
Abstract
Introduction
The uncompetitive N-methyl-D-aspartate (NMDA) receptor (NMDAR) antagonist ketamine has been proposed to model symptoms of psychosis. Inhibitory deficits in the schizophrenia spectrum have been reliably reported using the antisaccade task. Interestingly, although similar antisaccade deficits have been reported following ketamine in non-human primates, ketamine-induced deficits have not been observed in healthy human volunteers.
Methods
To investigate the effects of ketamine on brain function during an antisaccade task, we conducted a double-blind, placebo-controlled, within-subjects study on n = 15 healthy males. We measured the blood oxygen level dependent (BOLD) response and eye movements during a mixed antisaccade/prosaccade task while participants received a subanesthetic dose of intravenous ketamine (target plasma level 100 ng/ml) on one occasion and placebo on the other occasion.
Results
While ketamine significantly increased self-ratings of psychosis-like experiences, it did not induce antisaccade or prosaccade performance deficits. At the level of BOLD, we observed an interaction between treatment and task condition in somatosensory cortex, suggesting recruitment of additional neural resources in the antisaccade condition under NMDAR blockage.
Discussion
Given the robust evidence of antisaccade deficits in schizophrenia spectrum populations, the current findings suggest that ketamine may not mimic all features of psychosis at the dose used in this study. Our findings underline the importance of a more detailed research to further understand and define effects of NMDAR hypofunction on human brain function and behavior, with a view to applying ketamine administration as a model system of psychosis. Future studies with varying doses will be of importance in this context.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The phencyclidine (PCP) derivate ketamine is receiving intense interest as a model system of psychosis (Javitt et al. 2012; Kantrowitz and Javitt 2010; Poels et al. 2014). Translational model systems are a well-established method to investigate the pathophysiology of psychosis (Stone 2011; Stone et al. 2010) and possible new treatments for this condition (Javitt et al. 2012; Krystal et al. 2003; Stone et al. 2012). They may also help to explain the diverse reactions to available antipsychotic compounds (Stone 2011; Stone et al. 2010). Ketamine is an uncompetitive N-methyl-D-aspartate receptor (NMDAR) antagonist (Joules et al. 2015) that has less affinity to the NMDAR than PCP. Ketamine has a shorter plasma half-life than PCP and has an excellent safety record in both research and clinical settings (Doyle et al. 2013; Javitt et al. 2012; Krystal et al. 1994; Schmechtig et al. 2013).
The finding that NMDA antagonists like ketamine and PCP evoke transient symptoms that mimic a wide spectrum of psychotic symptoms has led to the emergence of glutamate theories of psychosis (Krystal et al. 1994; Lahti et al. 2001). These theories postulate that NMDAR hypofunction is fundamentally related to the etiology and pathophysiology of psychosis (Kantrowitz and Javitt 2010; Krystal et al. 2003; Poels et al. 2014). Psychotic symptoms are commonly divided into positive symptoms, such as suspiciousness and delusions; negative symptoms, such as social withdrawal and blunted affect; and disorganized symptoms, such as poor attention and conceptual disorganization (Javitt 2010). Symptoms from all three of these domains have been reported during ketamine administration (Krystal et al. 1994).
Despite this overlap, however, it appears that ketamine does not induce all aspects of psychosis (Murray et al. 2013; Steeds et al. 2015). To some extent this is true of all model systems, as psychotic disorders are complex and heterogeneous and it might be a near impossible challenge for one model to mimic all of their features (van Os and Kapur 2009).
Cognitive deficits are among those characteristics of psychosis that are for the most part resistant to currently available treatments (Carpenter and Koenig 2008). Given that cognitive deficits are associated with fundamental psycho-social impairments and reduced subjective well-being, they are an important target for possible new treatments (Carter and Barch 2007; Lesh et al. 2011; Morris and Cuthbert 2012). Model systems mimicking the cognitive symptoms of psychosis, especially when combined with cognitive biomarkers of psychosis, may further this important development (Javitt et al. 2012).
The antisaccade task is a widely studied biomarker of inhibitory control in schizophrenia (Gooding and Basso 2008; Koychev et al. 2011). Antisaccade performance is a measure of the integrity of cognitive and neural mechanisms of response inhibition (Hutton and Ettinger 2006). The neural correlates of antisaccade performance are well characterized (Leigh and Zee 2006; McDowell et al. 2002) and deficits in the psychosis spectrum are highly reliable (Reilly et al. 2008), which makes the task an ideal biomarker for the development and evaluation of new treatments (Ettinger and Kumar 2003; Hutton and Ettinger 2006). Deficits in antisaccade performance such as increased antisaccade latency and increased antisaccade error rate have been reported in schizophrenia patients (Hutton and Ettinger 2006), people with high levels of schizotypy (Ettinger et al. 2005; Gooding 1999; Holahan and O’Driscoll 2005; O’Driscoll et al. 1998), and first-degree relatives of schizophrenia patients (Calkins et al. 2008). On the brain functional level, schizophrenia patients were found to exhibit reduced activity in antisaccade-related areas, such as frontal eye fields (FEF), parietal eye fields (PEF), and supplementary eye fields (SEF), as well as in occipital areas that play a role in early visual processing while performing an antisaccade task (Camchong et al. 2008; Fukumoto-Motoshita et al. 2009; McDowell et al. 2002; Raemaekers et al. 2002).
Interestingly, although ketamine-induced deficits in antisaccade error rate and latency have been reported in studies on non-human primates (Condy et al. 2005; Ma et al. 2015; Skoblenick and Everling 2012, 2014) and in recreational ketamine users (Morgan et al. 2009), such deficits have not been reported for experimental ketamine administration in healthy humans (Radant et al. 1998; Schmechtig et al. 2013). One possible explanation for this discrepancy may be compensatory mechanisms on a brain functional level. Therefore, we used oculography to investigate the effects of ketamine on antisaccade performance and BOLD functional magnetic resonance imaging (fMRI) to identify possible brain functional effects of the substance in healthy human volunteers.
Experimental procedures
Participants
Right-handed non-smoking males were recruited via ads placed around the campus of the University of Bonn and online, as well as via existing participant pools. Before entering the study, participants gave written informed consent. Compensation for participation was a payment of €200. We obtained approval of the local ethics committee, and the study was conducted in agreement with the latest revision of the Code of Ethics of the World Medical Association (Declaration of Helsinki). The data reported here is based on the same sample investigated in earlier publications (Becker et al. 2017; Steffens et al. 2016).
Potential candidates were pre-screened via e-mail and telephone concerning general inclusion criteria, such as suitability for video-based eye-tracking and MRI. Suitable participants were then invited to a thorough screening in our lab. Exclusion criteria have been reported previously (Steffens et al. 2016). On the day of assessment, participants drank only water and stayed abstinent from food for 6 hours before the start of the infusion. Participants were instructed to take no medicines in the 24 h before assessment and to arrive well rested. Well before both assessment days, participants were informed that they would not be able to operate heavy machinery and that they should go home and rest after participation.
Study design
Participants who met all inclusion criteria during the screening session were invited to return for two fMRI assessments. Before the first fMRI assessment, the study anesthesiologist (C.N.) completed an additional medical screening. The anesthesiologist also monitored vital signs of participants throughout both assessments and in a supervised post-infusion period of at least 1 hour.
The study employed a double-blind, counter-balanced, placebo-controlled, and within-subjects design. An equal number of participants were randomly assigned to receive placebo first or ketamine first. Assessment took place at least 1 week apart and at the same time of day (maximum of a 90-min difference). Randomization and preparation of the infusion solution was not conducted by the study team carrying out the assessment.
On one of the assessment days, participants received racemic ketamine (Ketamin-ratiopharm 500 Injektionslösung, Ratiopharm, Ulm, Germany) via a computer-controlled infusion pump (Graseby 3500 infusion pump, Smith Medical Int. Ltd., Luton, UK) over the course of 1 hour. Ketamine was administered as a 2 mg/ml solution with a constant target plasma level of 100 ng/ml. On the other assessment day, participants received 0.9% saline solution following the same protocol employed during the ketamine assessment day. Both infusion solutions were prepared by the anesthesiologist shortly before infusion, and participants provided information about their height and weight, which was used to achieve the stable desired ketamine plasma concentration. Infusion started approximately 5 min before functional images were recorded. The solutions were each administered as a continuous bolus infusion, which was carried out with the STANPUMP program (Steven L. Shafer, MD, Anesthesiology Service (112A), PAVAMC, 3801 Miranda Ave., Palo Alto, CA. 94304) and was based on the three-compartment model by Domino et al. 1982 as described previously (Schmechtig et al. 2013).
Data collection
Eye movements
Movements of the right eye were recorded using the MRI-compatible EyeLink 1000 (SR Research, Ottawa, Ontario, Canada) eye-tracker with a sampling rate of 1000 Hz (minimal spatial resolution: 0.01°, average accuracy: 0.25° to 0.5°). A central pupil–tracking algorithm was used to detect the pupil and the corneal reflection. A three-point spatial calibration (horizontal, white calibration target on black background) was performed before the task. The task was presented on a NordicNeuroLab (Bergen, Norway) LCD monitor with an active area of 39.2 cm height × 52.35 cm width and a resolution of 1024 × 768 pixels. The distance from camera to eye (distance from eye to mirror and from mirror to eye) was 172 cm.
The prosaccade/antisaccade task (Fig. 1) was presented in an event-related mixed design consisting of 80 trials in total. There were 40 prosaccade (20 to the right, 20 to the left) and 40 antisaccade trials (20 to the right, 20 to the left). The order of trials was randomized once and this order was presented to each participant. The task was run after a smooth pursuit eye movements task (Steffens et al. 2016). Each trial began with the presentation of a fixation cross for 5000–18,000 ms (jittered, mean duration 11,450 ms) in the center of the screen. This was followed by a cue, which was presented for 500 ms. The cue was a vertical line to cue an antisaccade trial (gaze shift to mirror-inverted location of the target) or a horizontal line to cue a prosaccade trial (gaze shift to target location). The cue was followed by the presentation of the target, a circle (2.55° in diameter), which was presented for 1000 ms at 4.49° to the right or left of the center of the screen. All stimuli were presented in white on black background.

Schematic trial representation. Stimuli are not to scale, further detail on stimulus size can be found in the Experimental procedures section (figure created in GIMP)
fMRI
fMRI assessment was conducted using a Siemens (Erlangen, Germany) Trio MRI scanner at 3 Tesla field strength. Participants wore ear plugs to reduce the impact of scanner noise. In order to minimize head movements, participants were surrounded by foam paddings and were instructed to lie as still as possible.
After a brief initial scan to individually adjust the field of view, a high-resolution T1-weighted structural scan was acquired to exclude participants with apparent brain pathologies and to optimize normalization of functional MRI data. The scan parameters for this scan were as follows: repetition time = 1570 ms; echo time = 3.42 ms; inversion time = 800 ms; flip angle = 15°; field of view = 256 mm; matrix size = 256 × 256; 160 slices; slice thickness = 1 mm; sequential slice-order with no inter-slice gap; and voxel size = 1 × 1 × 1 mm. During the task, T2*-weighted images were collected with a standard 12 channel head coil employing a gradient-echo planar image sequence (repetition time = 2500 ms; echo time = 30 ms) to display the BOLD response. Slices were oriented parallel to the intercommissural plane. A total of 426 whole-brain images were collected for each participant. Additional scan parameters were: flip angle = 90°; field of view = 192 mm; matrix size = 64 × 64; 37 slices; slice thickness = 3 mm; sequential slice order with interslice gap of 0.3 mm; and voxel size = 3x3x3.3 mm.
Psychotomimetic states inventory, test of sustained attention, and blood samples
Participants completed the Psychotomimetic States Inventory (PSI) (Mason et al. 2008) before, during, and after infusion. To control for confounding effects of ketamine on basic attention, participants completed four lines of the d2 test (Brickenkamp 2002) of sustained attention during ketamine and four different lines during placebo infusion. The d2 test is a well-established measure of sustained attention and requires the crossing out of the letter d combined with two dashes amidst letters d and p with one, two, three, or four dashes. Samples of venous blood were drawn from the non-infusion arm (Niesters et al. 2012) before, during, and after infusion. They were immediately centrifuged for 10 min at 1300rcf and stored at − 80 °C.
Data processing and statistical analysis
Eye movements
Participant-level eye movement data were preprocessed using Data Viewer software (SR Research) and Matlab R2013a (The MathWorks, Natick, MA). Group-level statistical analysis was conducted in SPSS 24 (IBM Corp., Armonk, NY, USA). In each trial, the first saccade after target appearance was included in the analysis if its start position deviated no more than 2.04° visual angle from the center of the screen and its latency was between 80 ms and 3000 ms. Participants were included in the analysis if the eye movement recording of at least seven prosaccades and seven antisaccades was possible. The three main outcome measures were as follows: Antisaccade direction error rate was calculated as percent of antisaccade condition trials in which the first saccade was made in the direction of the peripheral target. Latency of saccades is given as the time between the detected start point and end point of a correct saccade (correct prosaccades, correct antisaccades) in milliseconds. Prosaccade direction errors very rarely occurred (2.41% overall); thus this variable is not considered further.
To analyze substance effects on antisaccade errors and pro- and antisaccade latency, repeated measures ANOVAs were conducted with the within-subjects factors substance (ketamine, placebo) and task condition (prosaccade, antisaccade). Effect sizes were calculated as partial eta squared (Cohen 1973). To ensure that assumptions for statistical analyses were met, data were screened for normality using skewness scores and Shapiro-Wilk tests.
fMRI
fMRI data analysis was performed using Statistical Parametric Mapping 12 software (SPM 12; http://www.fil.ion.ucl.ac.uk/spm/software) implemented in Matlab R2013a (The MathWorks, Natick, MA). Preprocessing included the following steps: To correct for head motion, images of each participant were realigned along the mean image in their time series using a least squares approach and a six-parameter rigid-body transformation. Functional scans were coregistered to the T1-weighted anatomical image. Normalization parameters were obtained from segmentation of the structural T1 images and used to transform the functional images into MNI standard space (Montreal Neurological Institute, MNI template). Functional images were resampled at 2 × 2 × 2 mm and smoothed with an 8 mm Gaussian filter (full width at half maximum).
At the first (single-subject) level, the BOLD response was modeled as a canonical hemodynamic response-function (hrf). The first-level model consisted of the following regressors of interest: correct prosaccades, correct antisaccades, missing, antisaccade errors, prosaccade errors. The six motion regressors were included as regressors of no interest. The onset of each trial was defined as the onset of the cue. Three contrasts were computed on the single-subject level: prosaccades > baseline, antisaccades > baseline, and antisaccades > prosaccades.
On the second (group) level, a conjunction analysis was performed to identify the saccadic network. This analysis aimed to identify consistently high and jointly significant structures for prosaccade and antisaccade performance compared to baseline in the placebo condition. We performed a conjunction analysis of the minimum T-statistic over the contrasts prosaccades > baseline and antisaccades > baseline as implemented in SPM12. Significant clusters describe effects that are significant in both task conditions against baseline in the placebo condition. To investigate effects of task and substance as well as their interaction, we performed a flexible factorial design random-effects model. In addition to the between-subjects subject factor, we defined substance (placebo vs. ketamine) and task condition (prosaccades vs. antisaccades) as within-subjects factors. The analysis was conducted on the whole-brain level, not masked for any region of interest.
For all second-level analyses, the statistical height threshold was set to p < .001, and significant clusters were inferred if the peak voxel of the cluster survived a statistical threshold of p < .05 family-wise error (FWE) corrected (cluster-level correction). Signal change scaled to local mean signal for relevant clusters was extracted using MarsBaR (http://marsbar.sourceforge.net). Anatomical labels were obtained using the anatomy toolbox (Eickhoff et al. 2005), and functional localizations were identified from previous literature (Binkofski et al. 2002; Leigh and Zee 2006; McDowell et al. 2002; Meyhöfer et al. 2017).
To test if substance had an effect on head motion during task completion, mean scores of each of the six motion parameters were calculated for ketamine and placebo, respectively, and compared using paired t tests. Results were corrected for multiple comparisons using the Bonferroni method (corrected α = .008). Effect sizes were calculated as Cohen’s d (Cohen 1992).
Psychotomimetic states inventory
PSI data were analyzed using SPSS 24 (IBM Corp., Armonk, NY, USA). The PSI consists of six subscales, namely delusional thinking, perceptual distortions, cognitive disorganization, anhedonia, mania, and paranoia. To ensure that assumptions for statistical analyses were met, data were screened for normality of distribution, sphericity, and homogeneity of variances using skewness scores, Shapiro-Wilk tests, Mauchly’s tests, and Levene’s statistics. Scores were transformed (ln(x)), as they violated normality assumptions. Where sphericity assumption was violated, results were Greenhouse-Geisser corrected, and uncorrected degrees of freedom and Greenhouse-Geisser epsilon are reported (Jennings 1987). Data were analyzed using mixed-design ANOVA with the within-subjects factors substance (ketamine, placebo) and application stage (pre-infusion, during infusion, and post-infusion). Effect sizes were calculated as partial eta-squared (Cohen 1973). Alpha level for post hoc t tests was Bonferroni corrected (corrected α = .01), and effect sizes for post hoc tests were calculated as Cohen’s d (Cohen 1992).
Correlations among measures
We calculated Pearson correlations of difference scores (ketamine – placebo) for antisaccade error rate, prosaccade latency, and antisaccade latency with difference scores (ketamine – placebo) for the six PSI scales. Alpha level for these correlations was Bonferroni corrected (α = 0.004).
We also calculated Pearson correlations between difference scores (ketamine – placebo) for antisaccade direction error rate, prosaccade latency, and antisaccade latency with the difference scores (ketamine – placebo) for the local BOLD signal change in the two clusters with substance effects. Alpha level for these correlations was Bonferroni corrected (corrected α = 0.006). Additionally, we calculated Pearson correlations between the local BOLD signal changes in the two clusters with substance effects with the six PSI scales. Alpha level for these correlations was Bonferroni corrected (corrected α = .004).
Results
Data prescreening
Of N = 43 individuals initially screened for participation, N = 29 met all inclusion criteria and were included in the study. Two participants did not participate in the second fMRI assessment and were therefore excluded from all analyses. Normalization failed for one participant, who was subsequently excluded from all data analyses. Due to technical limitations of eye tracking during fMRI, behavioral eye movement data could not be acquired for all participants (see discussion for details). Final results are thus based on N = 15 participants. Mean age of the final sample was M = 25.93 (SD = 3.65) years, and the mean number of years in education was M = 17.26 (SD = 2.43).
Test of sustained attention and blood samples
Results of the d2 test of sustained attention and the blood samples in the subset of participants examined here were similar to those reported for the entire sample elsewhere (Steffens et al. 2016). In short, ketamine had no significant effect on sustained attention task performance (p > .05). Regarding blood levels, measured ketamine concentration indicated that assignment to substance conditions was carried out as planned. The mean ketamine plasma level for during-ketamine infusion was M = 89.69 ng/mL (SD = 13.27 ng/mL) with a minimum of 54.30 ng/ mL and a maximum of 114.0 ng/mL, indicating that the plasma-levels were close to our target level.
Eye movements—behavioral results
Descriptive statistics of eye movement variables are reported in Table 1. Antisaccade error rate did not differ between ketamine and placebo conditions (p > .05), and there was no main effect of substance on saccade latency (p > .05). There was a main effect of task condition on saccade latency (F(1,14) = 18.42, p < .001, ηp2 = 0.56) with prosaccades having shorter latencies than antisaccades. This effect was not mediated by the task condition by substance interaction (p > .05).
Eye movements—fMRI results
Saccade network
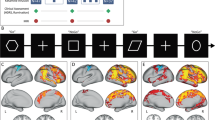
Table 2 and Fig. 2 show the saccade network (prosaccades and antisaccades) during placebo. The saccade network consisted of three large bilateral statistical clusters: The first cluster extended across primary visual, motion processing, and somatosensory areas (lingual gyrus, calcarine gyrus, superior occipital gyrus, middle occipital gyrus, inferior occipital gyrus, inferior parietal lobule, and inferior temporal gyrus). The second cluster extended across frontal regions associated with oculomotor control and executive function (precentral gyrus, middle frontal gyrus, inferior frontal gyrus, posterior medial frontal lobe, medial cingulate cortex, and insula). A third smaller cluster consisted of the bilateral medial cingulate cortex. There were no significant clusters for the reverse contrast.

Left: Activation pattern during anti- and prosaccades compared to baseline (whole-brain cluster-level FWE corrected p < .001) during placebo infusion illustrated in yellow. Interaction between substance (placebo, ketamine) and task condition (antisaccades, prosaccades) (whole-brain cluster-level FWE corrected p < .001) illustrated in blue. Right: Local signal change for substance by task condition interaction (top: MNI = − 38, − 24, 4, bottom: 40, − 18, 32; whole-brain cluster-level FWE corrected p < .001) (figure created in MRIcon and GIMP)
Task effects
There was a main effect of task condition, with clusters that differed between prosaccade and antisaccade conditions located in the saccade network as described above. Activation was higher in the antisaccade condition than the prosaccade condition in three clusters. The first of these was a large bilateral cluster that extended across primary visual, motion processing, and somatosensory areas (precuneus, superior parietal lobule, superior, and middle occipital gyrus, area V3A). The second cluster encompassed visual processing areas in the left lingual gyrus and the third cluster comprised the supplementary motor cortex in the left superior frontal gyrus. Activation was higher in the prosaccade condition than the antisaccade condition in two clusters, which consisted of early visual processing areas located in the right and left inferior occipital gyrus respectively. These task condition effects are shown in Table 3.
Drug effects
There was no main effect of substance. However, there was an interaction between substance and task condition in a bilateral cluster in the primary somatosensory cortex located in the postcentral gyrus. Post hoc tests indicated that signal change was higher for placebo than ketamine in the prosaccade condition (left: t(14) = 2.26, p = .04, d = 0.76; right: t(14) = 2.64, p = .001, d = .28) and higher for ketamine than placebo in the antisaccade condition (left: t(14) = −2.50, p = 0.02, d = 1.07; right: t(14) = −3.07, p = .008, d = 0.53). These drug effects are shown in Table 4 and Fig. 2. The local signal change in both clusters is illustrated in Fig. 2.
Motion parameters (total movement) were not affected by substance (all p > .05), arguing against confounding effects of motion differences during placebo and ketamine administration.
Psychotomimetic effects
PSI mean scores are in Table 5. Ketamine increased the PSI scales delusional thinking (F(2,14) = 9.82, p < .001, ηp2 = 0.41) and perceptual distortion (F(1,14) = 12.05, p < .001, ηp2 = 0.46) in interaction with application stage, with scores being higher only during substance infusion (delusional thinking: t(14) = 3.57, p = .003, d = 0.94; perceptual distortion: t(14) = 4.65, p < .001, d = 1.14).
Application stage had an effect on the PSI scales cognitive disorganization and mania, independently of substance, with cognitive disorganization scores being higher during infusion than before infusion (t(14) = 4.56, p < .001, d = 1.00) or after infusion (t(14) = 6.16, p < .001, d = 1.22) and mania scores being higher during infusion than before infusion (t(14) = 3.60, p = .003, d = 0.53).
Correlations among measures
There were no correlations of difference scores for antisaccade direction error rate, prosaccade, and antisaccade latency with signal change difference scores in the two clusters that showed a significant drug effect (all p > .05). Difference scores in these two clusters also did not correlate with difference scores of any of the PSI scales (all p > .05). Finally, difference scores of PSI scales also did not correlate with difference scores of eye movement variables (all p > .05).
Discussion
We investigated the effects of the uncompetitive NMDA-receptor antagonist ketamine on brain function during response inhibition using an antisaccade task. To do so, we conducted a double-blind, placebo-controlled, within-subjects study. We measured BOLD response and eye movements during a mixed design antisaccade task, while participants received a subanesthetic dose of intravenous ketamine on one and placebo on the other assessment day. Additionally, we obtained measures of psychotomimetic states, sustained attention, and ketamine blood levels. Our key findings are as follows.
There were no significant ketamine effects on saccadic latency or antisaccade error rate. However, ketamine had an effect on BOLD response in a small bilateral cluster in the somatosensory cortex located in the postcentral gyrus. This effect of ketamine was dependent on task condition, with BOLD response being significantly higher during placebo than ketamine administration in the prosaccade condition and significantly higher for ketamine than placebo in the antisaccade condition. Concerning task effects, in line with previous literature (Schmechtig et al. 2013), participants showed significantly shorter prosaccade than antisaccade latency overall. Also compatible with previous findings, execution of prosaccades and antisaccades was associated with higher BOLD response in a large bilateral cluster that encompassed areas associated with early visual processing, sensorimotor integration, and oculomotor response (McDowell and Clementz 2001). Prosaccade performance was associated with higher BOLD response in some areas involved with primary visual processing, whereas antisaccade performance was associated with higher BOLD response in some areas of primary visual processing and sensorimotor integration.
The lack of acute ketamine effects on antisaccade performance is consistent with previous literature on healthy human volunteers. Earlier studies observed no significant effects on antisaccade error rate or latency (Radant et al. 1998; Schmechtig et al. 2013) in contrast to the consistent antisaccade impairments in schizophrenia (Hutton and Ettinger 2006). A possible explanation for this dissociation of findings is that while ketamine influences the magnocellular visual pathway, which plays an important role in antisaccade performance, antisaccade deficits in schizophrenia are likely to be based on cognitive dysfunction rather than dysfunction of the magnocellular pathway (Javitt 2009; Leonard et al. 2013).
Interestingly, increased antisaccade error rate and latency under the influence of ketamine have been reported in studies on non-human primates (Condy et al. 2005; Ma et al. 2015; Skoblenick and Everling 2012, 2014). One possible explanation for the inconsistencies between results from human and non-human primate studies are methodological differences, which include that in studies on non-human primates, ketamine is usually given via intramuscular injection and that titration studies are usually performed to determine animal-specific concentrations of ketamine (Skoblenick and Everling 2012). This may complicate comparisons with human studies, where ketamine is usually given via intravenous infusion without previous titration studies (Radant et al. 1998; Schmechtig et al. 2013).
It is also worth noting that an adverse effect of ketamine on response inhibition has previously been shown in a study which employed the Hayling task (Morgan et al. 2004). That study showed a significantly increased rate of response errors in performance on the Hayling task under ketamine administration. In light of the heterogeneity of measures of inhibitory control (Aichert et al. 2012), further research will be necessary to more thoroughly describe the effect of ketamine on different facets of inhibition.
In evaluating ketamine as a psychosis model, the lack of antisaccade performance deficits means that the drug is seemingly unable to model all aspects of psychosis, at least at this dose, since antisaccade performance deficits are a robust and well-validated key finding in psychosis (Calkins et al. 2003; Hutton and Ettinger 2006; McDowell et al. 2002). Glutamate theories of psychosis predict that some symptoms of psychosis are directly caused by NMDAR hypofunction, while other symptoms may be less direct downstream consequences of these alterations (Javitt 2009; Javitt et al. 2012; Nelson et al. 2014; Pynn and DeSouza 2013). Since NMDAR shows a wide distribution in the brain, these theories predict regionally diffuse and widespread cortical dysfunction of processes mediated by NMDAR (Javitt et al. 2012). It is likely that a one-off administration of ketamine would model primary effects of NMDAR hypofunction, such as deficits in smooth pursuit eye movements (Meyhöfer et al. 2015; O’Driscoll and Callahan 2008) and mismatch negativity (Heekeren et al. 2008; Umbricht and Krljes 2005), but not the consequences of these alterations, such as impaired top-down attentional control and facial recognition (Javitt 2009). This is in line with findings of long-term ketamine users exhibiting psychosis-like symptoms such as hallucinations, which are a key symptom of schizophrenia. In contrast, hallucinations have not typically been reported in one-off ketamine administration studies of healthy volunteers (Muetzelfeldt et al. 2008).
On the brain functional level, we observed increased BOLD response in the saccadic task network replicating previous fMRI studies (McDowell et al. 2008; Raemaekers et al. 2002). The network included areas associated with primary visual, motion, and somatosensory processing and frontal regions associated with oculomotor control and executive function, as described extensively (Leigh and Zee 2006). We also observed an effect of ketamine on BOLD in a bilateral cluster in the postcentral gyrus that depended on task condition. BOLD response in this cluster, which was not located in the saccadic task network, was significantly higher during placebo than ketamine in the prosaccade condition, but significantly higher for ketamine than placebo in the antisaccade condition. While such effects in this somatosensory cortex cluster were unexpected, they may be related to the significant perceptual distortion induced by ketamine, an effect that has been described consistently in previous literature (Krystal et al. 1994; Lahti et al. 2001; Schmechtig et al. 2013; Stone et al. 2008, 2015). The observed interaction of the drug effect with task condition may be related to perceptual differences between the pro- and antisaccade conditions (gaze moving towards a visible target vs. towards blank space on screen) and their possible influence on proprioception (Balslev et al. 2011, 2013). The higher activation for ketamine compared to placebo in the antisaccade condition may also represent the recruitment of additional neuronal resources, particularly since this cluster decreased activity during antisaccade condition under placebo (Becker et al. 2013; Grady 2012).
Importantly, ketamine administration induced psychosis-like symptoms (namely delusional thinking and perceptual distortion) that correspond with the positive symptom domain in psychosis, as has been reported in previous studies (Krystal et al. 1994; Lahti et al. 2001; Schmechtig et al. 2013). Interestingly, we did not see a significant increase in disorganized or negative psychosis-like symptoms. It is worth noting that the results reported here are based on a subsample of the sample reported in our previous paper (Steffens et al. 2016), and that we did see a ketamine-induced increase in disorganized psychosis-like symptoms in the total sample. Negative symptoms, such as anxiety and withdrawal, have previously been described under ketamine infusion (Krystal et al. 1994; Lahti et al. 2001; Malhotra et al. 1997; Schmechtig et al. 2013). A possible explanation for the lack of negative symptoms in our study, in addition to issues of limited statistical power, may be that effects of ketamine on mood appear to be dose dependent, as higher doses of ketamine have been reported to be anxiogenic (Krystal et al. 1994), and one study reported elevated scores of anhedonia with a target ketamine plasma concentration of 150 ng/ml but not with a lower dose (Pollak et al. 2015).
While there were pronounced effects of ketamine on psychotomimetic states, there were no correlations between these effects on state measures and effects on eye movements or BOLD signal. These negative findings, while of course limited by issues of statistical power, indicate that ketamine effects on different levels of measurement within the same sample tend to be rather heterogeneous.
Limitations
The most important methodological limitation of the current study is that due to the technical challenges of eye tracking in MRI, behavioral data could only be acquired for a subset of our sample. fMRI data for the mixed pro- and antisaccade task can only be analyzed when behavioral data are available, because they are needed to identify direction errors and missed trials. Therefore, we could not include participants whose eye movements could not be recorded. However, recordings failed for anatomical reasons (position of eyes in the head, head size), but not due to variables that may have a significant impact on our analyses, thus not leading to any selection effects. Additionally, results of saccadic variables are broadly in agreement with those observed in laboratory studies, confirming the validity of our recordings. A general limitation of ketamine as a psychosis model concerns the ecological validity of the intervention. Participants are aware that they are in a safe environment, monitored by study staff, and know what is causing their symptoms when they are participating in a ketamine administration study. These meta-cognitive factors may be a key difference between the development of psychosis symptoms and the ketamine state model of psychosis (Steeds et al. 2015). As such, they represent a key limitation of the ketamine model, given that interpretation and appraisal of early perceptual dysfunctions have been discussed as important factors in the development of psychosis (Corlett et al. 2007; Nelson et al. 2014). Another limitation of this work is that although ketamine is among the most selective NMDAR antagonists available for human studies (Anis et al. 1983; Driesen et al. 2013; Moghaddam et al. 1997; Wood et al. 2012), it has secondary sites of action unrelated to NMDAR (Kohrs and Durieux 1998). It should furthermore be noted that the transient symptoms induced by experimental application of ketamine generally do not induce the entire spectrum of schizophrenia symptoms and impairments, particularly pre-disposing or vulnerability markers, neither in this study nor in previous studies (Krystal et al. 1994; Radant et al. 1998; Schmechtig et al. 2013; Steeds et al. 2015). Finally, another limitation of the current study is that we only applied a single dose of ketamine. Multi-dose studies would be of considerable interest, particularly given that we did not observe significant effects of the drug on saccadic performance. It remains unanswered whether higher doses of ketamine would cause schizophrenia-like impairments in antisaccades.
Conclusion
In summary, despite pronounced psychotomimetic effects of ketamine, we did not find antisaccade or prosaccade performance deficits under the influence of the drug. At the level of brain function, we observed BOLD differences between ketamine and placebo administration in somatosensory cortex, which depended on task condition. In evaluating ketamine as a model system, our findings overall suggest that the drug does not mimic all features of psychosis, at least at the dose investigated here. To an extent, this is true for all model systems (Carhart-Harris et al. 2013), and considering the complexity of psychosis, it might be challenging for one single model to mimic all features of this heterogeneous disorder (van Os and Kapur 2009). Defining the limitations of existing model systems may further our understanding of specific etiological mechanisms underlying psychotic symptoms. Our findings underline the importance of a more detailed research to further delineate effects of NMDAR hypofunction on human brain function and behavior, which will also be of importance when applying ketamine administration as a model system of psychosis.
References
Aichert DS, Wöstmann NM, Costa A, Macare C, Wenig JR, Möller H, Rubia K, Ettinger U (2012) Associations between trait impulsivity and prepotent response inhibition. J Clin Exp Neuropsychol 34:1016–1032. https://doi.org/10.1080/13803395.2012.706261
Anis NA, Berry SC, Burton NR, Lodge D (1983) The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol 79:565–575
Balslev D, Albert NB, Miall C (2011) Eye muscle proprioception is represented bilaterally in the sensorimotor cortex. Hum Brain Mapp 32:624–631. https://doi.org/10.1002/hbm.21050
Balslev D, Odoj B, Karnath H-O (2013) Role of somatosensory cortex in visuospatial attention. J Neurosci 33:18311–18318. https://doi.org/10.1523/JNEUROSCI.1112-13.2013
Becker B, Androsch L, Jahn RT, Alich T, Striepens N, Markett S, Maier W, Hurlemann R (2013) Inferior frontal gyrus preserves working memory and emotional learning under conditions of impaired noradrenergic signaling. Front Behav Neurosci 7(197):1–12. https://doi.org/10.3389/fnbeh.2013.00197
Becker B, Steffens M, Zhao Z, Kendrick KM, Neumann C, Weber B, Schultz J, Mehta MA, Ettinger U, Hurlemann R (2017) General and emotion-specific neural effects of ketamine during emotional memory formation. Neuroimage 150. https://doi.org/10.1016/j.neuroimage.2017.02.049
Binkofski F, Fink GR, Geyer S, Buccino G, Gruber O, Shah NJ, Taylor JG, Seitz RJ, Zilles K, Freund H-J (2002) Neural activity in human primary motor cortex areas 4a and 4p is modulated differentially by attention to action. J Neurophysiol 88:514–519. https://doi.org/10.1152/jn.00947.2001
Brickenkamp R (2002) Test d2 Aufmerksamkeits-Belastungs-Test
Calkins ME, Iacono WG, Curtis CE (2003) Smooth pursuit and antisaccade performance evidence trait stability in schizophrenia patients and their relatives. Int J Psychophysiol 49:139–146
Calkins ME, Iacono WG, Ones DS (2008) Eye movement dysfunction in first-degree relatives of patients with schizophrenia: a meta-analytic evaluation of candidate endophenotypes. Brain Cogn 68:436–461. https://doi.org/10.1016/j.bandc.2008.09.001
Camchong J, Dyckman KA, Austin BP, Clementz BA, McDowell JE (2008) Common neural circuitry supporting volitional saccades and its disruption in schizophrenia patients and relatives. Biol Psychiatry 64:1042–1050. https://doi.org/10.1016/j.biopsych.2008.06.015.Common
Carhart-Harris RL, Brugger S, Nutt DJ, Stone JM (2013) Psychiatry’s next top model: cause for a re-think on drug models of psychosis and other psychiatric disorders. J Psychopharmacol 27:771–778. https://doi.org/10.1177/0269881113494107
Carpenter WT, Koenig JI (2008) The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology 33:2061–2079. https://doi.org/10.1038/sj.npp.1301639
Carter CS, Barch DM (2007) Cognitive neuroscience-based approaches to measuring and improving treatment effects on cognition in schizophrenia: the CNTRICS initiative. Schizophr Bull 33:1131–1137. https://doi.org/10.1093/schbul/sbm081
Cohen J (1973) Eta-squared and partial eta-squared in fixed factor anova designs. Educ Psychol Meas 33:107–112. https://doi.org/10.1177/001316447303300111
Cohen J (1992) A power primer. Psychol Bull 112:155–159
Condy C, Wattiez N, Rivaud-Péchoux S, Gaymard B (2005) Ketamine-induced distractibility: an oculomotor study in monkeys. Biol Psychiatry 57:366–372. https://doi.org/10.1016/j.biopsych.2004.10.036
Corlett PR, Honey GD, Fletcher PC (2007) From prediction error to psychosis: ketamine as a pharmacological model of delusions. J Psychopharmacol 21:238–252. https://doi.org/10.1177/0269881107077716
Domino EF, Zsigmond EK, Domino LE, Domino KE, Kothary SP, Domino SE (1982) Plasma levels of ketamine and two of its metabolites in surgical patients using a gas chromatographic mass fragmentographic assay. Anesth Analg 61:87–92
Doyle OM, De Simoni S, Schwarz AJ, Brittain C, O’Daly OG, Williams SCR, Mehta MA (2013) Quantifying the attenuation of the ketamine pharmacological magnetic resonance imaging response in humans: a validation using antipsychotic and glutamatergic agents. J Pharmacol Exp Ther 345:151–160. https://doi.org/10.1124/jpet.112.201665
Driesen NR, McCarthy G, Bhagwagar Z, Bloch MH, Calhoun VD, D’Souza DC, Gueorguieva R, He G, Leung H-C, Ramani R, Anticevic A, Suckow RF, Morgan PT, Krystal JH (2013) The impact of NMDA receptor blockade on human working memory-related prefrontal function and connectivity. Neuropsychopharmacology 38:2613–2622. https://doi.org/10.1038/npp.2013.170
Eickhoff SB, Stephan KE, Mohlberg H, Grefkes C, Fink GR, Amunts K, Zilles K (2005) A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage 25:1325–1335. https://doi.org/10.1016/j.neuroimage.2004.12.034
Ettinger U, Kumari V (2003) Pharmacological studies of smooth pursuit and antisaccade eye movements in schizophrenia: current status and directions for future research. Curr Neuropharmacol 1:285–300. https://doi.org/10.2174/1570159033477017
Ettinger U, Kumari V, Crawford TJ, Flak V, Sharma T, Davis RE, Corr PJ (2005) Saccadic eye movements, schizotypy, and the role of neuroticism. Biol Psychol 68:61–78. https://doi.org/10.1016/j.biopsycho.2004.03.014
Fukumoto-Motoshita M, Matsuura M, Ohkubo T, Ohkubo H, Kanaka N, Matsushima E, Taira M, Kojima T, Matsuda T (2009) Hyperfrontality in patients with schizophrenia during saccade and antisaccade tasks: a study with fMRI. Psychiatry Clin Neurosci 63:209–217. https://doi.org/10.1111/j.1440-1819.2009.01941.x
Gooding DC (1999) Antisaccade task performance in questionnaire-identified schizotypes. Schizophr Res 35:157–166. https://doi.org/10.1016/S0920-9964(98)00120-0
Gooding DC, Basso MA (2008) The tell-tale tasks: a review of saccadic research in psychiatric patient populations. Brain Cogn 68:371–390. https://doi.org/10.1016/j.bandc.2008.08.024
Grady C (2012) The cognitive neuroscience of ageing. Nat Rev Neurosci 13:491–505. https://doi.org/10.1038/nrn3256
Heekeren K, Daumann J, Neukirch A, Stock C, Kawohl W, Norra C, Waberski TD, Gouzoulis-Mayfrank E (2008) Mismatch negativity generation in the human 5HT2A agonist and NMDA antagonist model of psychosis. Psychopharmacology 199:77–88. https://doi.org/10.1007/s00213-008-1129-4
Holahan A-LVA-LV, O’Driscoll GA (2005) Antisaccade and smooth pursuit performance in positive- and negative-symptom schizotypy. Schizophr Res 76:43–54. https://doi.org/10.1016/j.schres.2004.10.005
Hutton SB, Ettinger U (2006) The antisaccade task as a research tool in psychopathology: a critical review. Psychophysiology 43:302–313. https://doi.org/10.1111/j.1469-8986.2006.00403.x
Javitt DC (2009) When doors of perception close: bottom-up models of disrupted cognition in schizophrenia. Annu Rev Clin Psychol 5:249–275. https://doi.org/10.1146/annurev.clinpsy.032408.153502
Javitt DC (2010) Glutamatergic theories of schizophrenia. Isr J Psychiatry Relat Sci 47:4–16
Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D (2012) Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull 38:958–966. https://doi.org/10.1093/schbul/sbs069
Jennings JR (1987) Editorial policy on analyses of variance with repeated measures. Psychophysiology 24:474–475. https://doi.org/10.1111/j.1469-8986.1987.tb00320.x
Joules R, Doyle OM, Schwarz AJ, O’Daly OG, Brammer M, Williams SC, Mehta MA (2015) Ketamine induces a robust whole-brain connectivity pattern that can be differentially modulated by drugs of different mechanism and clinical profile. Psychopharmacology 232:4205–4218. https://doi.org/10.1007/s00213-015-3951-9
Kantrowitz JT, Javitt DC (2010) Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses. https://doi.org/10.3371/CSRP.4.3.6
Kohrs R, Durieux ME (1998) Ketamine: teaching an old drug new tricks. Anesth Analg 87:1186–1193. https://doi.org/10.1097/00000539-199811000-00039
Koychev I, Barkus E, Ettinger U, Killcross S, Roiser JP, Wilkinson L, Deakin B (2011) Evaluation of state and trait biomarkers in healthy volunteers for the development of novel drug treatments in schizophrenia. J Psychopharmacol 25:1207–1225. https://doi.org/10.1177/0269881111414450
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Charney DS (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214. https://doi.org/10.1001/archpsyc.1994.03950030035004
Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R (2003) NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology 169:215–233. https://doi.org/10.1007/s00213-003-1582-z
Lahti AC, Weiler MA, Michaelidis T, Parwani A, Tamminga CA, Tamara Michaelidis BA, Parwani A, Tamminga CA (2001) Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25:455–467. https://doi.org/10.1016/S0893-133X(01)00243-3
Leigh RJ, Zee DS (2006) The neurology of eye movements. Oxford University Press, Inc., New York
Leonard CJ, Robinson BM, Kaiser ST, Hahn B, McClenon C, Harvey AN, Luck SJ, Gold JM (2013) Testing sensory and cognitive explanations of the antisaccade deficit in schizophrenia. J Abnorm Psychol 122:1111–1120. https://doi.org/10.1037/a0034956
Lesh TA, Niendam TA, Minzenberg MJ, Carter CS (2011) Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology 36:316–338. https://doi.org/10.1038/npp.2010.156
Ma L, Skoblenick K, Seamans JK, Everling S (2015) Ketamine-induced changes in the signal and noise of rule representation in working memory by lateral prefrontal neurons. J Neurosci 35:11612–11622. https://doi.org/10.1523/JNEUROSCI.1839-15.2015
Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A (1997) Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 17:141–150. https://doi.org/10.1016/S0893-133X(97)00036-5
Mason OJ, Morgan CJM, Stefanovic A, Curran HV (2008) The psychotomimetic states inventory (PSI): measuring psychotic-type experiences from ketamine and cannabis. Schizophr Res 103:138–142. https://doi.org/10.1016/j.schres.2008.02.020
McDowell JE, Clementz BA (2001) Behavioral and brain imaging studies of saccadic performance in schizophrenia. Biol Psychol 57:5–22. https://doi.org/10.1016/S0301-0511(01)00087-4
McDowell JE, Brown GG, Paulus M, Martinez A, Stewart SE, Dubowitz DJ, Braff DL (2002) Neural correlates of refixation saccades and antisaccades in normal and schizophrenia subjects. Biol Psychiatry 51:216–223. https://doi.org/10.1016/S0006-3223(01)01204-5
McDowell JE, Dyckman KA, Austin BP, Clementz BA (2008) Neurophysiology and neuroanatomy of reflexive and volitional saccades: evidence from studies of humans. Brain Cogn 68:255–270. https://doi.org/10.1016/j.bandc.2008.08.016
Meyhöfer I, Steffens M, Kasparbauer A, Grant P, Weber B, Ettinger U (2015) Neural mechanisms of smooth pursuit eye movements in schizotypy. Hum Brain Mapp 36:340–353. https://doi.org/10.1002/hbm.22632
Meyhöfer I, Steffens M, Faiola E, Kasparbauer AM, Kumari V, Ettinger U (2017) Combining two model systems of psychosis: the effects of schizotypy and sleep deprivation on oculomotor control and psychotomimetic states. Psychophysiology. https://doi.org/10.1111/psyp.12917
Moghaddam B, Adams B, Verma A, Daly D (1997) Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17:2921–2927
Morgan, C.J.A., Mofeez, A., Brandner, B., Bromley, L., Curran, H Valerie, 2004. Ketamine impairs response inhibition and is positively reinforcing in healthy volunteers: a dose-response study Psychopharmacology (Berl) 172, 298–308. https://doi.org/10.1007/s00213-003-1656-y
Morgan CJA, Huddy V, Lipton M, Curran HV, Joyce EM (2009) Is persistent ketamine use a valid model of the cognitive and oculomotor deficits in schizophrenia? Biol Psychiatry 65:1099–1102. https://doi.org/10.1016/j.biopsych.2008.10.045
Morris SE, Cuthbert BN (2012) Research domain criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin Neurosci 14:29–37
Muetzelfeldt L, Kamboj SK, Rees H, Taylor J, Morgan CJA, Curran HV (2008) Journey through the K-hole: phenomenological aspects of ketamine use. Drug Alcohol Depend 95:219–229. https://doi.org/10.1016/j.drugalcdep.2008.01.024
Murray RM, Paparelli A, Morrison PD, Marconi A, Di Forti M (2013) What can we learn about schizophrenia from studying the human model, drug-induced psychosis? Am J Med Genet B Neuropsychiatr Genet 162:661–670. https://doi.org/10.1002/ajmg.b.32177
Nelson B, Whitford TJ, Lavoie S, Sass LA (2014) What are the neurocognitive correlates of basic self-disturbance in schizophrenia?: integrating phenomenology and neurocognition. Part 2 (Aberrant salience). Schizophr Res 152:20–27. https://doi.org/10.1016/j.schres.2013.06.033
Niesters M, Khalili-Mahani N, Martini C, Aarts L, van Gerven J, van Buchem MA, Dahan A, Rombouts S (2012) Effect of subanesthetic ketamine on intrinsic functional brain connectivity: a placebo-controlled functional magnetic resonance imaging study in healthy male volunteers. Anesthesiology 117:868–877. https://doi.org/10.1097/ALN.0b013e31826a0db3
O’Driscoll GA, Callahan BL (2008) Smooth pursuit in schizophrenia: a meta-analytic review of research since 1993. Brain Cogn 68:359–370. https://doi.org/10.1016/j.bandc.2008.08.023
O’Driscoll GA, Lenzenweger MF, Holzman PS (1998) Antisaccades and smooth pursuit eye tracking and schizotypy. Arch Gen Psychiatry 55:837–843. https://doi.org/10.1001/archpsyc.55.9.837
Poels EMP, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman J a, Abi-Dargham A, Girgis RR (2014) Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry 19:20–29. https://doi.org/10.1038/mp.2013.136
Pollak TA, De Simoni S, Barimani B, Zelaya FO, Stone JM, Mehta MA (2015) Phenomenologically distinct psychotomimetic effects of ketamine are associated with cerebral blood flow changes in functionally relevant cerebral foci: a continuous arterial spin labelling study. Psychopharmacology 232:4515–4524. https://doi.org/10.1007/s00213-015-4078-8
Pynn LK, DeSouza JFX (2013) The function of efference copy signals: implications for symptoms of schizophrenia. Vis Res 76:124–133. https://doi.org/10.1016/j.visres.2012.10.019
Radant AD, Bowdle TA, Cowley DS, Kharasch ED, Roy-Byrne PP (1998) Does ketamine-mediated N-methyl-D-aspartate receptor antagonism cause schizophrenia-like oculomotor abnormalities? Neuropsychopharmacology 19:434–444. https://doi.org/10.1016/S0893-133X(98)00030-X
Raemaekers M, Jansma JM, Cahn W, Van der Geest JN, van der Linden JA, Kahn RS, Ramsey NF (2002) Neuronal substrate of the saccadic inhibition deficit in schizophrenia investigated with 3-dimensional event-related functional magnetic resonance imaging. Arch Gen Psychiatry 59:313–320. https://doi.org/10.1001/archpsyc.59.4.313
Reilly JL, Lencer R, Bishop JR, Keedy SK, Sweeney JA (2008) Pharmacological treatment effects on eye movement control. Brain Cogn 68:415–435. https://doi.org/10.1016/j.bandc.2008.08.026.Pharmacological
Schmechtig A, Lees J, Perkins A, Altavilla A, Craig KJ, Dawson GR, William Deakin JF, Dourish CT, Evans LH, Koychev I, Weaver K, Smallman R, Walters J, Wilkinson LS, Morris R, Williams SCR, Ettinger U (2013) The effects of ketamine and risperidone on eye movement control in healthy volunteers. Transl Psychiatry 3:e334. https://doi.org/10.1038/tp.2013.109
Skoblenick K, Everling S (2012) NMDA antagonist ketamine reduces task selectivity in macaque dorsolateral prefrontal neurons and impairs performance of randomly interleaved prosaccades and antisaccades. J Neurosci 32:12018–12027. https://doi.org/10.1523/JNEUROSCI.1510-12.2012
Skoblenick K, Everling S (2014) N-methyl-d-aspartate receptor antagonist ketamine impairs action-monitoring activity in the prefrontal cortex. J Cogn Neurosci 26:577–592. https://doi.org/10.1162/jocn_a_00519
Steeds H, Carhart-Harris RL, Stone JM (2015) Drug models of schizophrenia. Ther Adv Psychopharmacol 5:43–58. https://doi.org/10.1177/2045125314557797
Steffens M, Becker B, Neumann C, Kasparbauer AM, Meyhöfer I, Weber B, Mehta MA, Hurlemann R, Ettinger U (2016) Effects of ketamine on brain function during smooth pursuit eye movements. Hum Brain Mapp 37:4047–4060. https://doi.org/10.1002/hbm.23294
Stone JM (2011) Glutamatergic antipsychotic drugs: a new dawn in the treatment of schizophrenia? Ther Adv Psychopharmacol. https://doi.org/10.1177/2045125311400779
Stone JM, Erlandsson K, Arstad E, Squassante L, Teneggi V, Bressan RA, Krystal JH, Ell PJ, Pilowsky LS (2008) Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy - a [123I]CNS-1261 SPET study. Psychopharmacology 197:401–408. https://doi.org/10.1007/s00213-007-1047-x
Stone JM, Raffin M, Morrison P, McGuire PK (2010) Review: the biological basis of antipsychotic response in schizophrenia. J Psychopharmacol 24:953–964. https://doi.org/10.1177/0269881109106959
Stone JM, Dietrich C, Edden R, Mehta M a, De Simoni S, Reed LJ, Krystal JH, Nutt D, Barker GJ (2012) Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry 17:664–665. https://doi.org/10.1038/mp.2011.171
Stone J, Kotoula V, Dietrich C, De Simoni S, Krystal JH, Mehta MA (2015) Perceptual distortions and delusional thinking following ketamine administration are related to increased pharmacological MRI signal changes in the parietal lobe. J Psychopharmacol 29:1025–1028. https://doi.org/10.1177/0269881115592337
Umbricht D, Krljes S (2005) Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res 76:1–23. https://doi.org/10.1016/j.schres.2004.12.002
van Os J, Kapur S (2009) Schizophrenia. Lancet 374:635–645. https://doi.org/10.1016/S0140-6736(09)60995-8
Wood J, Kim Y, Moghaddam B (2012) Disruption of prefrontal cortex large scale neuronal activity by different classes of psychotomimetic drugs. J Neurosci 32:3022–3031. https://doi.org/10.1523/JNEUROSCI.6377-11.2012
Acknowledgements
The authors thank Sam Hutton, Marcel Bartling, and Peter Trautner for their excellent technical support. The authors would like to thank Helen Röhrig and Inken Salhofen for their assistance in data collection and all volunteers who participated in the study.
Funding
The study was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft; Et 31/2-1). The funding body had no role in the design of the study, data analysis, data interpretation, or publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Steffens, M., Neumann, C., Kasparbauer, AM. et al. Effects of ketamine on brain function during response inhibition. Psychopharmacology 235, 3559–3571 (2018). https://doi.org/10.1007/s00213-018-5081-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-018-5081-7