
Abstract
Rationale
Bifeprunox is a partial dopamine agonist with a unique receptor-binding profile and potential antipsychotic properties.
Objectives
The current study evaluated the efficacy and safety of bifeprunox in patients with an acute exacerbation of schizophrenia.
Materials and methods
In this 6-week, double-blind, placebo-controlled study, 589 patients were randomly assigned to once-daily treatment with bifeprunox 5, 10, or 20 mg, placebo, or risperidone 6 mg. Efficacy was assessed by changes in symptom rating scales [Positive and Negative Syndrome Scale (PANSS) total and subscale scores; PANSS-derived BPRS scores; Clinical Global Impression—Severity (CGI—S) and Clinical Global Impression—Improvement (CGI—I) scores]. Safety and tolerability were assessed by monitoring adverse events, extrapyramidal symptoms (EPS), laboratory values, electrocardiograms, prolactin levels, and weight.
Results
Compared with placebo, bifeprunox 20 mg produced a statistically significantly greater reduction from baseline to last assessment in the primary efficacy variable (PANSS total score; effect size = −0.339), as well as most secondary efficacy measures. No statistically significant differences in efficacy were seen with lower doses of bifeprunox. The most common treatment-emergent adverse events (TEAEs) noted with bifeprunox were gastrointestinal; no clear dose-related trend in the incidence of any TEAE was observed in the bifeprunox groups. Compared to placebo, treatment with bifeprunox led to small but statistically significant decreases in weight and prolactin levels. EPS were comparable between bifeprunox and placebo. The active reference in this study, risperidone 6 mg, showed statistically significant differences from placebo for the primary efficacy parameter (effect size = −0.628) and all secondary efficacy parameters.
Conclusions
These data suggest that 20 mg of bifeprunox may be efficacious in improving symptoms in patients with an acute exacerbation of schizophrenia. Bifeprunox appeared to be safe and well tolerated by patients in this 6-week study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Antipsychotic medications are a cornerstone of treating schizophrenia, a chronic, disabling mental illness. Typical or first-generation antipsychotic compounds, first discovered in the early 1950s, work principally through blockade of the dopamine D2 receptor. These typical compounds, such as chlorpromazine and other phenothiazines, revolutionized treatment options for patients by alleviating the positive symptoms associated with schizophrenia (Dixon et al. 1995). Unfortunately, these agents are associated with extrapyramidal symptoms (EPS) such as akathisia, parkinsonism, dystonia, and tardive dyskinesia (Gerlach 2002).
More recently developed antipsychotics such as risperidone, olanzapine, quetiapine, and ziprasidone are antagonists for both serotonin (5-hydroxytryptophan or 5-HT) and dopamine receptors. These atypical antipsychotics display at least similar efficacy compared with the typical antipsychotics and are associated with decreased risks for EPS (e.g., Davis et al. 2003; Høiberg and Nielsen 2006; Lieberman et al. 2005; Leucht et al. 1999; Miller et al. 1998; Tandon et al. 2008), although atypical antipsychotics are not a homogenous group. In fact, the available atypical antipsychotics may differ in terms of efficacy (e.g., Davis et al. 2003; Lieberman et al. 2005), although some analyses have suggested there is little or no difference in efficacy across atypical agents (Tandon et al. 2008; Tandon and Fleischhacker 2005). The evidence is clearer that meaningful differences exist in terms of individual tolerability profiles. The use of certain atypical antipsychotics is associated with adverse metabolic effects, including weight gain, hyperglycemia, insulin resistance, diabetes, and elevation in triglyceride and cholesterol levels (e.g., ADA et al. 2004; Newcomer 2006; Meyer and Koro 2004; Tandon et al. 2008). For example, increased weight and central body fat deposition, as well as plasma glucose dysregulation, have been associated with olanzapine use, and an elevated risk for dyslipidemia has been associated with the use of quetiapine and olanzapine (Lieberman et al. 2005; Graham et al. 2005; Newcomer et al. 2002).
An adverse metabolic profile associated with the use of certain antipsychotics is particularly undesirable in patients with schizophrenia who are already burdened with elevated cardiometabolic risk due primarily to a clustering of risk factors such as abdominal obesity, atherogenic dyslipidemia, hypertension, and insulin resistance or glucose intolerance (Casey 2005; Newcomer 2005). In fact, patients with schizophrenia have demonstrated a significantly higher 10-year coronary heart disease (CHD) risk versus controls (Goff et al. 2005) and an increased incidence of metabolic syndrome (Ford et al. 2002; Heiskanen et al. 2003; McEvoy et al. 2005), highlighting the urgent need for novel, efficacious antipsychotic compounds that do not exacerbate metabolic risk.
Partial dopamine agonists represent a distinct subgroup of atypical antipsychotic compounds, with a mechanism of action different from that of other antipsychotics. These partial dopamine agonists have lower intrinsic activity than full agonists at dopamine receptors, allowing them to act either as functional agonists or antagonists, depending on the surrounding levels of endogenous neurotransmitter (Potkin et al. 2003; Lieberman 2004; Hirose et al. 2004; Simpson 2005). Bifeprunox (7-[4-([1,1′-biphenyl]-3-ylmethyl)-1-piperazinyl]-2(3H)-benzoxazolone monomethanesulphonate) is a partial dopamine D2 agonist and 5-HT1A partial agonist, with high affinity for both the D2 and 5-HT1 receptors (Long et al. 2000; Van Vliet et al. 2000; Hesselink et al. 2003; De Vries et al. 2003; Cosi et al. 2006; Wolf 2003; Newman-Tancredi et al. 2005). Bifeprunox also displays a high affinity for D3 and D4 receptors, low affinity for α1 and muscarinic receptors, and no appreciable affinity for 5-HT2A, 5HT2C, or histaminergic receptors (Marquis et al. 2005).
In preclinical studies, bifeprunox demonstrated a low propensity to induce EPS in rats and induced mild dystonia and EPS in monkeys (Van Vliet et al. 2000; Yang et al. 2003; Casey et al. 2000). Furthermore, in both in vitro and animal models, bifeprunox showed minimal impact on prolactin levels. Finally, bifeprunox has a unique receptor-binding profile, especially its low affinity for H1, 5HT2C, and muscarinic receptors. Activity at these receptors, particularly H1 and 5-HT2C, has been implicated as a mechanism in antipsychotic-induced weight gain (Kim et al. 2007; Kroeze et al. 2003; Matsui-Sakata et al. 2005). Weight gain is a risk factor for multiple morbidities, including glucose dysregulation and diabetes (Newcomer 2005), and the activity of some antipsychotics, particularly at H1 or serotonergic receptors, may also play a direct role in the development of diabetes, independent of weight gain (Matsui-Sakata et al. 2005; Newcomer 2005; Schwenkreis and Assion 2004).
The study presented here was designed to evaluate the efficacy and safety of different doses of bifeprunox in patients with an acute exacerbation of schizophrenia.
Materials and methods
This 6-week, randomized, double-blind, fixed-dose, placebo-controlled, parallel-group study to assess the efficacy, tolerability, and safety of bifeprunox in patients with an acute exacerbation of schizophrenia was conducted at 35 US and 2 Canadian centers from June 2002 to June 2003. Risperidone was used as an active reference for assay sensitivity.
The study was reviewed by all appropriate governing ethical committees before recruitment and was performed under the ethical principles laid down by Good Clinical Practices and the Declaration of Helsinki. Informed consent by the patient or a legally acceptable representative was required.
Patient population
Male and nonpregnant, nonlactating female patients 18 to 65 years of age with a current diagnosis of schizophrenia (American Psychiatric Association [DSM-IV-TR] 2000) were eligible to enter the study. Patients were required to have a total Positive and Negative Syndrome Scale (PANSS) score between 70 and 120; a baseline (day 1) score ≥4 on at least two of the following PANSS items: conceptual disorganization, hallucinatory behavior, suspiciousness, or unusual thought content; and a Clinical Global Impression—Severity (CGI—S) score ≥4, indicating at least moderate illness.
Patients were excluded from the study if they had a current psychiatric diagnosis other than schizophrenia that was expected to interfere with the study, had a current diagnosis or history of substance abuse (except for cannabinoid, nicotine, and caffeine) or alcohol abuse within 6 months before baseline (day 1), were at significant risk of suicide or violent behavior, had failed to respond to adequate courses of treatment (with reference to dose and duration) with two or more antipsychotic compounds belonging to different classes, were hospitalized with the current psychotic episode for >4 weeks, or showed clinically relevant abnormal vital signs or laboratory values. Other exclusion criteria included uncontrolled major medical illnesses, ischemic heart disease, history of myocardial infarction, coronary bypass surgery, coronary angioplasty, or clinically relevant electrocardiographic (ECG) abnormalities, or current treatment with any disallowed medications or continuous anticholinergic therapy for EPS.
Study design
The study included a 6-week, double-blind treatment period and a 1-week follow-up visit (week 7) after treatment discontinuation. Patients with an acute exacerbation of schizophrenia were hospitalized, starting from the screening period until at least 2 weeks after baseline evaluation. Patients could be hospitalized longer than 2 weeks, up to the full length of the study duration, if investigators deemed it necessary. Eligible patients were randomized to one of five treatment arms: bifeprunox 5, 10, or 20 mg, risperidone 6 mg, or placebo. After completing a single-blind placebo lead-in period (minimum of 3 days), subjects were titrated up according to a forced-dose titration schedule (Table 1). Once the target dose was reached, patients were maintained on that dose throughout the remainder of the study (approximately 5 weeks for patients receiving bifeprunox and 5 1/2 weeks for those receiving risperidone). Study compounds were administered orally, once daily, at approximately the same time of day; inpatient dosing was supervised.
Measurements of psychiatric efficacy and movement disorders were conducted at day 1 (baseline) and weekly intervals (excluding week 5). Prolactin levels were assessed on day 1 (baseline) and at weeks 1, 2, 4, 6, and 7 (post-study follow-up). Vital signs were measured at the screening visit, baseline, and weekly over 6 weeks. Electrocardiograms were performed at screening, baseline, week 1, and week 6. If any significant abnormalities were observed at week 6, an ECG was repeated at week 7. Laboratory measurements were performed at the screening and baseline visits and at weeks 1, 2, 4, and 6. If any significant findings were noted at week 6, laboratory measurements were repeated at week 7.
Concomitant medications
Specific adjunctive rescue compounds for the control of psychiatric symptoms and EPS included lorazepam, chloral hydrate, benztropine, and biperiden. The following compounds were allowed to manage medical conditions that might occur during the study: zolpidem or zaleplon for insomnia, milk of magnesia or docusate sodium for constipation, attapulgite for diarrhea, aluminum and magnesium hydroxide for gastrointestinal upset, and acetaminophen or ibuprofen for pain. In addition, prochlorperazine could be used as needed for nausea and vomiting; the duration of such treatment was to be kept to a minimum.
Efficacy measures
The primary efficacy measure was the change from baseline to last assessment in PANSS total 30-item score. All PANSS items were measured on a scale ranging from 1 (absent) to 7 (extreme). Patients were considered to be PANSS responders if their PANSS total score was reduced by 20% or more from baseline. In exploratory analyses, responder rates were also assessed using 25%, 30%, and 35% definitions of response. Secondary efficacy analyses included the change from baseline to last assessment on the PANSS positive, negative, and general psychopathology subscales, as well as the Brief Psychiatric Rating Scale (BPRS) total score derived from PANSS items P2 to P7, N1 and N2, and G1 to G10 (this BPRS score can range from 18 to 126; Kay et al. 1987). The change in psychosis cluster scores from the BPRS [defined as the mean score from four PANSS items (conceptual disorganization, hallucinatory behavior, suspiciousness, and unusual thought content)] was also analyzed. Finally, secondary efficacy analyses were performed using the CGI—S and Clinical Global Impression—Improvement (CGI—I) scores.
Safety measures
The following safety assessments were performed at designated study visits: physical examination, adverse event (AE) monitoring, monitoring of vital signs (including pulse, systolic/diastolic blood pressure, and oral temperature), clinical laboratory values (including hematology, biochemistry, and urinalysis), 12-lead ECG, and documentation of weight, concomitant medications, and use of anticholinergic treatment. EPS were evaluated with the Simpson-Angus Scale (SAS), Abnormal Involuntary Movement Scale (AIMS), and the Barnes Akathisia Scale (BAS).
Statistical analysis
Descriptive statistics were used to summarize and present patient demographic information and efficacy and safety measures.
Efficacy analyses were performed on the intent-to-treat (ITT) population, using both last observations carried forward (LOCF) and observed case (OC) data sets. LOCF analyses are presented in this report unless otherwise noted. The ITT population was defined as all randomized patients who took at least one dose of double-blind study compound and had at least one post-baseline measure of the primary efficacy parameter (PANSS total score). For the primary study measure (the change in PANSS total score from baseline to last assessment), analysis of covariance (ANCOVA) was performed, with treatment (excluding risperidone) and center as fixed factors and baseline total PANSS score as covariate. In addition, for the primary efficacy analyses, multiple comparisons among bifeprunox 5-, 10-, and 20-mg doses versus placebo were performed using step-down Dunnett’s procedure at a family-wise error rate <0.0499 (reduced per penalty for an interim analysis). All secondary efficacy measures were analyzed without correction for multiple comparisons using this same ANCOVA model, with baseline value of each measure as the covariate, except for CGI—I and responder rates. CGI—I scores and PANSS responder rates were analyzed using the Cochran–Mantel–Haenszel (CMH) chi-square test with pooled center as strata, when appropriate, or Fisher’s exact test.
Post hoc significance testing between risperidone and placebo on efficacy endpoints was based on an ANCOVA model separate from that used for bifeprunox versus placebo; these analyses were based on a model that included all treatment groups, but only tested risperidone versus placebo. Effect sizes for the primary efficacy endpoint (total PANSS) and key secondary efficacy endpoints (PANSS positive and negative scores; CGI—S and CGI—I) were each computed as the difference between treatment group and placebo group in mean change from baseline, divided by the common SD. For CGI—I, analyses were based on mean values at last assessment rather than on mean change from baseline.
Safety analyses were performed by summarizing adverse events, concomitant medications, and SAS, AIMS, and BAS scores for each treatment arm. Post hoc analyses were also completed for changes in weight and prolactin levels. Confidence intervals and pairwise comparisons for these analyses were based on an ANCOVA model including all treatment groups.
Sample size and power calculation
The sample size calculation was based on the primary efficacy measure. A sample size of approximately 115 patients per treatment group was required to detect with 85% power an effect size of 0.4 (i.e., a treatment difference versus placebo of at least 40% of the pooled standard deviation) at week 6 (LOCF) using a two-sided t test at a 0.05 level of significance. Assuming a 40% dropout rate, this sample size ensured approximately 80% power in detecting the requisite effect size for the OC data set at week 6.
Interim analysis
One unblinded interim analysis was performed by an independent statistician when approximately one-half of enrolled patients had completed the study. The purpose of this interim analysis was to permit the design of subsequent studies. No early study termination was allowed on the basis of the results of this analysis, which were confidential and not disseminated to study sites or any study personnel.
Results
Patient disposition and demographics
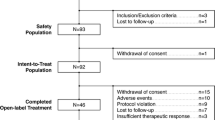
Of the 836 patients who were screened, 589 patients (male and female) were randomized, as shown in Table 2. A total of 258 patients (44%) completed the study, ranging from 36% on bifeprunox 10 mg to 50% on bifeprunox 20 mg (Table 2). Reasons for discontinuation are also summarized in Table 2; the most common reasons for subject discontinuation were lack of efficacy and withdrawal of consent (each occurred in 18% of the entire sample). Patients’ baseline demographics and clinical characteristics, including disease severity as rated using PANSS total scores, were similar for all treatment groups (Table 3). Most of the patients in this study were male (75%); the median patient age was 42 years.
Primary efficacy measure
Treatment with bifeprunox 20 mg produced a statistically significant greater mean reduction in PANSS total score from baseline to last assessment (P value adjusted for multiple comparisons = 0.031) compared with placebo (Table 4). Bifeprunox 20 mg began to separate from placebo on the primary efficacy measure as early as week 1 (Fig. 1). At last assessment, neither bifeprunox 5 or 10 mg produced mean changes (−9.7 and -5.0 points, respectively) that were statistically different from those for placebo. The risperidone arm showed a statistically significant mean change in PANSS total score from baseline to last assessment compared with placebo (P < 0.0001)Footnote 1, further confirming the responsiveness of the patient population. Effect sizes for the primary efficacy endpoint (and key secondary endpoints) based on LOCF are also shown in Table 4.

Mean change in baseline Positive and Negative Syndrome Scale (PANSS) total scores over time, last observation carried forward analyses. Patients were treated with bifeprunox 5 (n = 110), 10 (n = 118), or 20 mg (n = 111), placebo (n = 114), or risperidone 6 mg (n = 116) and followed for 6 weeks. P values shown represent mean change in an active treatment group that statistically significantly differed from mean change in placebo group at the same time point. P values for the comparison of risperidone versus placebo are derived from an ANCOVA analysis separate from that used to determine P values for the comparison between placebo and bifeprunox; the analysis of risperidone versus placebo was based on an ANCOVA model that included all treatment groups, but only tested risperidone versus placebo. *P < 0.05, **P < 0.01, ***P < 0.0001 compared to placebo
Analysis of the OC data showed greater reduction in PANSS total scores for all treatment groups compared with placebo at week 6; however, these reductions in the bifeprunox groups did not statistically significantly differ from placebo. In this regard, it is important to note that the OC data represents a smaller sample than the LOCF data, and therefore, analyses based on OC have less statistical power (mean change in PANSS score ± SD for bifeprunox: 5 mg, −18.9 ± 15.6; 10 mg, −18.2 ± 15.6; 20 mg, −18.9 ± 15.9; placebo, −11.1 ± 13.8; and risperidone, −20.0 ± 14.3).
Secondary efficacy measures
Treatment with bifeprunox 20 mg produced statistically significant changes at last assessment versus placebo in the PANSS-positive (P = 0.037), PANSS-negative (P = 0.026), and general psychopathology subscales (P = 0.016), the PANSS-derived BPRS (P = 0.012), and the BPRS psychosis cluster scores (P = 0.044) (Table 4). In addition, statistically significant reductions from baseline were observed with bifeprunox 20 mg compared with placebo in the PANSS negative subscale score at week 3 (mean change ± SD = −2.7 ± 4.4; P = 0.013), PANSS general psychopathology score at week 2 (−4.4 ± 7.5; P = 0.029) and week 3 (−5.2 ± 7.9; P = 0.032), and PANSS-derived BPRS total score at week 2 (−6.1 ± 8.8; P = 0.042), week 3 (−7.5 ± 9.1; P = 0.020), and week 4 (−7.7 ± 9.7; P = 0.024).
Treatment with bifeprunox 20 mg produced improvements in CGI—S scores during the study, although no statistically significant changes from baseline versus placebo were observed at last assessment (Table 4). Statistically significant changes in CGI—S scores were observed, however, with the 20 mg dose at week 2 (mean change ± SD = −0.50 ± 0.85; P = 0.008), week 3 (−0.57 ± 0.90; P = 0.020), and week 4 (−0.57 ± 0.89; P = 0.032). A statistically significant difference in CGI—S was noted with bifeprunox 5 mg at week 3 (−0.47 ± 0.90; P = 0.049), week 4 (−0.52 ± 0.033; P = 0.033), and last assessment (−0.58 ± 1.10; P = 0.013). No statistically significant differences between bifeprunox 10 mg and placebo were observed at any time point.
Treatment with bifeprunox 20 mg was associated with statistically significant better CGI—I scores at week 1 (mean score ± SD = 3.54 ± 0.99; P = 0.040) and week 2 (3.30 ± 1.14; P = 0.016) versus placebo (mean score ± SD = 3.77 ± 0.93 and 3.65 ± 1.18, weeks 1 and 2, respectively); CGI—I scores did not statistically significantly differ between bifeprunox 20 mg and placebo at last assessment (Table 4). No other bifeprunox treatment group showed statistically significant differences from placebo at any point on this measure.
Treatment with risperidone 6 mg resulted in statistically significant improvements at last assessment versus placebo in PANSS positive (P < 0.0001)Footnote 2, PANSS negative (P < 0.0001)Footnote 3, and PANSS general psychopathology scores (P < 0.0001)Footnote 4; BPRS total (P = 0.0001)Footnote 5 and BPRS psychosis cluster scores (P < 0.0001)Footnote 6; and CGI—S (P < 0.0001)Footnote 7 scores (Table 4). CGI—I scores at last assessment were also statistically significantly better in the risperidone group than in the placebo group (P = 0.0023)Footnote 8.
PANSS responder rates
PANSS responder rates were higher in each of the bifeprunox treatment groups than in the placebo group (5 mg: 28%; 10 mg: 24%; 20 mg: 34%; and placebo: 22%) using the response criterion of 20% (i.e., reduction from baseline in total PANSS score of 20%); however, the differences were not statistically significant (Table 5). Statistically significant differences were observed between bifeprunox 20 mg and placebo when the data were analyzed using the 25% PANSS responder rate (P = 0.008), the 30% rate (P = 0.042), and the 35% rate (P = 0.013). The 5 mg group showed a statistically significant difference versus placebo when the 25% responder criterion was used (Table 5). Responder rates in the risperidone group ranged from 40% at the 20%-reduction criterion to 13% at the 35%-reduction criterion (Table 5).
Safety
Adverse events
A total of 588 patients were included in the safety analysis (bifeprunox 5 mg: n = 115; 10 mg: n = 120; 20 mg: n = 114; placebo: n = 119; risperidone: n = 120). The majority of treatment-emergent adverse events (TEAEs) were transient and considered to be mild to moderate in severity. The percentage of patients with at least one TEAE was similar across treatment groups (Table 6). The most common TEAEs (reported in ≥5% of any treatment group and with an incidence at least twice that of placebo) noted with bifeprunox were gastrointestinal (Table 6). Concomitant prochlorperazine was used sparingly to treat nausea and vomiting (used by 3%, 3%, 4%, <1%, and <1% of the bifeprunox 5, 10, and 20 mg, placebo, and risperidone groups, respectively). No dose-related trend for an increase in the incidence of any TEAE was observed in the bifeprunox groups. The incidence of adverse events leading to discontinuation of study medication was similar among the different treatment groups (Table 2). The reported incidence of adverse events involving the reproductive system were low (e.g., erectile dysfunction was reported as an adverse event in one patient (1%) treated with risperidone and no other patient; galactorrhea was reported in one patient (<1%) treated with bifeprunox 10 mg and no other patient), although sexual functioning and the relationship between sexual functioning and prolactin levels (see Serum Prolactin Levels below) were not specifically addressed during the study.
The incidence of serious adverse events (SAEs) was also similar across all treatment groups and higher than that observed with placebo (bifeprunox arms: 12–15%; risperidone: 16%; and placebo: 9%). Most SAEs were not considered to be treatment related and resolved without discontinuation. The most frequent SAEs (reported in ≥5% of subjects in any treatment group) were aggravated psychosis (occurring in 3%, 8%, 4%, 3%, and 4% of the bifeprunox 5, 10, and 20 mg groups, the placebo group, and the risperidone group, respectively) and aggravated schizophrenia (occurring in 5%, 2%, 7%, 4%, and 4% of the bifeprunox 5, 10, and 20 mg groups, the placebo group, and the risperidone group, respectively). There was no clear indication of a dose-related increase in the incidence of any SAE in the bifeprunox groups.
Of the three deaths that occurred during this study, none was considered by the study investigators to be related to study compounds. One patient, a screening failure because of uncontrolled hypertension, died of cardiovascular disease before receiving any study compound. One patient in the placebo group experienced cardiorespiratory arrest after allegedly using crack cocaine, while one patient in the bifeprunox 5 mg group died from a cocaine overdose.
Extrapyramidal symptoms
Overall, rates of EPS were comparable among the bifeprunox and placebo arms. Minimal changes were observed for the bifeprunox and placebo groups with respect to movement disorder measures, including SAS total scores, BAS score, and AIMS score (Fig. 2). Akathisia occurred in one to three patients (<1% to 3%) across bifeprunox treatment groups, six patients (5%) in the risperidone group, and no patients in the placebo group. Use of anticholinergic medication in patients treated with bifeprunox occurred less frequently than in patients on risperidone and was similar in frequency to that in patients on placebo. For example, the incidence of benztropine use, the most commonly prescribed anticholinergic, was lower in the bifeprunox groups [bifeprunox 5 mg, five patients (5%); 10 mg, three patients (3%); 20 mg, eight patients (7%)], and placebo (six patients (5%)] than it was in the risperidone group [22 patients (19%)].

Mean change in movement disorder scores from baseline to last assessment (week 6, last observation carried forward) by treatment group. Patients were treated with bifeprunox 5 (n = 110), 10 (n = 118), or 20 mg (n = 111), placebo (n = 114), or risperidone 6 mg (n = 116) and followed for 6 weeks. Minimal change in scores on movement disorder scales designed to assess extrapyramidal symptoms were noted
Body weight
Small but statistically significant decreases in mean weight were observed at last assessment for all three bifeprunox treatment arms compared with placebo (bifeprunox 5 mg, −0.45 kg, P = 0.025; 10 mg, −0.59 kg, P = 0.009; and 20 mg, −0.27 kg, P = 0.031; placebo, +0.86 kg; Fig. 3). Patients in the risperidone-treated group had a statistically significant mean weight increase of +2.2 kg (P = 0.0057Footnote 9) versus placebo. Clinically relevant weight increase, defined as an increase of ≥7% in body weight, was experienced by 2% to 4% of bifeprunox patients, 5% of placebo patients, and 13% of risperidone patients. Weight decreases of ≥7% were observed in 6% to 8% of patients in the bifeprunox groups and 3% in the placebo group. No patient treated with risperidone had a weight decrease of ≥7%.

Mean weight change in kilograms from baseline to last assessment (week 6, last observation carried forward). Patients were treated with bifeprunox 5 (n = 110), 10 (n = 118), or 20 mg (n = 111), placebo (n = 114), or risperidone 6 mg (n = 116) and followed for 6 weeks. P values shown are against placebo. Small but statistically significant decreases in mean weight were observed for all three bifeprunox treatment arms compared with placebo; patients in the risperidone-treated group had a statistically significant mean weight increase versus placebo. Pairwise comparisons were based on a model including all treatment groups. Using observed case data instead of LOCF at week 6, weight change in each group was −2.0 kg, bifeprunox 5 mg (n = 74); −2.2 kg, bifeprunox 10 mg (n = 70); −0.18 kg, bifeprunox 20 mg (n = 95); +0.59 kg, placebo (n = 81); +2.27 kg, risperidone 6 mg (n = 96)
Lipids
Decreases from baseline in non-fasting total cholesterol levels were observed for all bifeprunox treatment groups at last assessment (mean change ranging from −11.8 to −14.1 mg/dL; Fig. 4a); a smaller decrease from baseline to last assessment in non-fasting total cholesterol levels occurred with placebo (−10.5 mg/dL) and risperidone (−4.80 mg/dL). For the majority of patients, total cholesterol levels fell within the same category (low, normal, or high) at baseline and at last assessment (79%, 67%, 76%, 73%, and 81% of patients in the bifeprunox 5, 10, and 20 mg, placebo, and risperidone groups, respectively); the most common individual shift in each treatment group was from high cholesterol at baseline to normal cholesterol at last assessment (14%, 27%, 17%, 16%, and 11% of patients in the bifeprunox 5, 10, and 20 mg, placebo, and risperidone arms, respectively).

Mean change in total cholesterol from baseline to last assessment (week 6, last observation carried forward; a) and mean change in triglycerides from baseline to last assessment (week 6, last observation carried forward; b), both assessed under non-fasting conditions. Patients were treated with bifeprunox 5 (n = 110), 10 (n = 118), or 20 mg (n = 111), placebo (n = 114), or risperidone 6 mg (n = 116) and followed for 6 weeks
Mean decreases in non-fasting triglycerides from baseline to last assessment ranged from −37.80 to −61.48 mg/dL in the bifeprunox groups. The decrease was −34.47 mg/dL in the placebo group and −10.8 in the risperidone group (Fig. 4b). Triglyceride levels were in the same category (low, normal, or high) at baseline and again at last assessment for the majority of patients (77%, 76%, 79%, 83%, and 75% of patients in the bifeprunox 5, 10, and 20 mg, placebo, and risperidone groups, respectively). The most common shift in each treatment group was from high triglycerides at baseline to normal triglycerides at last assessment (15%, 17%, 20%, 14%, and 16% of patients in the bifeprunox 5, 10, and 20 mg, placebo, and risperidone groups, respectively).
Serum prolactin levels
Mean prolactin levels decreased in the bifeprunox treatment groups from baseline to last assessment, were essentially unchanged in the placebo group, and increased in the risperidone group. The decreases in prolactin levels were observed in all bifeprunox treatment arms as early as week 1 (Fig. 5). At last assessment, mean prolactin values had statistically significantly decreased in the bifeprunox 5 mg (mean decrease = −6.70 ng/mL, P = 0.030)Footnote 10), 10 mg (−9.77 ng/mL, P = 0.006)Footnote 11, and 20 mg (−8.37 ng/mL, P = 0.012)Footnote 12 groups compared with placebo (−0.96 ng/mL)Footnote 13. The risperidone arm had a statistically significant increase in serum prolactin (mean increase = +27.81 ng/mL, P < 0.0001)Footnote 14 versus placebo. A majority of patients in each of the bifeprunox treatment groups shifted from normal prolactin levels at baseline to low prolactin levels at last assessment (56%, 73%, and 67% of patients in the 5, 10, and 20 mg bifeprunox groups, respectively); in the risperidone treatment arm, a majority of patients (61%) shifted from normal prolactin levels at baseline to high prolactin levels at last assessment. In the placebo group, 87% of patients presented with normal prolactin levels at both baseline and last assessment.

Mean change in prolactin levels over time, last observation carried forward analyses. Patients were treated with bifeprunox 5 (n = 110), 10 (n = 118), or 20 mg (n = 111), placebo (n = 114), or risperidone 6 mg (n = 116) and followed for 6 weeks. P values shown represent mean change in an active treatment group that statistically significantly differed from mean change in placebo group at last assessment (week 6, LOCF). At last assessment, mean prolactin values had statistically significantly decreased versus placebo in the bifeprunox treatment groups and statistically significantly increased versus placebo in the risperidone treatment group. Pairwise comparisons were based on a model including all treatment groups
Electrocardiography
There were minimal changes in mean values for pulse rate or for QTc and QRS intervals between baseline and last assessment in any treatment arm. Mean changes for QTc and QRS intervals ranged from approximately −2 to 4 ms. There were no trends in mean changes by treatment group. Of the patients with normal ECGs at baseline, only one patient (<1%) in the bifeprunox 5 mg group had a shift to an abnormal, clinically significant result at last assessment. The percentage of patients who categorically shifted from normal to abnormal (not clinically significant) ECGs in the bifeprunox treatment arms (5 mg, 16%; 10 mg, 15%; 20 mg, 13%) was similar to that observed in the placebo (17%) and risperidone (14%) treatment groups.
Discussion
There is a compelling need for more well-tolerated and efficacious treatments for schizophrenia (Lieberman et al. 2005). The results of this study suggest that bifeprunox 20 mg may be more efficacious in treating symptoms of schizophrenia than placebo, as shown by statistically significant improvements in PANSS total scores (the primary efficacy measure) at last assessment and statistically significant improvement in many secondary efficacy measures (i.e., the PANSS positive, negative, and general psychopathology subscale scores as well as the BPRS total and BPRS psychosis cluster scores). Doses lower than bifeprunox 20 mg were not effective in this study: there were no statistically significant differences from placebo on the primary efficacy endpoint at last assessment for lower doses. However, the 5 mg dose of bifeprunox did demonstrate an advantage compared with placebo on some secondary measures. It is noteworthy, although perhaps a chance finding, that the 5 mg dose of bifeprunox appeared to lead to greater improvement in clinical rating scale scores than did the 10 mg dose of bifeprunox.
Assay sensitivity was confirmed with risperidone 6 mg, which showed statistically significant differences from placebo for all efficacy parameters. The magnitude of the improvements seen in the 20 mg bifeprunox group was smaller than that seen in the risperidone group for most efficacy endpoints based on LOCF analyses; for example, the effect sizes for change in PANSS total score (the primary efficacy endpoint) were −0.339 and −0.628 for bifeprunox 20 mg and risperidone, respectively. As this trial was not designed as a direct comparison of risperidone and bifeprunox, any comparison of efficacy between the two compounds is post hoc and limited by the constraints of such analyses. In addition, the improvements seen with bifeprunox 20 mg are considered to be clinically relevant. Still, the appropriate use of bifeprunox in the acute treatment of schizophrenia with the doses tested in this trial requires further study.
In this study, bifeprunox appeared to be safe and well tolerated at all doses administered to subjects, with the majority of reported TEAEs being transient and mild to moderate in severity. The incidence of SAEs in the bifeprunox treatment groups was similar to that in the placebo and risperidone groups. Bifeprunox was titrated over a 7- to 8-day period because clinical data suggest a shorter titration period is associated with more intolerable effects such as orthostatic hypotension or dizziness (data on file, Solvay Pharmaceuticals). Risperidone was titrated as rapidly as its product label permits; a shorter or longer titration schedule may result in a different side effect profile for this agent.
Extrapyramidal symptoms, a common correlate of treatment with antipsychotics, are associated with a substantial reduction in a patient’s quality of life and with poor therapeutic compliance (Pierre 2005). In this study, the occurrence of EPS with bifeprunox was comparable to that with placebo. In addition, the incidence of anticholinergic use in patients treated with bifeprunox was similar to that used with placebo and less than that used with risperidone.
Patients with schizophrenia are at increased risk for metabolic disturbances such as dyslipidemia, partly because of poor diet and lifestyle issues (Hennekens et al. 2005). This baseline condition can be exacerbated by the use of certain antipsychotic compounds (ADA et al. 2004; Newcomer 2005). In this study, patients treated with bifeprunox experienced decreases in non-fasting total cholesterol and triglyceride levels that were similar to those seen with placebo and numerically greater than those seen with risperidone.
Obesity, an independent risk factor for cardiovascular disease (Poirier and Eckel 2002; Ogden et al. 2006), has been documented in as many as 75% of patients with schizophrenia, compared with 36% to 38% of the general population (Allison and Casey 2001; Dickerson et al. 2002; McCreadie and Scottish Schizophrenia Lifestyle Group 2003). Especially when manifested as intra-abdominal weight gain, obesity compromises long-term health because of its association with insulin resistance (Banerji et al. 1997). Insulin resistance is not only linked to type 2 diabetes, it is also associated with physiologic changes that include atherogenic dyslipidemia (comprising increases in plasma triglycerides and more-oxidized low-density lipoprotein particles), increased blood pressure, an increased risk of blood clotting, and increases in markers of inflammation, all of which are associated with an increased risk for cardiovascular disease (Avogaro 2006). In this study, patients treated with bifeprunox showed small but statistically significant reduction in weight compared with patients treated with placebo. Switching from an antipsychotic that causes or exacerbates weight gain or dyslipidemia to a more metabolically neutral antipsychotic may lead to weight loss and an improved lipid profile (Weiden 2007). Whether the metabolic improvements associated with bifeprunox treatment in this study more accurately reflect a switch away from less metabolically neutral compounds or the intrinsic metabolic activity of bifeprunox deserves further exploration.
Hyperprolactinemia, a condition that has been associated with sexual dysfunction, amenorrhea and infertility, galactorrhea, decreased bone mineral density, osteoporosis, breast cancer, and cardiovascular disorders may be a concern in the treatment of patients with schizophrenia (Haddad and Wieck 2004; O’Keane and Meaney 2005). Treatment with risperidone in this study was associated with statistically significantly increased mean prolactin levels versus placebo, consistent with previous studies (Halbreich and Kahn 2003; Lieberman et al. 2005; Melkersson 2005). There is no established literature on the clinical implications of low prolactin levels in the schizophrenic population. In this study, patients treated with bifeprunox exhibited a decrease in prolactin levels compared with the placebo group.
Conclusions
This study suggests that bifeprunox administered once daily for 6 weeks may have efficacy in controlling symptoms in patients with an acute exacerbation of schizophrenia. A dose of 20 mg may be the minimally effective dose, as defined by statistically significant improvements in total PANSS scores compared with placebo at study end. Neither 5 nor 10 mg of bifeprunox showed statistical separation from placebo for the primary efficacy variable. Overall, doses of bifeprunox appeared to be safe and well tolerated by the patients with schizophrenia in this study.
Notes
Post hoc analyses between risperidone and placebo were based on an ANCOVA model separate from that for bifeprunox versus placebo: these analyses were based on a model that included all treatment groups, but only tested risperidone versus placebo.
See Footnote 1.
See Footnote 1.
See Footnote 1.
See Footnote 1.
See Footnote 1.
See Footnote 1.
See Footnote 1.
Confidence intervals and pairwise comparisons were based on a model including all treatment groups.
See Footnote 9.
See Footnote 9.
See Footnote 9.
See Footnote 9.
See Footnote 9.
References
Allison DB, Casey DE (2001) Antipsychotic-induced weight gain: a review of the literature. J Clin Psychiatry 62(suppl 7):22–31
American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity (2004) Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 27(2):596–601
American Psychiatric Association (2000) Diagnostic and statistical manual of mental disorders, 4th edition, text revision (DSM-IV-TR). American Psychiatric, Washington, DC
Avogaro A (2006) Insulin resistance: trigger or concomitant factor in the metabolic syndrome. Panminerva Med 48:3–12
Banerji MA, Lebowitz J, Chaiken RL, Gordon D, Kral JG, Lebovitz HE (1997) Relationship of visceral adipose tissue and glucose disposal is independent of sex in black NIDDM subjects. Am J Physiol 273(suppl 2, pt 1):E425–E432
Casey DE (2005) Metabolic issues and cardiovascular disease in patients with psychiatric disorders. Am J Med 118(suppl 2):15S–22S
Casey DE, Van Vliet BJ, Feenstra R, Kruse CG, Long SK (2000) DU 127090: a highly potent, atypical dopamine receptor ligand—behavioral effects of DU 127090 in Cebus non-human primates. Eur Neuropsychopharmacol 10(suppl 3):333
Cosi C, Carilla-Durand E, Assié MB, Ormière AM, Maraval M, Leduc N, Newman-Tancredi A (2006) Partial agonist properties of the antipsychotics SSR181507, aripiprazole and bifeprunox at dopamine D2 receptors: G protein activation and prolactin release. Eur J Pharmacol 535:135–144
Davis JM, Chen N, Glick ID (2003) A meta-analysis of the efficacy of second-generation antipsychotics. Arch Gen Psychiatry 60:553–564
De Vries MH, Udo de Haes J, Grahnén A, Nyman L, Bergström M, Wall A, Langström B (2003) DU 127090: a novel partial dopamine agonist with antipsychotic activity: pilot study of dopamine D2 receptor occupancy after multiple oral administration of DU 127090 to healthy male volunteers, using 11C-raclopride by means of positron emission tomography. Schizophrenia Res 60(1 suppl 1):S239–S240
Dickerson FB, Pater A, Origoni AE (2002) Health behaviors and health status of older women with schizophrenia. Psychiatr Serv 53(7):882–884
Dixon LB, Lehman AF, Levine J (1995) Conventional antipsychotic medications for schizophrenia. Schizophr Bull 21(4):567–577
Ford ES, Giles WH, Dietz WH (2002) Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287(3):356–359
Gerlach J (2002) Improving outcome in schizophrenia: the potential importance of EPS and neuroleptic dysphoria. Ann Clin Psychiatry 14(1):47–57
Goff DC, Sullivan LM, McEvoy JP, Meyer JM, Nasrallah HA, Daumit GL, Lamberti S, D’Agostino RB, Stroup TS, Davis S, Lieberman JA (2005) A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res 80:45–53
Graham KA, Perkins DO, Edwards LJ, Barrier RC Jr, Lieberman JA, Harp JB (2005) Effect of olanzapine on body composition and energy expenditure in adults with first-episode psychosis. Am J Psychiatry 162:118–123
Haddad PM, Wieck A (2004) Antipsychotic-induced hyperprolactinaemia: mechanisms, clinical features and management. Drugs 64(20):2291–2314
Halbreich U, Kahn LS (2003) Hyperprolactinemia and schizophrenia: mechanisms and clinical aspects. J Psychiatr Pract 9(5):344–353
Heiskanen T, Niskanen L, Lyytikäinen R, Saarinen PI, Hintikka J (2003) Metabolic syndrome in patients with schizophrenia. J Clin Psychiatry 64:575–579
Hennekens CH, Hennekens AR, Hollar D, Casey DE (2005) Schizophrenia and increased risks of cardiovascular disease. Am Heart J 150:1115–1121
Hesselink MB, Van Vliet BJ, Ronken E, Tulp M, Long SK, Feenstra RW, Kruse CG (2003) DU 127090: a novel partial dopamine agonist with antipsychotic activity. High potency but low efficacy at dopamine D2 receptors in vitro. Schizophrenia Res 60:S108
Hirose T, Uwahodo Y, Yamada S, Miwa T, Kikuchi T, Kitagawa H, Burris KD, Altar CA, Nabeshima T (2004) Mechanism of action of aripiprazole predicts clinical efficacy and a favourable side-effect profile. J Psychopharm 18(3):375–383
Høiberg MP, Nielsen B (2006) Antipsychotic treatment and extrapyramidal symptoms amongst schizophrenic inpatients. Nord J Psychiatry 60:207–212
Kay SR, Fiszbein A, Opler LA (1987) The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13(2):261–276
Kim SF, Huang AS, Snowman AM, Teuscher C, Snyder SH (2007) Antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. PNAS 104:3456–3459
Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, Jayathilake K, Meltzer HY, Roth BL (2003) H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 28:519–526
Leucht S, Pitschel-Walz G, Abraham D, Kissling W (1999) Efficacy and extrapyramidal side-effects of the new antipsychotics olanzapine, quetiapine, risperidone, and sertindole compared to conventional antipsychotics and placebo. A meta-analysis of randomized controlled trials. Schizophr Res 35(1):51–68
Lieberman JA (2004) Dopamine partial agonists. CNS Drugs 18(4):251–267
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RSE, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK, for the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators (2005) Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353(12):1209–1223
Long SK, Feenstra R, Kruse CG, Van Vliet BJ (2000) DU 12790: a highly potent, atypical dopamine receptor ligand: partial agonist character in neurochemistry assays in vivo. Eur Neuropsychopharmacol 10(suppl 3):S295
Marquis KL, Hertel P, Reinders JH, van der Neut M, Ronken E, Hesselink MB (2005) Bifeprunox: a novel atypical antipsychotic sharing dopamine D2 receptor partial agonism and serotonin 5-HT1A receptor agonism. Schizophr Bull 31:305
Matsui-Sakata A, Ohtani H, Sawada Y (2005) Receptor occupancy-based analysis of the contributions of various receptors to antipsychotics-induced weight gain and diabetes mellitus. Drug Metab Pharmacokinet 20:368–378
McCreadie RG, Scottish Schizophrenia Lifestyle Group (2003) Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry 183:534–539
McEvoy JP, Meyer JM, Goff DC, Nasrallah HA, Davis SM, Sullivan L, Meltzer HY, Hsiao J, Stroup TS, Lieberman JA (2005) Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the clinical antipsychotic trials of intervention effectiveness (CATIE) schizophrenia trials and comparison with national estimates from NHANES III. Schiz Research 80:19–32
Melkersson K (2005) Differences in prolactin elevation and related symptoms of atypical antipsychotics in schizophrenic patients. J Clin Psychiatry 66(6):761–767
Meyer JM, Koro CE (2004) The effects of antipsychotic therapy on serum lipids: a comprehensive review. Schizophr Res 70:1–17
Miller CH, Mohr F, Umbricht D, Woerner M, Fleischhacker WW, Lieberman JA (1998) The prevalence of acute extrapyramidal signs and symptoms in patients treated with clozapine, risperidone, and conventional antipsychotics. J Clin Psychiatry 59(2):69–75
Newcomer JW (2005) Second generation (atypical) antipsychotics and metabolic effects. A comprehensive literature review. CNS Drugs 19(suppl 1):1–93
Newcomer JW (2006) Medical risk in patients with bipolar disorder and schizophrenia. J Clin Psychiatry 67(suppl 9):25–30
Newcomer JW, Haupt DW, Fucetola R, Melson AK, Schweiger JA, Cooper BP, Selke G (2002) Abnormalities in glucose regulation during antipsychotic treatment of schizophrenia. Arch Gen Psychiatry 59(4):337–345
Newman-Tancredi A, Assié MB, Leduc N, Ormière AM, Danty N, Cosi C (2005) Novel antipsychotics activate recombinant human and native rat serotonin 5-HT1A receptors: affinity, efficacy and potential implications for treatment of schizophrenia. Int J Neuropsychopharmacol 8:341–356
Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM (2006) Prevalence of overweight and obesity in the United States, 1999–2004. JAMA 295(13):1549–1555
O’Keane V, Meaney AM (2005) Antipsychotic drugs: a new risk factor for osteoporosis in young women with schizophrenia? J Clin Psychopharmacol 25(1):26–31
Pierre JM (2005) Extrapyramidal symptoms with atypical antipsychotics: incidence, prevention and management. Drug Saf 28(3):191–208
Poirier P, Eckel RH (2002) Obesity and cardiovascular disease. Curr Atheroscler Rep 4:448–453
Potkin SG, Saha AR, Kujawa MJ, Carson WH, Ali M, Stock E, Stringfellow J, Ingenito G, Marder SR (2003) Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs. placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry 60:681–690
Schwenkreis P, Assion H-J (2004) Atypical antipsychotics and diabetes mellitus. World J Biol Psychiatry 5:73–82
Simpson GM (2005) Atypical antipsychotics and the burden of disease. Am J Managed Care 11(suppl 8):S235–S241
Tandon R, Fleischhacker WW (2005) Comparative efficacy of antipsychotics in the treatment of schizophrenia: a critical assessment. Schizophr Res 79:145–155
Tandon R, Belmaker RH, Gattaz WF, Lopez-Ibor JJ Jr, Okasha A, Singh B, Stein DJ, Olie JP, Fleischhacker WW, Moeller HJ, for the Section of Pharmacopsychiatry, World Psychiatric Association (2008) World Psychiatric Association Pharmacopsychiatry Section statement on comparative effectiveness of antipsychotics in the treatment of schizophrenia. Schiz Res 100:20–38
Van Vliet BJ, Mos J, Van der Heijden JAM, Feenstra R, Kruse CG, Long SK (2000) DU 127090: a highly potent atypical dopamine receptor ligand—a putative potent full spectrum antipsychotic with low EPS potential. Eur Neuropsychopharmacol 10(suppl 3):293
Weiden PJ (2007) Switching antipsychotics as a treatment strategy for antipsychotic-induced weight gain and dyslipidemia. J Clin Psychiatry 68(suppl 4):34–39
Wolf W (2003) DU-127090 Solvay/H Lundbeck. Curr Opin Investig Drugs 4(1):72–76
Yang H, Casey DE, Feenstra RW, Kruse CG, Long SK (2003) DU127090: a novel partial dopamine agonist with antipsychotic activity behavioral effects of DU127090 in Cebus non-human primates. Schizophrenia Res 60(1 suppl 1):370
Acknowledgements
This work was sponsored by H. Lundbeck A/S, Solvay Pharmaceuticals, Inc., and Wyeth Pharmaceuticals. Editorial support was provided by Centron.
Financial disclosure
Dr Casey is a consultant to Abbott Laboratories, Bristol-Myers Squibb, Janssen Pharmaceuticals, Pfizer Inc., Solvay Pharmaceuticals, Inc., Dainippon Sumitomo Pharmaceuticals, and Wyeth Pharmaceuticals. He is on the speakers bureau for Abbott Laboratories, Bristol-Myers Squibb, Janssen Pharmaceuticals, and Pfizer Inc. and receives financial support from the Danicas Foundation. Earl Sands and Hwa-Ming Yang are employees of Solvay Pharmaceuticals, Inc. Jens Heisterberg is an employee of H. Lundbeck A/S.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Casey, D.E., Sands, E.E., Heisterberg, J. et al. Efficacy and safety of bifeprunox in patients with an acute exacerbation of schizophrenia: results from a randomized, double-blind, placebo-controlled, multicenter, dose-finding study. Psychopharmacology 200, 317–331 (2008). https://doi.org/10.1007/s00213-008-1207-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-008-1207-7