
Abstract
Introduction
The antisaccade task provides a laboratory analogue of situations in which execution of the correct behavioural response requires the suppression of a more prepotent or habitual response. Errors (failures to inhibit a reflexive prosaccade towards a sudden onset target) are significantly increased in patients with damage to the dorsolateral prefrontal cortex and patients with schizophrenia. Recent models of antisaccade performance suggest that errors are more likely to occur when the intention to initiate an antisaccade is insufficiently activated within working memory. Nicotine has been shown to enhance specific working memory processes in healthy adults.
Materials and methods
We explored the effect of nicotine on antisaccade performance in a large sample (N=44) of young adult smokers. Minimally abstinent participants attended two test sessions and were asked to smoke one of their own cigarettes between baseline and retest during one session only.
Results and conclusion
Nicotine reduced antisaccade errors and correct antisaccade latencies if delivered before optimum performance levels are achieved, suggesting that nicotine supports the activation of intentions in working memory during task performance. The implications of this research for current theoretical accounts of antisaccade performance, and for interpreting the increased rate of antisaccade errors found in some psychiatric patient groups are discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It is well established that administration of the cholinergic agonist nicotine results in improvements in basic, or low-level psychomotor performance in humans. For example, nicotine has been shown to increase finger tapping rate (West and Jarvis 1986) and decrease reaction times (Bates et al. 1994; Witte et al. 1997; Griesar et al. 2002). Nicotine also improves performance on tests involving sustained attention, such as the Rapid Visual Information Processing task (e.g. Warburton and Arnall 1994; Foulds et al. 1996) and Continuous Performance test (Levin et al. 1998).
A number of studies have suggested that nicotine may additionally improve performance on tasks that require high-level cognitive control processes such as error detection and correction, planning, updating working memory and active response inhibition. For example, administration of nicotine can lead to better performance on the n-back task (Ernst et al. 2001; Kumari et al. 2003) and random letter generation (Mancuso et al. 2001), both of which require monitoring and updating information held in active or ‘working’ memory.
The ability to inhibit the processing of irrelevant information and withhold prepotent or habitual responses to external stimuli is a key function of working memory (Roberts et al. 1994). Nicotinic enhancement of inhibition of irrelevant or conflicting material has been demonstrated using the Stroop test (Della Casa et al. 1999) and the retrieval-induced forgetting paradigm (Edginton and Rusted 2003). The antisaccade task (Hallett 1978) also provides a laboratory measure of the ability to inhibit prepotent responses. The sudden appearance of an object in the visual periphery typically captures attention and elicits a ‘reflexive’ prosaccade in its direction (Findlay and Walker 1999).
In the antisaccade task, participants are required to inhibit the prosaccade towards the target and instead initiate a voluntary eye-movement (an antisaccade) to the opposite hemifield. As a tool with which to study the effects of nicotine on cognitive function, the antisaccade task has a number of advantages over neuropsychological measures of inhibition, such as Stroop. The neural mechanisms underlying saccadic control are comparatively well charted (e.g. Leigh and Kennard 2004), and the oculomotor system has a limited output, which can easily be measured with a high degree of precision using modern oculographic recording equipment. In addition, the prosaccade task (in which participants are asked to make a saccade towards a target) provides a useful control condition.
Antisaccade errors are significantly increased in patients with damage limited to the dorsolateral prefrontal cortex (DLPFC) but not frontal eye fields (FEF) (Pierrot-Deseilligny et al. 2003). Accordingly, increased antisaccade errors have been taken as evidence of dysfunctional dorsolateral prefrontal cortex in a number of clinical populations (for a review, see Munoz and Everling 2004), most notably schizophrenia. Importantly, antisaccade errors are also significantly increased in the first-degree relatives of patients with schizophrenia, and, as such, are considered a potentially important marker of genetic vulnerability to the disorder (Calkins et al. 2004).
According to recent models of antisaccade performance (Massen 2004; Munoz and Everling 2004; Reuter and Kathmann 2004), the sudden appearance of the peripheral target triggers a ‘race’ between two separate saccade programs—an exogenously driven prosaccade towards the target, and an internally generated (endogenous) antisaccade to the opposite hemifield. If the antisaccade can be programmed fast enough, it ‘wins’ the race, and the prosaccade is cancelled. Alternatively, if the prosaccade is programmed fast enough (or the computation for the antisaccade is too slow), an erroneous prosaccade is made first, and the correct antisaccade follows. Parallel programming of saccades has been demonstrated in several other tasks (e.g. Godijn and Theeuwes 2002), but it is worth noting that the extent to which correct and incorrect responses are always programmed in parallel is unclear—on some antisaccade trials, errors may be compounded by one or more further saccades toward the target before being corrected.
Within the framework outlined above, error rates can be considered to be a function of the levels of activity in the neural systems responsible for initiating the two competing saccades—the higher the baseline activity or the faster the rate of rise, the sooner the threshold required to trigger a saccade is reached. Thus, Massen (2004) argues that any experimental manipulation that non-selectively influences activity in both systems (e.g. increases or decreases activity equally in both) will not result in a change in error rate. In contrast, a manipulation that either selectively increases activity in the neural systems responsible for the prosaccade, or decreases activity in the neural systems responsible for the antisaccade, should result in increased errors. Similarly, an experimental manipulation that does the opposite (e.g. either decreases activity in the neural systems responsible for the prosaccade or increases activity in the neural systems responsible for the antisaccade) would be expected to result in a decrease in antisaccade errors.
According to this model, if nicotine is acting simply to increase general arousal, and this increase impacts equally on activity in the neural systems underlying both the endogenous and exogenously driven processes, then the likelihood of either reaching threshold before the other would be unchanged, and there would be no change in antisaccade error rate. However, if the effects of nicotine are greater on high-level endogenous processes (such as the ability to adequately maintain the intention to initiate an antisaccade within working memory) than on lower level exogenously driven processes, then nicotine ought to result in a decrease in errors (as increased activity in the neural system underlying the endogenous antisaccade would increase the likelihood of it reaching threshold before the exogenous prosaccade).
Two studies have reported that nicotine decreases antisaccade errors in patients with schizophrenia (Larrison-Faucher et al. 2004; Depatie et al. 2002). However, schizophrenia is associated with increased rates of smoking and alpha 7 nicotinic receptor abnormalities (de Leon and Diaz 2005; Martin-Ruiz et al. 2003). Findings in healthy populations are less consistent (Depatie et al. 2002; Larrison-Faucher et al. 2004; Larrison et al. 2004; Roos et al. 1995; Powell et al. 2002). Both Depatie et al. (2002) and Powell et al. (2002) used overnight abstinent smokers, so the reduction in antisaccade errors they report could be due to a reversal of a withdrawal-induced deficit in performance. The only study to use minimally abstinent smokers delivered 4 mg nicotine gum (Larrison et al. 2004) to task naïve subjects participating in two sessions. They reported a trend towards fewer errors but no effect on saccade latencies on single-task blocks of the antisaccade task. However, Larrison et al. (2004) did not address the confounding of practice effects and novelty effects, and thus did not compare performance amongst those administered nicotine during their first experimental session to those receiving nicotine on the second session.
In this study, we used a crossover design that allows an easy differentiation between improvements resulting from practice and those resulting from enhancement by nicotine. Nicotine was delivered through smoking, and the volunteers were moderate smokers (10–20 cigarettes per day) who were minimally (2 h) abstinent before testing. As a delivery system for nicotine in habitual users, smoking provides better opportunity for self-titrated ‘optimal’ delivery than recently available systems such as nasal spray (Myers et al. 2004) nicotine patch (Poltavski and Petros 2005) and gum (Harris et al. 2004). This avoids negative side effects such as nausea, and the experiential differences associated with unfamiliar delivery systems, which can significantly change the outcome (Dar and Frenk 2004). The 2-h deprivation procedure minimises the likelihood of subjective experience of ‘withdrawal’ or ‘craving’ in moderate smokers during the test session and thus militates against an interpretation of any cognitive effects in terms of deprivation reinstatement.
Materials and methods
Participants
Participants were recruited for two separate studies with identical inclusion criteria. These were that participants should be aged between 18–35, smoke 10–20 cigarettes a day, habitually smoke before lunchtime and have normal or corrected to normal vision. Both studies were part of the first author’s PhD programme. The second study was identical to the first but contained an additional third testing session and participants performed another variant of the AS task at baseline. We do not present this additional data in this paper. Twenty volunteers (seven men) took part in study 1, mean (SD) age 22.3 (4.06) years. These participants scored 3.95 (1.61) on the Fagerström (1978) measure of nicotine dependence, had been smoking on average for 6.13 (3.63) years and were 183 (88.1) min abstinent at the start of the experiment. Twenty four volunteers (four men) took part in study 2. These participants were aged 20.6 (1.93), scored 4.46 (1.91) on the Fagerström (1978) questionnaire, had been smoking for 5.38 (2.18) years and were 149 (34.3) min abstinent at the start of the experiment.
The larger mean and standard deviation in the time-to-last cigarette data from study 1 is due to one participant choosing not to smoke in the morning before her second session. Independent t tests showed no differences (p>0.1) between participants in the two studies on the above demographics and smoking characteristics. The fact that the majority of participants were female reflects the gender bias in the undergraduate psychology populations. Research has demonstrated that acute effects of nicotine are not significantly mediated by gender-related issues (see Perkins et al. 1999 for a review). To increase experimental power, data were collapsed across both studies and the combined data are reported in this paper. All participants were volunteers from the existing pool of subjects at the University of Sussex; they gave informed consent at the start of the first session and were paid £10 (study 1) or £15 (study 2) or received Psychology course credits for their participation. The University of Sussex School of Life Sciences Ethics Committee gave approval for this experiment.
Tests
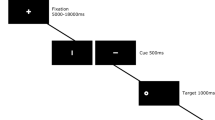
Participants were seated approximately 70 cm from a 21-inch monitor, and eye movements were recorded with an Eyelink II eye tracker (SR-Research, Ontario, Canada). The antisaccade task required participants to fixate a small red circle (subtending approximately 0.5°) in the centre of the screen. To increase the potential for observing facilitatory effects of nicotine, we manipulated the length of the gap between the offset of the fixation stimulus and the onset of the peripheral target. Previous research has demonstrated that antisaccade errors are significantly increased when a 200-ms gap is introduced compared to the 0-ms gap or ‘step’ version of the antisaccade task that is traditionally used.
We, therefore, used a 200-ms-gap condition and also a 500-ms-gap condition that results in similar error rates as the standard 0-ms “step” version (Fischer and Weber 1997), but does not require attention to be disengaged from the fixation stimulus at the time of target onset. After a random interval between 1,000 and 1,500 ms, the central fixation stimulus disappeared and, after a gap (200 or 500 ms), was replaced by a peripheral target (a red circle of the same diameter). The peripheral target appeared at one of four possible locations, +/−4 and 8° from fixation. Participants were instructed to look as quickly and as accurately as possible to the mirror image location of the target. Two blocks of 72 trials were performed at each baseline and retest. Within each block an equal number of trials had 200- and 500-ms gaps, and the target appeared at each location an equal number of times. Target location and gap length were varied pseudorandomly such that no gap length or target location was used more than three times in a row. An 800-Hz tone sounded for 50 ms at exactly the same time as the target appeared.
Procedure
All participants were tested on two separate sessions, separated by 2–7 days. Participants were requested to abstain from smoking for at least 2 h before arrival, and compliance with this request was monitored with end-tidal CO readings taken on arrival. Mean (SD) end-tidal CO measures of 10.1 (5.12) for session 1 and 8.09 (4.56) for session 2 were taken as compliance with this request. Both experimental sessions involved a baseline test of two 72-trial blocks, a short break and a retest of two further 72-trial blocks. In counter-balanced sessions, participants were asked to either smoke one of their own, preferred brand of cigarettes during the break, or abstain throughout. Thus, 22 of the participants smoked between baseline and retesting in their first session and abstained during the second. The remaining 22 participants abstained during the first session and smoked between baseline and retesting in the second session. During the first session, an eight-trial practice block of the antisaccade task was performed to ensure that all participants had understood the task instructions.
Analysis
The performance measures were percentage errors, latency for correct antisaccades and correct antisaccade gain (the ratio of correct saccade amplitude and target amplitude). At each baseline and retest in the two sessions, mean scores were calculated from the two 72-trial blocks combined. To explore between session effects, baseline data were entered into a mixed ANOVA with session (session 1 vs session 2), gap length (200-ms or 500-ms gap), smoking order (smoked in session 1 vs smoked in session 2) and study (participation in study 1 or 2) entered as factors. Less than 6% of the cells in the ANOVA on error data had 0% errors. To explore the effects of nicotine, we calculated difference scores (retest minus baseline) and entered these into a mixed ANOVA with nicotine (smoked vs abstained), gap length, smoking order and study entered as factors. Less than 4% of the cells in the ANOVA on error data had 0% errors.
Results
There were no significant differences on any baseline measure between those who took part in study 1 or study 2 (p>0.1 for all main effects). There was a trend towards faster latencies for correct antisaccades [F(1, 40)=3.23, p=0.08] and fewer errors [F(1, 40)=2.93, p=0.09] at the 200-ms gap length amongst those who took part in study 2. These reflect practice effects due to the additional block of antisaccade trials performed in experiment 2. There were no main effects of, or interactions with study (p>0.1) for the difference scores.
Baseline data
Significant main effects of session revealed practice effects for percent errors and correct antisaccade latencies. Percentage errors [F(1, 40)=22.4, p<0.001] and latencies for correct antisaccades [F(1, 40)=43.1, p<0.001] were lower at the baseline test in the second session compared to the baseline test in the first session. Participants made more errors overall for the 200-ms-gap trials compared to the 500-ms-gap trials [F(1, 40)=9.76, p<0.01)] and were also generally faster to initiate correct antisaccades for 500-ms-gap trials compared to 200-ms-gap trails [F(1, 40)=57.1, p<0.001]. These main effects of session and gap length were qualified by significant interactions between session and gap length (see Fig. 1). These revealed that the improvement in percentage errors occurred only for the 200-ms-gap length [F(1, 40)=24.8, p=0.003] and that the reduction in correct antisaccade latency between the sessions was greater for the 500-ms gap compared to the 200-ms-gap trials [F(1, 40)=29.7, p<0.001].

Percentage errors (a) and latencies for correct antisaccades (b) at the baseline tests for both gap lengths
In the baseline data, before delivery of nicotine, a significant interaction between session and smoking order for antisaccade errors revealed a greater reduction in errors from the first to the second session amongst those who had smoked in session 1 [F(1, 40)=5.94, p<0.02], see Table 1. Paired t tests confirmed that the difference in error rates between sessions 1 and 2 is significant for those who smoked in session 1 (t=4.56, df=21, p<0.001) and significant at a trend level for those who smoked in session 2 (t=1.87, df=21, p=0.077). An independent t test revealed that the apparent difference in session 1 error rates between the two smoking order groups is not significant (p=0.36).
For correct antisaccade amplitude, the main effect of session was not significant [F(1, 40)=0.24, p=0.63]. However, a significant session by gap interaction [F(1, 40)=4.37, p<0.05] arose because correct antisaccade amplitudes are more hypometric at the baseline of the second session for 200-ms-gap trials, but not 500-ms-gap trials. This was further qualified by a significant three-way interaction between session, gap and the between-subjects factor smoking order [F(1, 40)=14.6, p<0.01]. This unexpected interaction reflects the fact that amplitudes were reduced for both gap lengths at the second session compared to the first for participants who smoked in session 2, whereas participants who smoked in session 1 showed a reduction in amplitude for 200-ms-gap trials but an increase in amplitude for 500-ms-gap trials.
Nicotine effects
Smoking significantly reduced the number of antisaccade errors made [F(1, 40)=11.2, p<0.01] and the latency with which correct antisaccades were initiated [F(1, 40)=5.61, p<0.05] compared to abstaining. As is clear from Fig. 2, these main effects of nicotine were qualified by significant nicotine by smoking order interactions (percent errors, [F(1, 40)=9.6, p<0.01]; correct antisaccade latencies [F(1, 40)=5.78, p<0.05]). The interaction for percent errors reflects the fact that errors were reduced after nicotine if the cigarette was smoked during the first session (t=−4.31, df=21, p<0.01) but not during the second session (t=−0.31, df=21, p=0.76). The interaction between nicotine and smoking order for correct antisaccade latencies occurred because for the participants who smoked in session 1, smoking resulted in significantly greater reduction in correct antisaccade latencies than abstaining (t=−4.32, df=21, p<0.01), whereas for those participants who smoked in session 2, both smoking and abstaining resulted in small reductions that were equivalent (t=−0.01, df=21, p=0.99). A three-way interaction between nicotine, gap length and smoking order for antisaccade errors [F(1, 40)=5.2, p<0.05] occurred because nicotine, when smoked in session 1, reduced errors on trials with a 200-ms gap to a greater extent than errors on trials with a 500-ms gap. Paired t tests performed on data from each smoking order group separately confirm that errors on 200-ms (t=−4.3, df=21, p<0.001) and 500-ms (t=−2.58, df=21, p<0.02) gap trials were reduced after nicotine amongst those who smoked in session 1, while there was no reduction in errors after nicotine at either gap length for those who smoked in session 2 (p’s>0.1).

Change in percentage errors (a) and antisaccade latency (b) after smoking and abstinence for both smoking order groups
There was a very weak overall effect of nicotine on correct saccade amplitude [F(1, 40)=3.06, p=0.09], with nicotine generally resulting in a slight increase in amplitude, whereas abstinence resulted in a slight decrease. As with error rate and correct antisaccade latency, the nicotine by smoking order interaction was significant [F(1, 40)=7.07, p<0.01]. However, unlike the equivalent interactions for error rate and correct antisaccade latency, nicotine increased correct saccade amplitude only among those who smoked in session 2. This interaction is not readily interpretable and suggests that the trend for an overall effect of nicotine should be treated with caution. The nicotine by gap interaction was also significant [F(1, 40)=11.7, p<0.01]. The interaction occurs because correct antisaccade amplitude is not affected by nicotine or abstinence for 200-ms-gap trials, whereas nicotine increases amplitudes and abstinence decreases amplitudes for 500-ms-gap trails. In general, the effects of nicotine on correct antisaccade amplitude are complex, and are difficult to interpret in the light of the baseline differences that were observed.
Discussion
We investigated the effect of nicotine (administered in the form of a single preferred brand cigarette) on antisaccade performance in a non-clinical population. We found that nicotine led to a significant reduction in antisaccade errors when it was received during the first experimental session. Nicotine also led to a reduction in the latencies of correct antisaccades, and again, the reduction was greater for participants who smoked in the first session. These findings support previous work showing a nicotine-induced reduction in antisaccade errors (Depatie et al. 2002; Powell et al. 2002; Larrison et al. 2004) and latencies (Larrison et al. 2004) in healthy young adults. Further evidence for cholinergic modulation of antisaccade performance comes from reports of an increase in antisaccade errors amongst schizophrenic patients administered the cholinergic antagonist procyclidine (Ettinger et al. 2003a).
A number of studies using similar designs have observed effects of nicotine only when administered in the first session. Powell et al. (2002) reported fewer antisaccade errors after smoking in smokers permitted to smoke before the first testing session but not those who smoked in the second session. Also, using a complex visual search task, we found that nicotine reduced the number of fixations and refixations of stimuli made during the search only if the cigarette was smoked in the first session (Rycroft et al. 2005).
We found significant between sessions practice effects—average baseline error rates were 20.1% in the first session compared to 14.5% in the second session. Other researchers have also demonstrated significant between sessions practice effects for the antisaccade task (e.g. Ettinger et al. 2003b). One explanation of our findings, and those described above, is that any facilitatory effects of nicotine are more likely to be observed when performance is least optimal—as practice improves performance towards the higher end of the range of possible scores, ceiling effects reduce the potential for nicotine to induce any further improvements. Our finding that nicotine led to a greater reduction in errors for the 200-ms gap compared to the 500-ms gap trials supports this interpretation—baseline errors were higher for the 200-ms trials compared to the 500-ms trials. This interpretation also clarifies the failure to observe facilitatory effects of nicotine on antisaccade performance in a subgroup of schizophrenic patients who did not have abnormally increased antisaccade errors (Larrison-Faucher et al. 2004), and the fact that, in general, facilitatory effects of nicotine on antisaccade performance have been more consistently observed in patients with schizophrenia (who have high baseline levels of antisaccade errors) compared to healthy controls (Roos et al. 1995; Depatie et al. 2002). This interpretation is also consistent with the finding that participants with poor antisaccade performance benefit most from practice effects (Ettinger et al. 2003b).
If nicotine were acting to increase levels of arousal, one potential consequence would be faster processing of the target—in which case (according to the model of antisaccade performance outlined in the Introduction) an increase in error rates would be predicted. Alternatively, if a general increase in arousal led to faster processing of the visual stimulus and faster programming of the correct response, then no change in error rates would be expected. Our results support the suggestion that nicotine has a facilitatory effect on endogenous, but not exogenous, processes during antisaccade performance. In other words, nicotine may be increasing activity in the neural systems responsible for initiating the correct antisaccade response (Nieuwenhuis et al. 2004; Munoz and Everling 2004) over and above any influence they have on activity in the neural systems responsible for target detection.
Several converging lines of evidence confirm that working memory processes are important moderators of antisaccade performance. Secondary tasks that place demands on working memory capacity increase antisaccade errors, while tasks with the same motor or stimulus processing requirements (but no working memory requirements) do not (Stuyven et al. 2000; Mitchell et al. 2002; Roberts et al. 1994). Individuals with low working memory spans have slower latencies for correct antisaccades and more antisaccade errors than individuals with high working memory spans (Unsworth et al. 2004). Several studies have demonstrated increased antisaccade errors in populations with known working memory limitations. For example, antisaccade errors and correct antisaccade latencies are increased in patients with schizophrenia (Hutton et al. 1998, 2002), and the degree of impairment correlates significantly with working memory dysfunction in these patients (Hutton et al. 2004). Similarly, increased antisaccade errors reported in healthy elderly participants (e.g. Eenshuistra et al. 2004; Nieuwenhuis et al. 2004) have been attributed to lower activation of task goals within working memory.
In the context of these findings, our results are consistent with current models of antisaccade performance and suggest that nicotine increases the extent to which healthy participants are able to maintain the intention to initiate an antisaccade within working memory. This results in a reduction in the time taken to program a correct antisaccade, and, consequently, a reduction in the number of trials in which an erroneous prosaccade is programmed first. It is worth noting that another study exploring pharmacological manipulation of antisaccade performance found results that are difficult to interpret within the general model of antisaccade performance outlined in the introduction. Khan et al. (2003) administered ethanol to healthy participants and found that it increased correct antisaccade latency but reduced the number of errors. Activation models would predict that if the correct response is slowed, the erroneous response has a greater likelihood of reaching threshold first, and, therefore, errors should increase. The authors argued that the reduction in errors occurred because ethanol slowed down the processing of the target. Activation models would still be able to account for this pattern of results if the effect of ethanol was to slow the processing of the peripheral stimuli to a greater extent that it slowed the generation of the correct response. Further research using variants of pro- and antisaccade tasks and different pharmacological agents will provide important insights into the interactions between stimulus and goal-based behaviour.
In addition to a reduction in antisaccade errors and correct antisaccade latencies, we also found a novel ‘carryover’ effect of nicotine on antisaccade error rate—the improvement in baseline performance in the first to the second session was superior in those participants who had received nicotine in the first session compared to those who had abstained. In other words, those participants who benefited maximally from nicotine by receiving it in the first session maintained the improvements gained in that session for a period of up to a week. A similar effect in monkeys was reported by Buccafusco et al. (1995) after administration of nicotine or ABT-418, a centrally acting nicotinic cholinergic agonist. Both compounds improved performance on the delayed-matching to sample task 10 min post-administration, but only those given nicotine showed better performance 24 h later as well. Indeed, it has recently been suggested that such long-term effects of nicotine may reflect its action on cellular mechanisms underlying learning and memory, such as LTP (Buccafusco et al. 2005). These findings suggest that acute effects of nicotine on cognitive function may have consequences that last significantly longer than the pharmacokinetic properties of the compound.
The pharmacological effects of nicotine are extremely complex. As well as modulating the release of a variety of different neurotransmitters such as acetylcholine (Arnold et al. 2003), glutamate (Vidal and Changeux 1993), dopamine (Corrigall et al. 1994), serotonin (Reuben and Clarke 2000) and noradrenalin (Clarke and Reuben 1996), there are a number of different receptor subtypes with different affinities for nicotine binding (Paterson and Nordberg 2000). As these have different thresholds for nicotine effects, behavioural consequences of selective modulation of these subtypes are likely to be dose-dependent (Kumari and Postma 2005). Both of the major subtypes of nicotinic receptors, alpha-4 and alpha-7, reliably influence memory and attention (Nordberg 2001; Levin et al. 2006), but receptor subtype selectivity for specific cognitive processes has been difficult to establish. Both selective alpha-4 (Levin and Christopher 2002) and selective alpha-7 (Bettany and Levin 2001) compounds have been shown to modulate working memory performance in rat models, for example. Previously, alpha-7 receptors particularly have been linked to lower-level processes such as auditory gating, prepulse inhibition and priming (e.g. Leonard et al. 1998). Whether goal activation in working memory is mediated by effects on early perceptual processes or more direct prefrontal activation is a focus for further research.
In summary, this study found that nicotine reduces the number of antisaccade errors and increases the speed with which correct responses can be made. One interpretation of these findings is that nicotine increases the strength of activation in the memory representations supporting the goal to make antisaccades. As all participants were minimally abstinent and allowed to maintain a relatively naturalistic smoking pattern before the experiment, the effects of nicotine are unlikely to be due to a reversal of a withdrawal-induced deficit in performance. In addition, we have shown that the performance benefits derived from a single acute dose of nicotine persist over a period of at least a week, possibly reflecting the potential for nicotine to influence long-term learning processes.
References
Arnold HM, Nelson CL, Sarter M, Bruno JP (2003) Sensitization of cortical acetylcholine release by repeated administration of nicotine in rats. Psychopharmacology 165:346–358
Bates T, Pellett O, Stough C, Mangan G (1994) Effects of smoking on simple and choice reaction time. Psychopharmacology 114:365–368
Bettany JH, Levin ED (2001) Ventral hippocampal alpha-7 nicotinic receptor blockade and chronic nicotine effects on memory performance in the radial arm-maze. Pharmacol Biochem Behav 70:464–467
Buccafusco JJ, Jackson WJ, Terry AV Jr, Marsh KC, Decker MW, Arneric SP (1995) Improvement in performance of a delayed matching-to-sample task by monkeys following ABT-418: a novel cholinergic channel activator for memory enhancement. Psychopharmacology 120:256–266
Buccafusco JJ, Letchworth SR, Bencherif M, Lippiello PM (2005) Long lasting cognitive improvement with nicotine receptor agonists: mechanisms of pharmacokinetic–pharmacodynamic discordance. Trends Pharmacol Sci 26:352–360
Calkins ME, Curtis CE, Iacono WG, Grove WM (2004) Antisaccade performance is impaired in medically and psychiatrically healthy biological relatives of schizophrenia patients. Schizophr Res 71:167–178
Clarke PBS, Reuben M (1996) Release of [3H]-noradrenalin from rat hippocampal synaptosomes by nicotine: mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. Br J Pharmacol 117:595–606
Corrigall WA, Coen KM, Adamson L (1994) Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res 653:278–284
Dar R, Frenk H (2004) Do smokers self-administer pure nicotine? A review of the evidence. Psychopharmacology 173:18–26
de Leon J, Diaz FJ (2005) A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophr Res 76:135–157
Della Casa V, Hofer I, Feldon J (1999) Latent inhibition in smokers vs nonsmokers: interaction with number or intensity of preexposures. Pharmacol Biochem Behav 62:353–359
Depatie L, O’Driscoll GA, Holahan AV, Atkinson V, Thavundayil JX, Kin NNY, Lal S (2002) Nicotine and behavioural markers of risk for schizophrenia: a double-blind, placebo-controlled, cross-over study. Neuropsychopharmacology 27:1056–1070
Edginton T, Rusted JM (2003) Separate and combined effects of scopolamine and nicotine on retrieval-induced forgetting. Psychopharmacology 170:351–357
Eenshuistra RM, Ridderinkhof KR, van der Molen MW (2004) Age-related changes in antisaccade performance: inhibitory control or working-memory engagement? Brain Cogn 56:177–188
Ernst M, Matochik JA, Heishman SJ, Van Horn JD, Jons PH, Henningfield JE, London ED (2001) Effect of nicotine on brain activation during performance of a working memory task. Proc Natl Acad Sci U S A 98:4728–4733
Ettinger U, Kumari V, Zachariah E, Galea A, Crawford TJ, Corr PJ, Taylor D, Das M, Sharma T (2003a) Effects of procyclidine on eye movements in schizophrenia. Neuropsychopharmacology 28:2199–2208
Ettinger U, Kumari V, Crawford TJ, Davis RE, Sharma T, Corr PJ (2003b) Reliability of smooth pursuit, fixation, and saccadic eye movements. Psychophysiology 40:620–628
Fagerström KO (1978) Measuring degree of physical dependence to tobacco smoking with reference to individualization of treatment. Addict Behav 3:235–241
Fischer B, Weber H (1997) Effects of stimulus conditions on the performance of antisaccades in man. Exp Brain Res 116:191–200
Findlay JM, Walker R (1999) A model of saccade generation based on parallel processing and competitive inhibition. Behav Br Sci 22:661–721
Foulds J, Stapleton J, Swettenham J, Bell N, McSorley K, Russell, MAH (1996) Cognitive performance effects of subcutaneous nicotine in smokers and never-smokers. Psychopharmacology 127:31–38
Griesar WS, Zajdel DP, Oken BS (2002) Nicotine effects on alertness and spatial attention in non-smokers. Nicotine Tob Res 4:185–194
Godijn R, Theeuwes J (2002) Programming of endogenous and exogenous saccades: evidence for a competitive integration model. J Exp Psychol Hum Percept Perform 28:1039–1054
Hallett PE (1978) Primary and secondary saccades to goals defined by instructions. Vis Res 18:1279–1296
Harris JG, Kongs S, Allensworth D, Martin L, Tregellas J, Sullivan B, Zerbe G, Freedman R. (2004) Effects of nicotine on cognitive deficits in schizophrenia. Neuropsychopharmacology 29:1378–1385
Hutton SB, Crawford TJ, Puri BK, Duncan LJ, Chapman M, Kennard C, Barnes TRE, Joyce EM (1998) Smooth pursuit and saccadic abnormalities in first-episode schizophrenia. Psychol Med 28:685–692
Hutton SB, Joyce EM, Barnes TRE, Kennard C (2002) Saccadic distractibility in first-episode schizophrenia. Neuropsychologia 40:1729–1736
Hutton SB, Huddy V, Barnes TR, Robbins TW, Crawford TJ, Kennard C, Joyce EM (2004) The relationship between antisaccades, smooth pursuit, and executive dysfunction in first-episode schizophrenia. Biol Psychiatry 56:553–559
Khan SA, Ford K, Timney B, Everling F (2003) Effects of ethanol on anti-saccade task performance. Exp Brain Res 150:68–74
Kumari V, Postma P (2005) Nicotine use in schizophrenia: the self medication hypothesis. Exp Brain Res 29:1021–1034
Kumari V, Gray JA, Ffytche DH, Mitterschiffthaler MT, Das M, Zachariah E, Vythelingum GN, Williams SCR, Simmons A, Sharma T (2003) Cognitive effects of nicotine in humans: an fMRI study. NeuroImage 19:1002–1013
Larrison AL, Briand KA, Sereno AB (2004) Nicotine improves antisaccade performance without affecting prosaccades. Hum Psychopharmacol 19:409–419
Larrison-Faucher AL, Matorin AA, Sereno AB (2004) Nicotine reduces antisaccade errors in task impaired subjects. Prog Neuro-psychopharmacol Biol Psychiatry 28:505–516
Leigh RJ, Kennard C (2004) Using saccades as a research tool in the clinical neurosciences. Brain 127:460–477
Leonard S, Gault J, Adams C, Breese CR, Rollins Y, Adler LE, Olincy A, Freedman R (1998) Nicotinic receptors, smoking and schizophrenia. Restor Neurol Neurosci 12:195–201
Levin ED, Christopher NC (2002) Persistence of nicotinic agonist RJR 2403 induced working memory improvements in rats. Drug Dev Res 55:97–103
Levin ED, McClernon FJ, Rezvani AH (2006) Nicotinic effects on cognitive function: behavioural characterisation, pharmacological specification and anatomical localisation. Psychopharmacology 184:523–539
Levin ED, Conners CK, Silva D, Hinton SC, Meck WH, March J, Rose JE (1998) Transdermal nicotine effects on attention. Psychopharmacology 140:135–141
Mancuso G, Lejeune M, Ansseau M (2001) Cigarette smoking and attention: processing speed or specific effects? Psychopharmacology 155:372–378
Martin-Ruiz CM, Haroutunian VH, Long P, Young AH, Davis KL, Perry EK, Court JA (2003) Dementia rating and nicotinic receptor expression in the prefrontal cortex in schizophrenia. Biol Psychiatry 54:1222–1233
Massen C (2004) Parallel programming of exogenous and endogenous components in the antisaccade task. Q J Exp Psychol A 57:475–498
Mitchell JP, Macrae CN, Gilchrist ID (2002) Working memory and the suppression of reflexive saccades. J Cogn Neurosci 14:95–103
Munoz DP, Everling S (2004) Look away: the anti-saccade task and the voluntary control of eye movement. Nat Rev Neurosci 5:218–228
Myers CS, Robles O, Kakoyannis AN, Sherr JD, Avila MT, Blaxton TA, Thaker GK (2004) Nicotine improves delayed recognition in schizophrenic patients. Psychopharmacology 174(3):334–340
Nieuwenhuis S, Broerse A, Nielen MMA, de Jong R (2004) A goal activation approach to the study of executive function: An application to antisaccade tasks. Brain Cogn 56:198–214
Nordberg A (2001) Nicotinic receptor abnormalities in Alzheimer’s disease: therapeutic implications. Biol Psychiatry 49:200–210
Paterson D, Nordberg A (2000) Neuronal nicotinic receptors in the human brain. Biol Psychiatry 61:75–111
Perkins KA, Donny E, Caggiula AR (1999) Sex differences in nicotine effects and self-administration: review of human and animal evidence. Nicotine Tob Res 1(4):301–315
Pierrot-Deseilligny C, Müri RM, Ploner CJ, Gaymard B, Demeret S, Rivaud-Pechoux S (2003) Decisional role of the dorsolateral prefrontal cortex in ocular motor behaviour. Brain 126:1460–1473
Poltavski DV, Petros T (2005) Effects of transdermal nicotine on prose memory and attention in smokers and nonsmokers. Physiol Behav 83(5):833–843
Powell J, Dawkins L, Davis RE (2002) Smoking, reward responsiveness and response inhibition: tests of an incentive motivational model. Biol Psychiatry 51:151–163
Reuben M, Clarke PBS (2000) Nicotine evoked [3H]5-hydroxytryptamine release from rat striatal synaptosomes. Neuropharmacology 39:290–299
Reuter B, Kathmann N (2004) Using saccade tasks as a tool to analyze executive dysfunctions in schizophrenia. Acta Psychologica 115:255–269
Roberts RJ, Hager LD, Heron C (1994) Prefrontal cognitive processes: working memory and inhibition in the antisaccade task. Biol Psychiatry 123:374–393
Roos YBWEM, de Jongh FE, Crevits L (1995) The effects of smoking on anti-saccades. Neuro-ophthalmology 15:3–8
Rycroft N, Rusted JM, Hutton SB (2005) Acute effects of nicotine on visual search tasks in young adult smokers. Psychopharmacology 181:160–169
Stuyven E, Van der Goten K, Vandierendonck A, Claeys K, Crevits L (2000) The effect of cognitive load on saccadic eye movements. Acta Psychologica 104:69–85
Unsworth N, Schrock JC, Engle RW (2004) Working memory capacity and the antisaccade task: individual differences in voluntary saccade control. J Exp Psychol Learn Mem Cogn 30:1302–1321
Vidal C, Changeux JP (1993) Nicotinic and muscarinic modulations of excitatory synaptic transmission in the rat prefrontal cortex in vitro. Neuroscience 56:23–32
Warburton DM, Arnall C (1994) Improvements in performance without nicotine withdrawal. Psychopharmacology 115:539–542
West RJ, Jarvis MJ (1986) Effects of nicotine on finger tapping rate in non-smokers. Pharmacol Biochem Behav 25:727–731
Witte EA, Davidson MC, Marrocco RT (1997) Effects of altering brain cholinergic activity on covert orienting of attention: comparison of monkey and human performance. Psychopharmacology 132:324–334
Acknowledgements
This work was completed as part of the first authors Ph.D. thesis, funded by a bursary from the BBSRC. Thanks are due to Pennie Ingram for management of the database of volunteers.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rycroft, N., Hutton, S.B. & Rusted, J.M. The antisaccade task as an index of sustained goal activation in working memory: modulation by nicotine. Psychopharmacology 188, 521–529 (2006). https://doi.org/10.1007/s00213-006-0455-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-006-0455-7