffusion of the solution.
To verify the infusion site, animals were killed using CO2 and ink was infused icv before removal of the brain. Brains were cut coronally and checked for staining of the ventricle. Only animals with correct infusion sites were included in the statistical analyses. Drug doses and timing were selected based on previous studies in our laboratory (Waldherr and Neumann 2007; Bosch and Neumann 2008; Lukas et al. 2011).
Cued fear conditioning apparatus
The cued fear experiments were performed in two different contexts, A and B, which differed in visual, tactile, and olfactory cues as previously described (Toth et al. 2012a). Briefly, fear conditioning occurred in context A, which consisted of a transparent Perspex box (rats, 45 × 45 × 40 cm; mice, 23 × 23 × 36 cm) with an electric grid floor. Context A was cleaned with water containing a small amount of a neutral-smelling detergent before each trial. Extinction training and retention occurred in context B, which consisted of a black Perspex box (rats, 45 × 45 × 40 cm; mice, 23 × 23 × 36 cm) with a smooth floor. Context B was cleaned with water containing a small amount of a lemon-scented detergent before each trial. The boxes were enclosed in a wooden chamber to reduce external noise and visual stimulation. A low level of background noise was produced by ventilation fans within the chamber. Illumination (300 lx for context A and 20 lx for context B) was provided by four white light-emitting diodes. Auditory stimuli were delivered through a speaker attached 30 cm above the floor of the box. Freezing, defined as the absence of all movement except that required for respiration (Fanselow 1980), was measured with the TSE fear conditioning system (TSE System GmbH, Bad Homburg, Germany). The conditioning chamber contained two horizontal detection fields, each with 32 (rats) or 16 (mice) infrared light beams set 1.3 cm apart. Inactivity was measured by the infrared beams and defined as no light beam interruption for at least 3 s (rats) or 1 s (mice). We have previously shown that such measurements are comparable with hand-scoring by an experienced observed (Toth et al. 2012a).
Cued fear conditioning procedure
The procedure was adapted from the literature (Muigg et al. 2008) and has been shown to induce a robust cued fear conditioning in our laboratory (Toth et al. 2012a).
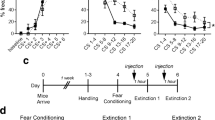
Fear conditioning (day 1)
Animals were placed in the conditioning chamber (context A) and, after a 5-min adaptation period, exposed to five CS–US pairings with a 2-min interstimulus interval. The CS was an 80-dB, 4.5-kHz (rats) or 8-kHz (mice), 30-s white noise, which co-terminated with a mild electric footshock (US, 0.7 mA; pulsed current, 2 s). Animals were returned to their home cage 5 min after the last CS–US pairing.
Extinction training (day 2)
One day after fear conditioning, animals were placed in context B and, after a 5-min adaptation period, exposed to 30 (rats) or 20 (mice) CS presentations (30 s white noise, 5 s interstimulus interval). Animals were returned to their home cage 5 min after the last CS presentation. Freezing during extinction training increased until the sixth CS; therefore, this period was defined as fear expression. After the sixth CS, freezing decreased; therefore, this period was defined as fear extinction. These CS presentations were collapsed into ten blocks with the mean freezing percentage during three or two CS presentations represented in each block for rats and mice, respectively.
Extinction retention (day 3)
One day after extinction training, animals were again placed in context B; after a 5-min adaptation period, they were exposed to five CS presentations (30 s white noise, 5 s interstimulus interval). Animals were returned to their home cage after the last CS presentation. These CS presentations were then collapsed into one block.
Home cage locomotion
In separate groups of rats and mice, locomotion was assessed immediately after OXT administration in the home cage for 1 h using the Noldus Ethovision XT 5.1 program (Noldus Information Technology, Wageningen, The Netherlands), as previously described (Slattery et al. 2012; Toth et al. 2012b).
Statistical analysis
PASW/SPSS (Version 17) was used to perform all statistical analyses. Fear conditioning and extinction training data were analyzed using repeated-measures analysis of variance (ANOVA), followed by a Bonferroni post hoc analysis whenever appropriate. Extinction retention and home cage locomotion data were analyzed using a one-way ANOVA, followed by a Bonferroni post hoc analysis whenever appropriate. The criterion for significance was p ≤ 0.05.
Results
Effects of preconditioning manipulation of the OXT system on cued fear in rats
To determine whether preconditioning manipulation of the OXT system influences cued fear, rats were infused icv with either vehicle (n = 12), OXT (1.0 μg/5 μl; n = 13), or OXTR-A (n = 13) 10 min before conditioning.
Fear conditioning
Fear conditioning was successful, as the level of freezing increased across trials (F (4,140) = 42.77; p < 0.001; Fig. 1a). There was no difference in conditioning between treatment groups (F (2,35) = 0.475; p = 0.63).

OXT facilitates, whereas OXTR-A impairs fear extinction when infused prior to conditioning in rats. a Rats were infused icv with either vehicle (5 μl; n = 12), OXT (1.0 μg/5 μl; n = 13), or OXTR-A (0.75 μg/5 μl; n = 13) 10 min before conditioning. b On day 2, extinction training was assessed. c On day 3, extinction retention was assessed. Data represent the mean time of CS-elicited freezing ± SEM. *p < 0.05 compared with vehicle-treated rats
Extinction training
There was a significant difference in fear extinction between treatment groups (F (2,35) = 11.50; p < 0.001), with OXT-treated rats showing lower CS-elicited freezing during blocks 2 and 3 compared with vehicle-treated rats, while OXTR-A-treated rats showed higher freezing during blocks 6–10 compared with vehicle-treated rats (p < 0.05; Fig. 1b).
Extinction retention
There was a significant difference in extinction retention between treatment groups (F (2,35) = 6.95; p = 0.003; Fig. 1c), with OXTR-A-treated rats showing higher CS-elicited freezing compared with both vehicle- and OXT-treated rats (p < 0.05). There was no difference between vehicle- and OXT-treated rats.
Effects of OXT prior to extinction training on cued fear in rats and mice
To determine whether OXT administered before extinction training influences cued fear, rats and mice were infused icv with either vehicle (rats: n = 9; mice: n = 21), a lower OXT dose (rats: n = 6; mice: n = 8), or a higher OXT dose (rats: n = 12; mice: n = 16) 10 min before extinction training.
Fear conditioning
Fear conditioning was successful in both rats and mice, as the level of freezing increased across trials (rats: F (4,96) = 14.84; p < 0.001; Fig. 2a; mice: F (4,168) = 22.15; p < 0.001; Fig. 3a). There was no difference in conditioning between groups the day before treatment (rats: F (2,24) = 0.065; p = 0.94; mice: F (2,42) = 0.081; p = 0.92).

OXT impairs fear extinction when infused prior to extinction training in rats. a On day 1, rats were fear conditioned. b On day 2, 10 min before extinction training, rats were infused icv with either vehicle (5 μl; n = 9), a lower OXT dose (0.1 μg/5 μl; n = 6), or a higher OXT dose (1.0 μg/5 μl; n = 12). c On day 3, extinction retention was assessed. Data represent the mean time of CS-elicited freezing ± SEM. *p < 0.05 compared with vehicle-treated rats

OXT impairs fear extinction when infused in a low dose before extinction training in mice. a On day 1, mice were fear conditioned. b On day 2, 10 min before extinction training, mice were infused icv with either vehicle (2 μl; n = 21), a lower OXT dose (0.1 μg/2 μl; n = 8), or a higher OXT dose (0.5 μg/2 μl; n = 16). c On day 3, extinction retention was assessed. Data represent the mean time of CS-elicited freezing ± SEM. *p < 0.05 compared with vehicle-treated mice
Extinction training
There was a significant difference in fear extinction between treatment groups in both rats (F (2,24) = 3.401; p = 0.05; Fig. 2b) and mice (F (2,42) = 24.33; p < 0.001; Fig. 3b). While both OXT doses increased CS-elicited freezing compared with vehicle in rats (0.1 μg, blocks 7, 10; 1.0 μg, blocks 7–10), OXT exhibited a dose-dependent effect in mice. More specifically, the lower OXT dose increased (p = 0.05), while the higher dose decreased (p < 0.001) CS-elicited freezing across the whole trial compared with the vehicle-treated group. Further post hoc analyses revealed that the lower dose increased (block 9; p < 0.05) and the higher dose decreased (blocks 1–7, 9; p < 0.05) CS-elicited freezing (Fig. 3b).
Extinction retention
There was a tendency towards an increased CS-elicited freezing during extinction retention in OXT-treated rats compared with vehicle-treated rats (F (2,24) = 2.881; p = 0.076; Fig. 2c), while no difference in extinction retention was found between treatment groups in mice (F (2,42) = 0.324; p = 0.73; Fig. 3c).
Effects of OXTR-A alone and on OXT-induced delay in fear extinction in rats
To determine whether OXTR-A infusion itself facilitates fear extinction and whether synthetic OXT impairs fear extinction by binding to the OXTR, rats were infused icv with either vehicle (n = 8) or OXTR-A (n = 16) 40 min before extinction training. Thirty minutes later, vehicle-treated rats were infused icv with vehicle, while OXTR-A-treated rats were infused with either vehicle (n = 8) or OXT (1.0 μg/5 μl; n = 8).
Fear conditioning
Fear conditioning was successful, as the level of freezing increased across trials (F (4,84) = 14.75; p < 0.001; Fig. 4a). There was no difference in conditioning between groups the day before treatment (F (2,21) = 0.023; p = 0.98).

OXT impairs fear extinction via OXTRs in rats. a On day 1, rats were fear conditioned. b On day 2, 40 min before extinction training, rats were infused icv with either vehicle (5 μl; n = 8) or OXTR-A (0.75 μg/5 μl; n = 16). Thirty minutes later, vehicle-treated rats were infused again with 5 μl vehicle (Veh/Veh), while OXTR-A-treated rats were infused with either vehicle (OXTR-A/Veh; 5 μl; n = 8) or OXT (OXTR-A/OXT; 1.0 μg/5 μl; n = 8). c On day 3, extinction retention was assessed. Data represent the mean time of CS-elicited freezing ± SEM
Extinction training
Extinction was successful in all treatment groups, as the high levels of freezing during the first trials decreased substantially by the last trial (F (9,189) = 8.29; p < 0.001; Fig. 4b). There was no difference in fear extinction between treatment groups (F (2,21) = 0.42; p = 0.66).
Extinction retention
There was no difference in extinction retention between treatment groups (F (2,21) = 0.99; p = 0.39; Fig. 4c).
Effects of OXT on home cage locomotion in rats and mice
To determine whether the doses of OXT used for the cued fear experiments affect locomotion, separate groups of rats and mice were infused icv with either vehicle (rats: n = 8; mice: n = 6), a lower OXT dose (rats: n = 7; mice: n = 7), or a higher OXT dose (rats: n = 7; mice: n = 7) and home cage locomotion was measured immediately for 1 h.
There was no difference in home cage locomotion between groups in rats (F (2,19) = 0.22; p = 0.80; Fig. 5a). In mice, however, there was a significant difference between groups (F (2,17) = 6.88; p = 0.006; Fig. 5b), with the higher OXT dose increasing locomotion compared with both vehicle (p = 0.029) and the lower OXT dose (p = 0.01). The lower OXT dose, however, did not affect home cage locomotion.

OXT effects on home cage locomotion in rats (a) and mice (b). Separate groups of rats and mice were infused icv with either vehicle (rats, 5 μl; n = 8; mice, 2 μl; n = 6), a lower OXT dose (rats, 0.1 μg/5 μl; n = 7; mice, 0.1 μg/2 μl; n = 7), or a higher OXT dose (rats, 1.0 μg/5 μl; n = 7; mice, 0.5 μg/2 μl; n = 7) immediately before home cage locomotion was monitored. Data represent the distance moved within 1 h ± SEM. *p < 0.05 compared with vehicle-treated mice
Discussion
The present study demonstrates that modulation of the central OXT system affects cued fear extinction in a timepoint-dependent manner. In more detail, we could show that, when administered before fear conditioning, OXT did not affect fear conditioning, but decreased fear expression during fear extinction training and facilitated fear extinction. In contrast, OXTR-A administered at the same timepoint did not affect fear conditioning or fear expression, but impaired fear extinction. In contrast, when administered before extinction training, OXT impaired fear extinction, while OXTR-A had no effect, suggesting a lack of involvement of the endogenous OXT system at this timepoint. These findings could be observed both in rats and mice, indicating that the effects of OXT on fear extinction are conserved across species, making the translation of these findings to humans more applicable. OXT impaired fear extinction by binding to the OXTR, as the inhibitory effect of icv OXT on fear extinction was abolished by prior treatment with icv OXTR-A. However, the impaired fear extinction was not a result of reduced locomotion, as neither rats nor mice showed changes in locomotion after OXT treatment. These findings suggest that, while elevated OXT levels at the time of a traumatic event prevent the formation of aversive memories, caution is needed before recommending OXT for the treatment of PTSD.
Preconditioning manipulation of the OXT system
According to our hypothesis, preconditioning administration of OXT decreased fear expression and facilitated fear extinction, without directly affecting fear conditioning. In contrast, OXTR-A administration impaired both fear extinction and extinction retention, indicating that an elevated activity of the endogenous OXT during conditioning is required for successful fear extinction.
A possible explanation for these effects is the modulatory effect of OXT on corticosterone (CORT) secretion. In female rats, chronic OXT reduced stress-induced CORT release (Windle et al. 1997), while OXTR-A increased CORT secretion into the blood in both male and female rats via an activation of the HPA axis (Neumann et al. 2000). Previous studies demonstrated that decreasing CORT concentration before conditioning by glucocorticoid synthesis inhibitors, such as metyrapone (Loscertales et al. 1997; Cordero et al. 2002) or dehydroepiandrosterone (Fleshner et al. 1997), or by blocking CORT activity through a glucocorticoid receptor antagonist (Cordero and Sandi 1998) attenuated fear expression. Although CORT activation before exposure to tasks that involve acquisition of information has been shown to impair cognitive processing (Conrad et al. 1996; Kirschbaum et al. 1996; Lupien and McEwen 1997), CORT release during the actual learning process facilitates cognitive processing (for reviews, see Sandi 1998; de Kloet et al. 1999). However, whether alterations in available CORT mediate the facilitatory effects of preconditioning OXT on fear extinction remain to be verified.
As OXT and OXTR-A treatment did not alter fear conditioning itself, the observed effects on extinction are unlikely to be due to the antinociceptive properties of OXT. However, several studies have shown that the OXT system modulates pain perception (Yang et al. 2007, 2011; Condés-Lara et al. 2009), with OXT increasing and OXTR-A decreasing the pain threshold in a dose-dependent manner (Uvnäs-Moberg et al. 1992; Lundeberg et al. 1994; Yang et al. 2011).
Although the mechanisms underlying the facilitatory effect of preconditioning OXT on cued fear extinction are yet unknown, these findings suggest that activation of the endogenous OXT system is beneficial during traumatic experiences to protect against the development of traumatic memory pathologies, such as PTSD.
Manipulation of the OXT system prior to extinction training
Contrary to our hypothesis, icv administration of OXT prior to extinction training impaired fear extinction as reflected by increased CS-elicited freezing. This was observed both in rats and mice, indicating that the inhibitory effects of OXT on fear extinction are conserved across species. However, while we could show that the impairing effects of OXT were mediated via the OXTR as preadministration of an OXTR-A blocked its effects, OXTR-A treatment alone did not facilitate fear extinction, indicating that the endogenous OXT system is not involved in fear extinction at this timepoint. The enhanced OXT-induced freezing to the CS was tone-specific and not generalized as neither rats nor mice froze before tone onset nor did they show increased freezing responses to the tone prior to its association with the shock. Taken together, these results suggest that OXT treatment before extinction training delays the extinction of cued fear. Considering that extinction training is regarded as a form of new learning (for reviews, see Cammarota et al. 2007; Quirk et al. 2010), when animals learn that the CS no longer predicts the US, drugs that interfere with the acquisition of fear learning should also block the acquisition of extinction memories when administered before extinction training (Myers and Davis 2002). This might explain why OXT decreased fear expression and facilitated fear extinction when administered before fear conditioning and impaired fear extinction when administered before extinction training.
We propose that CORT is a possible mediator of the pre-extinction training effects of OXT on fear extinction, similar to its preconditioning effects. While decreasing CORT concentrations before conditioning attenuates fear expression (Fleshner et al. 1997; Loscertales et al. 1997; Cordero and Sandi 1998; Cordero et al. 2002), decreasing CORT concentrations before extinction training by icv and basolateral amygdala (BLA) administration of metyrapone (Barrett and Gonzalez-Lima 2004; Yang et al. 2006) blocks fear extinction. In contrast, glucocorticoid receptor agonists were shown to facilitate fear extinction when administered before extinction training (Yang et al. 2006, 2007).
Several studies have shown that OXT facilitated, rather than impaired, fear extinction when administered before extinction training directly into the CeA (Roozendaal et al. 1992; Viviani et al. 2011), a brain region that coordinates the behavioral and physiological correlates of fear expression (LeDoux et al. 1988). In our study, however, OXT was administered icv, which is likely to explain the discrepant results. While OXT administered into the cerebral ventricles may reach the CeA, it may not do so in a concentration sufficient to facilitate fear extinction. Moreover, it is likely to reach brain areas which increase fear responses, such as the BLA. The BLA, a storage site for fear memories, is thought to mediate the initial acquisition of extinction (Herry et al. 2006, 2008; Sotres-Bayon et al. 2007) and the expression of extinction memory via inhibition of CeA output neurons (Quirk et al. 2003; Likhtik et al. 2008). However, whether OXT impairs fear extinction when administered into the BLA remains to be verified.
In support of this region-dependent hypothesis, several studies have shown that OXT facilitated the extinction of passive avoidance behavior when applied either icv into the hippocampal dentate gyrus or into the dorsal raphe nucleus immediately after the learning trial (Bohus et al. 1978; Kovács et al. 1979; de Wied et al. 1991). However, when applied into the dorsal septal nucleus, OXT impaired the extinction of passive avoidance (Kovács et al. 1979), suggesting that OXT affects extinction memory in a region-dependent manner. Although both passive avoidance and cued fear conditioning use footshocks as the aversive sensory stimuli, several studies utilizing knockout mice have shown deficits in cued fear conditioning, while passive avoidance behavior was normal (Weeber et al. 2000; Takao et al. 2010; Kaidanovich-Beilin et al. 2009). The subtle differences between the two paradigms and the different timepoints of OXT administration might also account for the different effects of central OXT on extinction of cued fear versus passive avoidance behavior.
Despite previous studies showing that OXT causes sedation at high doses in rats (Uvnäs-Moberg et al. 1994), neither dose of OXT used in the present study altered home cage locomotion in rats. This indicates that the impairment of fear extinction by OXT in rats is not due to nonspecific alterations in locomotion. In contrast, the higher OXT dose employed in mice resulted in behavioral hyperactivity, defined as increased home cage locomotion and excessive scratching and grooming, confirming previous findings (Delanoy et al. 1979; Meisenberg and Simmons 1982). This behavioral hyperactivity likely reflects the apparent decrease in fear expression and facilitation of fear extinction caused by the higher OXT dose in mice as such behaviors would mask any underlying fear-related behaviors. However, the lower OXT dose, which did not alter home cage locomotion, actually impaired fear extinction in mice. This is in agreement with the rat studies and strongly implies that OXT administered prior to extinction training has a detrimental outcome on fear extinction.
In summary, we have shown that icv OXT decreases fear expression and facilitates fear extinction when administered before fear conditioning, which might have a beneficial effect during traumatic events. In contrast, when applied before fear extinction training, which would be the comparable timepoint for psychotherapy in PTSD patients, OXT delays fear extinction. Considering that a more specific and local administration of OXT is not possible in patients, caution is needed before recommending OXT for the treatment of PTSD.
References
Barrett D, Gonzalez-Lima F (2004) Behavioral effects of metyrapone on Pavlovian extinction. Neurosci Lett 371(2–3):91–96
Bisson JI, Ehlers A, Matthews R, Pilling S, Richards D, Turner S (2007) Psychological treatments for chronic post-traumatic stress disorder. Systematic review and meta-analysis. Br J Psychiatry 190:97–104
Blume A, Bosch OJ, Miklos S, Torner L, Wales L, Waldherr M, Neumann ID (2008) Oxytocin reduces anxiety via ERK 1/2 activation: local effect within the rat hypothalamic paraventricular nucleus. Eur J Neurosci 27:1947–1956
Bohus B, Kovacs GL, de Wied D (1978) Oxytocin, vasopressin and memory: opposite effects on consolidation and retrieval processes. Brain Res 157:414–417
Bosch OJ, Neumann ID (2008) Brain vasopressin is an important regulator of maternal behavior independent of dams' trait anxiety. Proc Natl Acad Sci 105(44):17139–17144
Brunello N, Davidson JR, Deahl M, Kessler RC, Mendlewicz J, Racagni G, Shalev AY, Zohar J (2001) Posttraumatic stress disorder: diagnosis and epidemiology, comorbidity and social consequences, biology and treatment. Neuropsychobiology 43:150–162
Cammarota M, Bevilaqua LR, Vianna MR, Medina JH, Izquierdo I (2007) The extinction of conditioned fear: structural and molecular basis and therapeutic use. Rev Bras Psiquiatr 29(1):80–85
Carter CS, Altemus M, Chrousos GP (2001) Neuroendocrine and emotional changes in the post-partum period. Prog Brain Res 133:241–249
Condés-Lara M, Rojas-Piloni G, Martínez-Lorenzana G, López-Hidalgo M, Rodríguez-Jiménez J (2009) Hypothalamospinal oxytocinergic antinociception is mediated by GABAergic and opiate neurons that reduce A-delta and C fiber primary afferent excitation of spinal cord cells. Brain Res 1247:38–49
Conrad CD, Galea LAM, Kuroda Y, McEwen BS (1996) Chronic stress impairs spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav Neurosci 110:1321–1334
Cordero MI, Sandi C (1998) A role for brain glucocorticoid receptors in contextual fear conditioning: dependence upon training intensity. Brain Res 786(1–2):11–17
Cordero MI, Kruyt ND, Merino JJ, Sandi C (2002) Glucocorticoid involvement in memory formation in a rat model for traumatic memory. Stress 5(1):73–79
Davidson JR, Stein DJ, Shalev AY, Yehuda R (2004) Posttraumatic stress disorder: acquisition, recognition, course, and treatment. J Neuropsychiatry Clin Neurosci 16:135–147
de Kloet ER, Oitzl MS, Joels M (1999) Stress and cognition, are corticosteroids good or bad guys? Trends Neurosci 22:422–426
de Wied D, Elands J, Kovacs G (1991) Interactive effects of neurohypophyseal neuropeptides with receptor antagonists on passive avoidance behavior: mediation by a cerebral neurohypophyseal hormone receptor. Proc Natl Acad Sci USA 88:1494–1498
Delanoy RL, Dunn AJ, Walter R (1979) Neurohypophyseal hormones and behavior: effects of intracerebroventricularly injected hormone analogs in mice. Life Sci 24:651–658
Dilger S, Straube T, Mentzel HJ, Fitzek C, Reichenbach JR, Hecht H, Krieschel S, Gutberlet I, Miltner WH (2003) Brain activation to phobia-related pictures in spider phobic humans: an event-related functional magnetic resonance imaging study. Neurosci Lett 348:29–32
Ebner K, Bosch OJ, Krömer SA, Singewald N, Neumann ID (2005) Release of oxytocin in the rat central amygdala modulates stress-coping behavior and the release of excitatory amino acids. Neuropsychopharmacol 30(2):223–230
Fanselow MS (1980) Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci 15(4):177–182
Fleshner M, Pugh CR, Tremblay D, Rudy JW (1997) DHEA-S selectively impairs contextual-fear conditioning: support for the antiglucocorticoid hypothesis. Behav Neurosci 111(3):512–517
Gülpinar MA, Yegen BC (2004) The physiology of learning and memory: role of peptides and stress. Curr Protein Pept Sci 5(6):457–473
Heinrichs M, Meinlschmidt G, Neumann ID, Wagner S, Kirschbaum C, Ehlert U, Hellhammer DH (2001) Effects of suckling on hypothalamic–pituitary–adrenal axis responses to psychosocial stress in postpartum lactating women. J Clin Endocrinol Metab 86:4798–4804
Heinrichs M, Baumgartner T, Kirschbaum C, Ehlert U (2003) Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol Psychiatry 54:1389–1398
Herry C, Trifilieff P, Micheau J, Luthi A, Mons N (2006) Extinction of auditory fear conditioning requires MAPK/ERK activation in the basolateral amygdala. Eur J Neurosci 24:261–269
Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A (2008) Switching on and off fear by distinct neuronal circuits. Nature 454:600–606
Ipser J, Seedat S, Stein DJ (2006) Pharmacotherapy for post-traumatic stress disorder—a systematic review and meta-analysis. S Afr Med J 96:1088–1096
Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberté C, Khan M, Okamoto K, Chambers JW, Fletcher PJ, MacAulay K, Doble BW, Henkelman M, Miyakawa T, Roder J, Woodgett JR (2009) Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain 19:2–35
Kirsch P, Esslinger C, Chen Q, Mier D, Lis S, Siddhanti S, Gruppe H, Mattay VS, Gallhofer B, Meyer-Lindenberg A (2005) Oxytocin modulates neural circuitry for social cognition and fear in humans. J Neurosci 25(49):11489–11493
Kirschbaum C, Wolf OT, May M, Wippich W, Hellhammer DH (1996) Stress- and treatment-induced elevations of cortisol levels associated with impaired declarative memory in healthy adults. Life Sci 58:1475–1483
Kovács GL, Bohus B, Versteeg DHG, de Kolet ER, de Wied D (1979) Effect of oxytocin and vasopressin on memory consolidation: sites of action and catecholaminergic correlates after local microinjection into limbic–midbrain structures. Brain Res 175:303–314
Labuschagne I, Phan KL, Wood A, Angstadt M, Chua P, Heinrichs M, Stout JC, Nathan PJ (2010) Oxytocin attenuates amygdala reactivity to fear in generalized social anxiety disorder. Neuropsychopharmacol 35(12):2403–2413
Landgraf R, Neumann ID (2004) Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front Neuroendocrinol 25:150–176
Lattal KM, Abel T (2001) Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci 21(15):5773–5780
LeDoux JE, Iwata J, Cicchetti P, Reis DJ (1988) Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci 8:2517–2529
Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Paré D (2008) Amygdala intercalated neurons are required for expression of fear extinction. Nature 454:642–645
Loscertales M, Rose SP, Sandi C (1997) The corticosteroid synthesis inhibitors metyrapone and aminoglutethimide impair long-term memory for a passive avoidance task in day-old chicks. Brain Res 769(2):357–361
Lukas M, Toth I, Reber SO, Slattery DA, Veenema AH, Neumann ID (2011) The neuropeptide oxytocin facilitates pro-social behavior and prevents social avoidance in rats and mice. Neuropsychopharmacol 36(11):2159–2168
Lundeberg T, Uvnäs-Moberg K, Agren G, Bruzelius G (1994) Anti-nociceptive effects of oxytocin in rats and mice. Neurosci Lett 28;170(1):153–157
Lupien SJ, McEwen BS (1997) The acute effects of corticosteroids on cognition: integration of animal and human model studies. Brain Res Rev 24:1–27
Makkar SR, Zhang SQ, Cranney J (2010) Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacol 35:1625–1652
Manning M, Stoev S, Chini B, Durroux T, Mouillac B, Guillon G (2008) Peptide and non-peptide agonists and antagonists for the vasopressin and oxytocin V1a, V1b, V2 and OT receptors: research tools and potential therapeutic agents. Prog Brain Res 170:473–512
Marshall RD, Pierce D (2000) Implications of recent findings in posttraumatic stress disorder and the role of pharmacotherapy. Harv Rev Psychiatry 7(5):247–256
McCarthy MM, McDonald CH, Brooks PJ, Goldman D (1996) An anxiolytic action of oxytocin is enhanced by estrogen in the mouse. Physiol Behav 60(5):1209–1215
Meisenberg G, Simmons WH (1982) Behavioral effects of intracerebroventricularly administered neurohypophyseal hormone analogs in mice. Pharmac Biochem Behav 16:819–825
Muigg P, Hetzenauer A, Hauer G, Hauschild M, Gaburro S, Frank E, Landgraf R, Singewald N (2008) Impaired extinction of learned fear in rats selectively bred for high anxiety—evidence of altered neuronal processing in prefrontal–amygdala pathways. Eur J Neurosci 28(11):2299–2309
Myers KM, Davis M (2002) Behavioral and neural analysis of extinction. Neuron 36(4):567–584
Neumann ID (2007) Stimuli and consequences of dendritic release of oxytocin within the brain. Biochem Soc Trans 35(5):1252–1257
Neumann ID, Wigger A, Torner L, Holsboer F, Landgraf R (2000) Brain oxytocin inhibits basal and stress-induced activity of the hypothalamo-pituitary-adrenal axis in male and female rats: partial action within the paraventricular nucleus. J Neuroendocrinol 12(3):235–243
Olff M, Langeland W, Witteveen A, Denys D (2010) A psychobiological rationale for oxytocin in the treatment of posttraumatic stress disorder. CNS Spectr 15(8):522–530
Quirk GJ, Likhtik E, Pelletier JG, Paré D (2003) Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci 23:8800–8807
Quirk GJ, Paré D, Richardson R, Herry C, Monfils MH, Schiller D, Vicentic A (2010) Erasing fear memories with extinction training. J Neurosci 30(45):14993–14997
Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB, Orr SP, Pitman RK (2000) Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biol Psychiatry 47:769–776
Ring RH, Malberg JE, Potestio L, Ping J, Boikess S, Luo B, Schechter LE, Rizzo S, Rahman Z, Rosenzweig-Lipson S (2006) Anxiolytic-like activity of oxytocin in male mice: behavioral and autonomic evidence, therapeutic implications. Psychopharmacology (Berl) 185(2):218–225
Roozendaal B, Schoorlemmer GH, Wiersma A, Sluyter S, Driscoll P, Koolhaas JM, Bohus B (1992) Opposite effects of central amygdaloid vasopressin and oxytocin on the regulation of conditioned stress responses in male rats. Ann NY Acad Sci 652:460–461
Sandi C (1998) Role and mechanisms of action of glucocorticoids in memory formation. Neural Plasticity 6:39–49
Shin LM, Orr SP, Carson MA, Rauch SL, Macklin ML, Lasko NB, Peters PM, Metzger LJ, Dougherty DD, Cannistraro PA, Alpert NM, Fischman AJ, Pitman RK (2004) Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry 61:168–176
Slattery DA, Neumann ID (2008) No stress please! Mechanisms of stress hyporesponsiveness of the maternal brain. J Physiol 586(2):377–385
Slattery DA, Uschold N, Magoni M, Bär J, Popoli M, Neumann ID, Reber SO (2012) Behavioural consequences of two chronic psychosocial stress paradigms: anxiety without depression. Psychoneuroendocrinol 37(5):702–714
Sotres-Bayon F, Bush DE, LeDoux JE (2007) Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacol 32:1929–1940
Stein MB, Goldin PR, Sareen J, Zorrilla LT, Brown GG (2002) Increased amygdala activation to angry and contemptuous faces in generalized social phobia. Arch Gen Psychiatry 59:1027–1034
Stein DJ, Ipser JC, Seedat S (2006) Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database Syst Rev (1):CD002795
Stein DJ, Ipser J, McAnda N (2009) Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr 14:25–31
Takao K, Tanda K, Nakamura K, Kasahara J, Nakao K, Katsuki M, Nakanishi K, Yamasaki N, Toyama K, Adachi M, Umeda M, Araki T, Fukunaga K, Kondo H, Sakagami H, Miyakawa T (2010) Comprehensive behavioral analysis of calcium/calmodulin-dependent protein kinase IV knockout mice. PLoS One 5(3):e9460
Toth I, Dietz M, Peterlik D, Huber SE, Fendt M, Neumann ID, Flor PJ, Slattery DA (2012a) Pharmacological interference with metabotropic glutamate receptor subtype 7 but not subtype 5 differentially affects within- and between-session extinction of Pavlovian conditioned fear. Neuropharmacol 62(4):1619–1626
Toth I, Neumann ID, Slattery DA (2012b) Social fear conditioning: a novel and specific animal model to study social anxiety disorder. Neuropsychopharmacology. doi:10.1038/npp.2011.329
Uvnäs-Moberg K, Bruzelius G, Alster P, Bileviciute I, Lundeberg T (1992) Oxytocin increases and a specific oxytocin antagonist decreases pain threshold in male rats. Acta Physiol Scand 144(4):487–488
Uvnäs-Moberg K, Ahlenius S, Hillegaart V, Alster P (1994) High doses of oxytocin cause sedation and low doses cause an anxiolytic-like effect in male rats. Pharmacol Biochem Behav 49(1):101–106
Viero C, Shibuya I, Kitamura N, Verkhratsky A, Fujihara H, Katoh A, Ueta Y, Zingg HH, Chvatal A, Sykova E, Dayanithi G (2010) Oxytocin: crossing the bridge between basic science and pharmacotherapy. CNS Neurosci Ther 16(5):138–156
Viviani D, Charlet A, van den Burg E, Robinet C, Hurni N, Abatis M, Magara F, Stoop R (2011) Oxytocin selectively gates fear responses through distinct outputs from the central amygdala. Science 333(6038):104–107
Waldherr M, Neumann ID (2007) Centrally released oxytocin mediates mating-induced anxiolysis in male rats. Proc Natl Acad Sci USA 104(42):16681–16684
Weeber EJ, Atkins CM, Selcher JC, Varga AW, Mirnikjoo B, Paylor R, Leitges M, Sweatt JD (2000) A role for the beta isoform of protein kinase C in fear conditioning. J Neurosci 20(16):5906–5914
Windle RJ, Shanks N, Lightman SL, Ingram CD (1997) Central oxytocin administration reduces stress-induced corticosterone release and anxiety behavior in rats. Endocrinol 138:2829–2834
Wotjak CT, Landgraf R, Engelmann M (2008) Listening to neuropeptides by microdialysis: echoes and new sounds? Pharmacol Biochem Behav 90(2):125–134
Yang YL, Chao PK, Lu KT (2006) Systemic and intra-amygdala administration of glucocorticoid agonist and antagonist modulate extinction of conditioned fear. Neuropsychopharmacol 31(5):912–924
Yang YL, Chao PK, Ro LS, Wo YY, Lu KT (2007) Glutamate NMDA receptors within the amygdala participate in the modulatory effect of glucocorticoids on extinction of conditioned fear in rats. Neuropsychopharmacol 32(5):1042–1051
Yang J, Liang JY, Zhang XY, Qiu PY, Pan YJ, Li P, Zhang J, Hao F, Wang DX, Yan FL (2011) Oxytocin, but not arginine vasopressin is involving in the antinociceptive role of hypothalamic supraoptic nucleus. Peptides 32(5):1042–1046
Acknowledgments
We thank Dr. Maurice Manning for generously providing the oxytocin receptor antagonist. We also thank Rodrigue Maloumby, Andreas Thuy, and Doris Bayerl for the technical assistance. IT received financial support from the Bayerische Forschungsstiftung. IDN received financial support from BMBF and Deutsche Forschungsgemeinschaft.
Disclosure/conflict of interest
The authors report no biomedical financial interest or potential conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Toth, I., Neumann, I.D. & Slattery, D.A. Central administration of oxytocin receptor ligands affects cued fear extinction in rats and mice in a timepoint-dependent manner. Psychopharmacology 223, 149–158 (2012). https://doi.org/10.1007/s00213-012-2702-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-012-2702-4