
Abstract
Rationale
Schizophrenia patients display an excessive rate of smoking compared to the general population. Nicotine increases acoustic prepulse inhibition (PPI) in animals as well as healthy humans, suggesting that smoking may provide a way of restoring deficient sensorimotor gating in schizophrenia. No previous study has examined the neural mechanisms of the effect of nicotine on PPI in humans.
Objectives
To investigate whether nicotine enhances tactile PPI in healthy subjects and patients with schizophrenia employing a double-blind, placebo-controlled, cross-over design and, if so, what are the neural correlates of nicotine-induced modulation of PPI.
Materials and methods
In experiment 1, 12 healthy smokers, 12 healthy non-smokers and nine smoking schizophrenia patients underwent testing for tactile PPI on two occasions, 14 days apart, once after receiving (subcutaneously) 12 μg/kg body weight of nicotine and once after receiving saline (placebo). In experiment 2, six healthy subjects and five schizophrenia patients of the original sample (all male smokers) underwent functional magnetic resonance imaging (fMRI) under the same drug conditions and the same tactile PPI paradigm as in experiment 1.
Results
Nicotine enhanced PPI in both groups. A comparison of patterns of brain activation on nicotine vs placebo conditions showed increased activation of limbic regions and striatum in both groups after nicotine administration. Subsequent correlational analyses demonstrated that the PPI-enhancing effect of nicotine was related to increased hippocampal activity in both groups.
Conclusions
Nicotine enhances tactile PPI in both healthy and schizophrenia groups. Our preliminary fMRI findings reveal that this effect is modulated by increased limbic activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A key feature of schizophrenia is the inability to screen out irrelevant sensory input. The resulting cognitive overload is thought to give rise to some of the complex symptoms seen clinically. Prepulse inhibition (PPI) of startle appears to provide an index of this ability to screen input, as it is believed to be an operational measure of a sensorimotor filtering system (Geyer et al. 1990). PPI refers to a reduction in response to a startling sensory stimulus, when that stimulus is preceded shortly by a weak non-startling stimulus (Graham 1975). PPI has been found to be impaired in several independent studies of patients with schizophrenia (e.g. Braff et al. 1978, 2001; Kumari et al. 2000, 2004; Ludewig et al. 2003). In addition, animal models of disrupted PPI have proved valuable for the evaluation of antipsychotic substances (Swerdlow and Geyer 1998; Geyer et al. 2001; Geyer and Ellenbroek 2003).
Studies in rats demonstrate that nicotine increases PPI of the acoustic startle response (Acri et al. 1991, 1994; Curzon et al. 1994), though in some studies, this effect is noted to be strain dependent (Faraday et al. 1999). More recently, nicotine has been shown to attenuate the disruption of acoustic PPI produced by phencyclidine (PCP) in DBA/2J and C3H/HeJ but not in C57BL/6J mice (Spielewoy and Markou 2004). Psychotomimetic NMDA antagonists, such as PCP disrupt PPI in animals (Geyer et al. 1990), induce both positive and negative symptoms of schizophrenia in healthy subjects and exacerbate such symptoms in schizophrenia patients (Javitt and Zukin 1991; Lahti et al. 2001). Other data show that decreased PPI during nicotine withdrawal in DBA/2J mice can be reversed by nicotine self-administration (Semenova et al. 2003). More recently, the disruption of PPI produced by the dopamine agonist apomorphine has been shown to be blocked by nicotine in rats (Suemaru et al. 2004).
Nicotine also enhances PPI in healthy human smokers and non-smokers (Kumari et al. 1996, 1997; Della Casa et al. 1998; Duncan et al. 2001) and may produce transient PPI enhancement in patients with schizophrenia (Kumari et al. 2001). Improvement with nicotine/cigarette smoking is also seen in (impaired) sensory gating of the acoustically elicited P50 wave in patients with schizophrenia (Adler et al. 1993) and their relatives (Adler et al. 1992). The rate of smoking in patients with schizophrenia is estimated to be two- to fourfold of the rate seen in the general population (Hughes et al. 1986; Glassman 1993; de Leon et al. 1995, 2002; Diwan et al. 1998; Sacco et al. 2004; Uck et al. 2004). This observation together with the findings of improvement with nicotine on sensory gating paradigms have led to the suggestion that schizophrenia patients may smoke to transiently improve deficient sensory filtering (for a review, see Kumari and Postma 2005).
At the neural level, PPI is regulated by the cortico–striatal–pallido–thalamic circuitry (for a review of animal studies, see Swerdlow et al. 2001; for human studies, see Hazlett et al. 2001; Kumari et al. 2003a, 2005). Challenges to substrates of this circuitry reliably produce deficits in PPI, which is consistent with the hypothesis that the impaired gating seen in schizophrenia may arise from abnormalities in the neural interactions between limbic structures and basal ganglia. Indirect evidence strongly suggests involvement of hippocampal α7 subunit-containing nicotinic receptors in the nicotine-induced improvement of deficient P50 gating in humans (e.g. Freedman et al. 1995). Evidence from animal studies confirms that a similar mechanism may also be involved in the effect of nicotine in PPI in animals (Stevens et al. 1998; Bullock et al. 1997).
The present study used functional magnetic resonance imaging (fMRI) to investigate changes in blood-oxygen-level-dependent (BOLD) brain activity co-occurring with the effects of subcutaneous nicotine on PPI in healthy subjects and people with schizophrenia. As a necessary precursor to this study, we sought to establish whether the PPI-enhancing effect of nicotine occurred using a novel, intramodal tactile startle paradigm, specifically designed for the MRI environment by Kumari et al. (2003a) (Experiment 1: behavioral investigation). To date, such effects have been shown only using acoustic startle paradigms. Our hypotheses on taking the tactile paradigm to the scanner was that the modulatory effects of nicotine on PPI would be particularly evident in limbic and thalamic structures as these areas are rich in the α7 subunit-containing nicotinic receptors implicated in previous findings (Experiment 2: fMRI investigation).
Materials and methods
Experiment 1: behavioral investigation
Subjects
Twenty-two healthy male non-smokers and 19 smokers were recruited via advertisement. Fifteen male schizophrenic patients with a Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV (First et al. 1995) diagnosis of schizophrenia were recruited via the in- and outpatient services of the Maudsley Hospital, London, of whom ten were smokers and four were non-smokers. All patients had been on stable typical antipsychotic medication for a minimum of 6 weeks before taking part in this study.
Control subjects were screened by a clinician using the Structured Clinical Interview for DSM-IV axis I disorders, non-patient edition (First et al. 1996). Exclusion criteria included any history of significant medical illness and loss of consciousness as well as history of psychiatric illness in subjects or their first-degree relatives. All subjects were right-handed and were screened for illicit drug use via urine analysis (none were excluded). Smoking dependence in the smoking controls and patients was established using the Fagerstrom tolerance questionnaire (Fagerstrom and Schneider 1989) (see Table 1 for demographic characteristics of participant groups). All subjects gave written informed consent. This study was approved by the ethics committee of the Institute of Psychiatry and Maudsley Hospital, London.
Experimental design
Subjects were tested (double-blind) on two separate occasions, 14 days apart:
-
(1)
once after receiving 12 mg/kg bodyweight of nicotine and
-
(2)
once after receiving saline (placebo)
This dose of nicotine has been shown to elicit changes in PPI in healthy subjects (Kumari et al. 1997). The appearance of the saline and nicotine preparations was identical. Placebo or nicotine was administered via subcutaneous injection to the triceps region of the left upper arm. The doses of nicotine were prepared as 1 mg nicotine base in 1 ml of 0.9% saline with added sodium bicarbonate (8.52 g/l of prepared solution). Time of day of testing was held constant for each subject. Drug order was randomised such that half the subjects received saline and the other half nicotine on the first occasion. Startle testing commenced 10 min after injection to cover the period of maximum effects of nicotine given via this method (Russell et al. 1990).
Startle response measurement
Tactile stimuli comprised pulse-alone trials (a 40-ms presentation of 30 p.s.i. air puff) and prepulse trials (pulse preceded by a 20-ms presentation of 10 p.s.i. air puff) delivered via two plastic tubes to the neck above the sternum. Presentation of tactile startle stimuli was controlled by a commercially available computerised human startle response system (San Diego Instruments, San Diego, CA, USA). Measurement of the startle response (eyeblink amplitude) was achieved by recording the electromyographic (EMG) activity of the right orbicularis oculi muscle via two (6 mm) Ag/AgCl electrodes filled with Dracard electrode gel. After preparing the skin surface with Sterets sterile swabs, one electrode was positioned approximately 1 cm lateral to and 0.5 cm below the lateral canthus of the subject’s right eye, while the second electrode was placed 1.5 cm below and slightly medial to the first electrode, such that both electrodes were equidistant from the centre of the eye. In addition, a ground electrode was placed behind the right ear over the mastoid. The startle system recorded EMG activity for 250 ms (sample interval 1 ms) from the onset of the startle stimulus. Recorded EMG activity was band-pass filtered, as recommended by the SR-Lab. A 50-Hz notch filter was used to eliminate the 50-Hz interference. The data were scored off-line by the analytic programme of this system for response amplitude (in arbitrary analog-to-digit units) and latencies to response peak (in millisecond). The latency to response peak was determined as the point of maximal amplitude that occurred within 150 ms from the startle stimulus onset.
Experimental paradigm
Three 5-min experiments (experiments 1, 2 and 3) were conducted within which five 30-s blocks of six pulse-alone trials were alternated with five 30-s blocks of six prepulse + pulse trials (total of 60 trials per experiment). Within each block, the inter-trial intervals between the six trials were randomised. This A/B ‘box-car’ design, in which two blocks or conditions are alternated at a regular interval, is widely used in fMRI studies, enabling a comparison of BOLD activation between the control condition and condition of interest. The three experiments were distinguished by the different stimulus onset asynchronies (SOAs, i.e. time from onset of prepulse to onset of pulse) of the prepulse trials, these being set at 30 (experiment 1), 60 (experiment 2) and 120 ms (experiment 3).
General procedure
Upon recruitment, subjects were informed about the procedure and were told that the purpose of the investigation was to establish the effects of nicotine on reactivity to air puffs. All subjects were asked to refrain from alcohol for 24 h before their appointment. Smoking subjects were also requested to abstain from smoking for 12 h before testing. Non-smoking compliance (expired carbon monoxide level <12 ppm) was confirmed upon arrival using an expired carbon monoxide monitor (Bedfont Technical Instruments, EC 50).
Subjects were made comfortable on a hospital trolley with the back adjusted so that each subject was half-reclining. The procedure was explained again, after which electrodes were positioned. Subjects were told to relax as much as possible but to stay awake. No instructions were given regarding ignoring or attending to the stimuli. Subjects then received the injection. Startle testing began 10 min post-injection. Blood pressure and heart rate were monitored at three time points: before injection, after injection and after completion of the test.
Subjects showing low reactivity to the tactile stimuli as measured by eyeblink activity were excluded from the final data analysis. Subjects were deemed ‘non-responders’ if they failed to show a clearly visible blink response on 40% or more of pulse-alone trials.
Statistical analyses
Effects of nicotine on heart rate were assessed via a 2×3×2 [drug × occasion (before injection, 9 min after injection and post-testing) × smoking status (smokers/non-smokers)] analysis of variance (ANOVA, Greenhouse Geisser). Forty-three percent of the non-smokers, 37% of the smokers and 36% of the patients were classed as non-responders to the tactile stimuli, leaving 12 healthy non-smokers, 12 healthy smokers and nine patients (of whom two were non-smokers) for the final analysis. A chi-square test was performed to determine whether the rate of responding/ non-responding varied between groups. All analyses were performed using SPSS Windows (version 10).
Habituation of the startle response was assessed by subjecting the amplitude data from the pulse-alone trials to a 2×3×2×2 [drug × experiment (pulse-alone amplitudes from each 5 min experiment in order of presentation) × group (patients/controls) × smoking status] ANOVA (Greenhouse Geisser). Drug and experiment were entered as within-subjects factors.
Prepulse inhibition was calculated as percentage of PPI (%PPI), namely, as (a−b/a)×100, where a=pulse-alone amplitude and b=amplitude over prepulse + pulse trials. The effect of subcutaneous nicotine on PPI was examined through a comparison of %PPIs which were subjected to a 2×3×2×2 [drug×trial type (30-, 60- and 120-ms prepulse trials) × group × smoking status] ANOVA (Greenhouse Geisser) with drug and trial type entered as within-subjects factors. Where relevant, interactions were followed up with paired and independent t tests. Although the duration of the study (20 min) was intended to exploit the maximal effects of nicotine (Russell et al. 1990), the pharmacological effect of nicotine may have dissipated over time and, possibly, at different rates in different individuals. As the effects of nicotine were likely to be maximal during the first block of trials, data were also analyzed for the first block of trials only via a 2 (drug)×2 (group)×3 (trial type) ANOVA, with drug entered as a within-subjects factor and group and trial type as between-subjects factors.
The effects of subcutaneous nicotine on latency to response peak were considered via a 2×4×2×2 [drug × trial type (three SOAs and latency over pulse-alone trials) × group × smoking status] ANOVA (Greenhouse Geisser) with drug and trial type entered as within-subjects factors.
Experiment 2: fMRI investigation
Subjects
Six healthy male control subjects and six male schizophrenic patients (all smokers) were recruited from the original sample (experiment 1). Subjects were recruited for the fMRI study on the basis of their responsivity to the startling stimuli and whether their response to nicotine was representative of the overall group response seen in experiment 1. One of the patients subsequently withdrew from the study, limiting the number of patients to five. For all subjects, the data obtained from the off-line study served as the behavioural data for analysis in the present fMRI study, as the MRI methods in use at that time precluded measurement of EMG responses during scanning. This approach was deemed appropriate because PPI is a very stable neurobiological measure with high test–retest reliability both in healthy subjects (Schwarzkopf et al. 1993; Abel et al. 1998; Cadenhead et al. 1999; Flaten 2002) and patients with schizophrenia (Ludewig et al. 2002). To minimise disparities between the off-line and on-line tests, the placebo and nicotine administrations for each subject were given in the same order for both studies.
Experimental design
Subjects were scanned (double-blind) on two separate occasions a fortnight apart.
The procedure was the same as described in the first study. The injection was administered in the clinical room adjacent to the scanning room, where subjects were positioned in the scanner immediately after. Functional scanning commenced 10 min after injection.
Functional magnetic resonance imaging
Gradient echoplanar imaging data were acquired using a 1.5 T GE Signa System (General Electric, Milwaukee, WI, USA) fitted with advanced MRI hardware and software (ANMR, Woburn, MA, USA) in the Department of Neuroimaging at the Maudsley Hospital, London. 100 T2* weighted MR images depicting BOLD contrast (Ogawa et al. 1990) were acquired at each of 16 near-axial non-contiguous slices parallel to the anterior commisure (AC)–posterior commisure (PC) (intracommisural) line. In-plane resolution was 3.3 mm, slice thickness 7 mm and interslice gap 0.7 mm to include the whole brain. TE (echo time) was 40 ms; TR (repetition time)=3 s. Subjects’ heads were placed in a quadrature birdcage coil, which was used for radio frequency pulse transmission and reception, where head movement was minimised by foam padding and a restraining band across the forehead. After completion of functional data acquisition, a high contrast, high resolution 3-D inversion recovery prepared spoiled GRASS volume data set was acquired in the AC–PC plane with TE=5.3 ms, TI=300 ms, TR=12.2 s, in-plane resolution=0.94 mm and slice thickness=1.5 mm.
Statistical analysis
fMRI data were analysed using statistical parametric mapping (SPM99, http://www.fil.ion.ucl.ac.uk/spm). Movement-related artefacts were corrected by realigning all volumes to the first volume (head movement was <3 mm in all subjects). The mean image produced by the realignment was co-registered with the structural volume. The resulting images were spatially normalised to conform to the standardised anatomical space of Talairach and Tournoux (1988). The activity at each voxel was scaled, high-pass-filtered and modeled for each experiment with the covariate of interest, consisting of 30-s epochs each convolved with the haemodynamic response function. These data were then analysed by testing the activity differences between the prepulse + pulse (experimental) blocks and pulse-alone (control) blocks for the placebo and nicotine conditions for the two subject groups. To identify generic activations representative of those found over the entire population of each subject group, a one-way random effects analysis was used. The second stage of analysis tested for drug and group effects using repeated and independent measures t tests (P<0.01).
Finally, Spearman’s rho was used to assess correlations between the (off-line) behavioural changes and the (on-line) activation changes, as predicted by the hypotheses, i.e. (a) the calculated difference in PPI under placebo and nicotine and (b) the change in power of activation in the hippocampus and thalamus after nicotine. These analyses were repeated for each SOA.
Results
Experiment 1: behavioural investigation
Two subjects (one patient and one control) reported adverse side effects and had to be withdrawn from the study (no medication required for these two or any other subjects). Nine of the remaining 21 healthy non-smokers (43%), seven of the healthy 19 smokers (37%) and five of the 14 patients with schizophrenia (36%) were non-responders to the startle stimuli on pulse-alone trials. There was no difference in startle responsivity between the patient and healthy groups (P>0.05).
Heart rate
For the effects of nicotine on heart rate, we found a significant interaction between occasion and drug [F(1.23, 45)=3.69, P<0.031; partial eta squared (pe2)=0.091], which a posthoc t test confirmed to reflect an increase in heart rate 9 min after the nicotine injection [t(32)=2.21, P<0.034; pe2=0.132].
Habituation of the startle response
There was significant habituation of startle, as evidenced by the main effect of block [F(1.34, 39.06)=22.513, P<0.01; pe2=0.435]. Habituation was not affected by group, drug or smoking status (P>0.05).
PPI
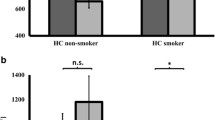
Analysis of %PPI showed main effects of trial type [F(1.65, 47.85)=9.853, P<0.01; pe2=0.244]. Supporting our hypothesis, there was a main effect of drug [F(1, 29)=9.459, P<0.005; pe2=0.214]. There was also a three-way interaction of drug × trial type × group [F(1.552, 58)=3.75, P<0.041; pe2=0.114]. Further analysis of this interaction revealed that nicotine administration enhanced PPI at all three SOAs in controls [drug F(1, 23)=9.00, P=0.006; pe2=0.28; drug × type F<2) but interacted with trial type in patients [drug × trial type F(1.780,16)=5.720, P=0.017; pe2=0.417] showing that change in PPI with nicotine was significant at the 30-ms SOA (t (pooled)(8)=3.97, P<0.004; pe2=0.66; Bonferroni-corrected P=0.024] but not at the 60- or 120-ms SOAs (see Fig. 1). The analysis of %PPI for the first block of trials, however, showed a significant effect of drug [F(1,27)=8.85, P<0.006; pe2=0.247], with no further effects or interactions. This pattern of results suggests that there was an enhancing effect of nicotine on PPI for both subject groups.

Average increases (error bar displays +1 SEM) in prepulse inhibition (%PPI) at 30, 60 and 120-ms SOAs in healthy controls and patients with schizophrenia after nicotine administration
Latencies to peak
An analysis of mean latencies to response peak showed a main effect of trial type [F(2.142, 66.39)=22.359, P<0.01] confirming shorter latencies in the prepulse + pulse conditions compared with pulse-alone trials (data not shown), with the greatest facilitation occurring at the 120-ms condition. No other main or interaction effects were found.
Experiment 2: fMRI investigation
Brain activation patterns
The initial random effects analyses identified significant changes in BOLD activation during the ‘on’ condition (i.e. PPI trials) relative to the ‘off’ condition (pulse-alone trials), i.e. those areas involved in the inhibition response. This analysis was performed for each subject group and for both the nicotine and placebo conditions. As performance during placebo was assumed sub-optimal due to nicotine withdrawal, however, the associated brain activation patterns are not very meaningful and, thus, are not being presented for the purpose of brevity. In general, activations during both the placebo and nicotine conditions mainly were located, as expected, in the limbic and striatal regions.
The areas showing the relative changes in activation under nicotine yielded by both the within- and between-groups comparisons are presented in Table 2. These results demonstrated that the effects of nicotine on PPI were modulated consistently by its actions in the striatal, thalamic and hippocampal regions across all SOAs in both the control and patient groups.
Correlational analysis of behavioural data with predicted regions of interest
For both subject groups combined, this analysis showed a significant positive relationship between change in %PPI after nicotine and change in power of activation of the left hippocampus after nicotine at the SOA of 30 ms where the enhancing effect of nicotine on PPI was strongest (rho=0.738, P=0.01; Fig. 2). No other relationships were found to be significant.

Nicotine-induced modulation of hippocampal activity during 30-ms prepulse + pulse (greater than pulse-alone) condition in controls and patients with schizophrenia
Discussion
Behavioural observations
We found that acute nicotine enhanced PPI in both healthy subjects and patients with schizophrenia using an intramodal tactile startle paradigm. This finding is consistent with previous reports of enhanced acoustic PPI after nicotine in both healthy smokers and non-smokers (Kumari et al. 1996, 1997; Della Casa et al. 1998; Duncan et al. 2001). In the present study, the nicotine-induced enhancement of PPI was most pronounced at 30 ms and only marginal at 60 ms. This pattern parallels findings reported earlier by Kumari et al. (2001), who investigated the relationship between PPI and time elapsed since the last cigarette in a group of schizophrenia patients and found a negative correlation between these two measures for the SOA of 30 ms, with a trend for a similar relationship at 60 ms.
The present study was designed to detect the maximal effects of nicotine by testing the subset of smoking subjects (patients and controls) under nicotine withdrawal on both the placebo and drug occasions. This requirement also controlled for the confounding effects of varying baseline levels of nicotine in smokers. The observation of increased PPI in both smokers and non-smokers indicate that the observed enhancement in PPI was a ‘true’ drug effect and not solely a restoration of a withdrawal-induced deficit. It should be noted, however, that PPI during placebo in our subjects was assumed to be sub-optimal due to nicotine withdrawal (e.g. Kumari et al. 1996; Kumari and Gray 1999) and did not provide an appropriate reference for comparisons between the patient and control groups.
The excessive use of tobacco amongst patients with schizophrenia has been hypothesised as an attempt at some form of self-medication (Spielewoy and Markou 2004; for a review, see Kumari and Postma 2005). The most convincing evidence to date supports a role for nicotine in improvement of more basic attentional abnormalities of the disorder as measured by paradigms tapping auditory sensory (P50 gating; Adler et al. 1993, 1992) and sensorimotor gating (PPI; Kumari et al. 1996, 1997). The present results add to the generality of these findings, showing nicotine to increase tactile PPI in patients as well as in healthy smokers and non-smokers.
fMRI observations
The fMRI data revealed significant involvement of limbic regions in the effect of nicotine on PPI at all SOAs. During the 30-ms condition, where the effect of nicotine on PPI was maximal for both groups, we observed significantly greater left hippocampal gyrus activation relative to the placebo condition in both patients and controls. Under nicotine, the same region was activated in both groups on the right side during the 60-ms condition. Nicotine induced greater right hippocampal activation during the 120-ms trials in control subjects and increased bilateral activation of this region in the patient group. Over all three SOA conditions, hippocampal gyrus activation was significantly greater in the patient group relative to controls, perhaps indicating a greater sensitivity to the effects of nicotine in the patient group. Chronic smoking upregulates nicotinic receptors in the hippocampus, caudate and thalamus in healthy smokers but not in schizophrenic smokers (Breese et al. 2000). Although rather speculative, it is possible that abnormalities in upregulation may reflect population differences in mechanisms involved in receptor desensitisation. Such a mechanism could explain the differential sensitivity we observed in these structures.
Under nicotine, activity was also observed in the thalamus under all three SOA conditions, although only significantly for the control group at the 30-ms SOA (left thalamus) compared to all three SOAs for the patient group (30 ms, right; 60 ms, right; 120 ms, left). It is possible that acute nicotine administration reverses thalamic hypo-activity considered to be associated with reduced PPI in schizophrenia (Hazlett et al. 2001; Kumari et al. 2003a). Both the hippocampal gyrus and thalamus harbour high concentrations of the low-affinity nicotinic α7 receptors, which are prominent in the areas near the dentate gyrus and CA3–CA4 region in the hippocampus (Freedman et al. 1993) and the reticular nucleus of the thalamus (Leonard et al. 1996, 1998; Adler et al. 1998). Post-mortem studies have established reduced numbers of this receptor subtype in the hippocampi of schizophrenic patients who show deficits in P50 auditory gating (Freedman et al. 1995). Selectively bred mice with low numbers of these receptors show reduced PPI, which is increased after nicotine administration (e.g. Stevens and Wear 1997; Bullock et al. 1997). The high concentrations of α7 receptors present in the thalamus implicate this structure in a similar way to the hippocampus in the cholinergic modulation of filtering of sensory input.
We observed increased activation of the striatum under nicotine with strongest activation of these areas occurring at the longer SOAs. Animal and human studies confirm involvement of the striatum in the regulation of PPI (animal studies; Kodsi and Swerdlow 1995a,b; for a review, see Swerdlow et al. 2001; human studies; Kumari et al. 2003b, 2005). We also observed activation in the insula (right) under nicotine, but only in the patient group at 120 ms. Relative to placebo, there was greater activation of the left cingulate gyrus at 30 (controls) and 120 ms (patients) during nicotine. The anterior cingulate is part of the cortico–striato–pallido–thalamic circuitry implicated in the modulation of PPI (Swerdlow and Geyer 1998; Swerdlow et al. 2001) and was also shown in the Hazlett et al. (1998) PET study to be involved in attention-enhanced PPI. Nicotine has been found to increase cingulate activity concomitant with cognitive improvement in recent fMRI studies of healthy subjects (Kumari et al. 2003b) as well as schizophrenia patients (Jacobsen et al. 2004).
Patients showed strong cerebellar activation at 30 and 120 ms and significantly greater cerebellar activity than control subjects under nicotine. McNamara et al. (1990) have observed increases in activity in the cerebellum after nicotine in the rat. In humans, a PET study by Nagata et al. (1995) also reported strong cerebellar activation after acute nicotine in overnight smoking withdrawn subjects. As our subjects were in a similar state to those in the study of Nagata et al. (1995), this result could suggest that the cerebellar effect seen in the present study reflects a restoration of withdrawal-induced lack of tolerance. However, our previous fMRI study in non-smokers also detected a nicotine-induced increase in cerebellar activity (Kumari et al. 2003b), suggesting that nicotine may have potentiated the previously suggested involvement of the cerebellum in PPI (Stitt et al. 1976).
The observed activation patterns may be compared with those seen in previous imaging studies of the sites of action of nicotine in human brain. For example, Nyback et al. (1989) used PET to visualise central sites of nicotine action, confirming large concentrations of binding sites in frontal, cingulate and insular lobe as well as in the thalamus and basal ganglia. The investigation of Stein et al. (1998) on the sites of action of intravenously administered nicotine showed significant activation in cingulate and dorsolateral, and orbital and medial frontal regions, all implicated in cognitive processes such as attention and vigilance known to be modified by nicotine (Warburton 1990). Also activated were those regions known to be involved in the reinforcing effects of nicotine, i.e. the nucleus accumbens, amygdala and thalamus. Visual cortex and temporal lobe areas were activated, an effect which was attributed to the extensive projections from the thalamic nuclei (which are particularly rich in nicotinic receptors) to these areas as well as the frontal, cingulate and parietal regions. These authors suggest that much of the cortical activation observed in their study might have reflected intense thalamic nicotinic receptor activation. Such activations could also explain some of the findings of the present study: in addition to the predicted areas of activation described earlier, nicotine-induced increases in PPI were accompanied by activation of medial and superior temporal, parietal and occipital cortices. The cortical activation observed in our study similarly could have been secondary to an intense thalamic effect.
An area not specifically hypothesized to be associated with nicotine or PPI but emerging in both subject groups as showing nicotine-induced increases in activity was the precuneus (BA7). Previous studies have reported activation of this region during the resting stage of block paradigms (Mazoyer et al. 2001), the speculation being that such activation might reflect an affective anticipatory response to impending active task conditions (Simpson et al. 2001). In the current study, the comparison condition of ‘pulse-alone’ stimuli served to elicit a strong startle response that was inhibited during the experimental prepulse phase. The increased inhibition under nicotine would have emphasized the contrast between the experimental and comparison conditions which could potentially have increased the anticipatory response to the pulse-alone trials, which in turn could explain the increased precuneal activation.
The present investigation was limited by the inability to measure behavioural performance at the time of scanning due to the metal content of the EMG electrodes. This constraint precluded the analysis of choice: namely, a factorial analysis in which the interaction term would have identified brain regions activated by the (active) PPI condition, where a change in neuronal activation had occurred as a result of the nicotine challenge. Therefore, an alternative strategy was adopted to substantiate the above findings. A correlational analysis was performed of the off-line behavioural changes with the areas of activation, i.e. (a) the difference in %PPI under placebo and nicotine and (b) the change in amount of activation after nicotine for each SOA. This analysis showed a significant positive relationship between increased PPI and increased activation of the hippocampus at the 30-ms SOA, where this behavioural change was strongest. This, we believe, is the strongest evidence to suggest that in humans, as with P50 gating, the enhancing effect of nicotine on PPI involves the hippocampus.
Limitations
Our fMRI investigation had some limitations. Firstly, it was limited by the small number of patients, all of whom were smokers and treated with typical antipsychotics. Nevertheless, this small number produced significant activations of predicted areas. Another issue was the high rate of non-responders to this tactile paradigm, as was evident in the off-line version of this experiment, which calls into question the representativeness of our final sample as only good responders were selected for the scanning component of the study. The possibilities deserving further investigation for a high rate of startle non-responding in this study include (a) non-random presentation of the stimuli in the box-car design (as opposed to random presentation generally employed in startle studies), (b) the chosen intensity of air puffs which was kept rather low to avoid excessive body movements and related artefacts during the scanning and (c) a genetic trait, perhaps even a variant of an allele, which confers vulnerablility to startle or lack of PPI. Finally, although the present study examined the effects of nicotine in the subset of smoking subjects after 12 h of smoking deprivation, there may have been some residual nicotine after this abstinence period especially in subjects with baseline levels exceeding 40 ng/ml (a level which is often seen in schizophrenia smokers, Sacco et al. 2005). Furthermore, no nicotine levels were obtained post-nicotine injection, so the levels of nicotine in the brain may have varied across subjects.
Conclusions
Our results support a role for nicotine in the restoration of deficits in schizophrenia as indexed by PPI. Our fMRI findings confirm involvement of the hippocampus and thalamus in modulation of PPI via nicotine in both healthy control subjects and schizophrenia patients. The correlational analysis between the calculated behavioural enhancement of PPI and change in neural activation after nicotine confirmed the hippocampus as the primary structure for the modulatory effect of nicotine. In addition, we observed basal ganglia involvement that appeared to be less consistent across conditions than the hippocampal and thalamic changes. Patients showed stronger activation of both hippocampal and thalamic areas across all SOA conditions, which may be interpreted as reflecting a greater sensitivity to the effects of nicotine in this group. However, given that our fMRI study involved only a small number of smoking schizophrenia patients treated with typical antipsychotics, the results should be considered preliminary and extended to a larger group of smoking and non-smoking antipsychotic-naive and medicated (with typical as well as atypical antipsychotics) schizophrenia patients.
References
Abel K, Waikar M, Pedro B, Hemsley D, Geyer M (1998) Repeated testing of prepulse inhibition and habituation of the startle reflex: a study in healthy human controls. J Psychopharmacol 12(4):330–337
Acri JB, Grunberg NE, Morse DE (1991) Effects of nicotine on the acoustic startle reflex amplitude in rats. Psychopharmacology 104(2):244–248
Acri JB, Morse DE, Popke EJ, Grunberg NE (1994) Nicotine increases sensory gating measured as inhibition of the acoustic startle reflex in rats. Psychopharmacology 114(2):369–374
Adler LE, Hoffer LJ, Griffith J, Waldo MC, Freedman R (1992) Normalization by nicotine of deficient auditory sensory gating in the relatives of schizophrenics. Biol Psychiatry 32(7):607–616
Adler LE, Hoffer LD, Wise A, Freedman R (1993) Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry 150(12):1856–1861
Adler LE, Olincy A, Waldo M, Harris JG, Griffith J, Stevens K, Flach K, Nagamoto H, Bickford P, Leonard S, Freedman R (1998) Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull 24:189–202
Braff DL, Stone C, Callaway E, Geyer MA, Glick ID, Bali L (1978) Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 15:339–343
Braff DL, Geyer MA, Swerdlow NR (2001) Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology 156(2–3):234–258
Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM, Marks MJ, Collins AC, Leonard S (2000) Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology 23:351–364
Bullock AE, Slobe BS, Vázquez V, Collins AC (1997) Inbred mouse strains differ in the regulation of startle and prepulse inhibition of the startle response. Behav Neurosci 111:1353–1360
Cadenhead KS, Carasso BS, Swerdlow NR, Geyer MA, Braff DL (1999) Prepulse inhibition and habituation are stable neurological measures in a normal male population. Biol Psychiatry 45:360–364
Curzon P, Kim DJ, Decker MW (1994) Effect of nicotine, lobeline, and mecamylamine on sensory gating in the rat. Pharmacol Biochem Behav 49(4):877–882
Della Casa V, Hofer I, Weiner I, Feldon J (1998) The effects of smoking on acoustic prepulse inhibition in healthy men and women. Psychopharmacology 137(4):362–368
de Leon J, Dadvand M, Canuso C, White AO, Stanilla JK, Simpson GM (1995) Schizophrenia and smoking: an epidemiological survey in a state hospital. Am J Psychiatry 152:453–455
de Leon J, Tracy J, McCann E, McGrory A, Diaz FJ (2002) Schizophrenia and tobacco smoking: a replication study in another US psychiatric hospital. Schizophr Res 56(1–2):55–65
Diwan A, Castine M, Pomerleau CS, Meador-Woodruff JH, Dalack GW (1998) Differential prevalence of cigarette smoking in patients with schizophrenic vs mood disorders. Schizophr Res 33(1–2):113–118
Duncan E, Madonick S, Chakravorty S, Parwani A, Szilagyi S, Efferen T, Gonzenbach S, Angrist B, Rotrosen J (2001) Effects of smoking on acoustic startle and prepulse inhibition in humans. Psychopharmacology 156(2–3):266–272
Faraday MM, O’Donoghue VA, Grunberg NE (1999) Effects of nicotine and stress on startle amplitude and sensory gating depend on rat strain and sex. Pharmacol Biochem Behav 2(2):273–284
First MB, Spitzer RL, Gibbon M, Williams JBW (1995) Structured clinical interview for DSM-IV Axis II disorders, patient edition (SCID-P), version 2. New York State Psychiatric Institute, Biometrics Research, New York
First MB, Spitzer RL, Gibbon M, Williams JBW (1996) Structured clinical interview for DSM-IV Axis I disorders, non-patient edition (SCID-1/NP), version 2. New York State Psychiatric Institute, Biometrics Research, New York
Fagerstrom KO, Schneider NG (1989) Measuring nicotine dependence: a review of the Fagerstrom tolerance questionnaire. J Behav Med 12:159–182
Flaten MA (2002) Test–retest reliability of the somatosensory blink reflex and its inhibition. Int J Psychophysiol 45(3):261–265
Freedman R, Wetmore C, Stromberg I, Leonard S, Olson L (1993) Alpha-bungarotoxin binding to hippocampal interneurons: immunocytochemical characterization and effects on growth factor expression. J Neurosci 13:1965–1975
Freedman R, Hall M, Adler LE, Leonard S (1995) Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry 38:22–33
Geyer MA, Ellenbroek B (2003) Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry 27(7):1071–1079
Geyer MA, Swerdlow NR, Mansbach RS, Braff DL (1990) Startle response models of sensorimotor gating and habituation deficits in schizophrenia. Brain Res Bull 25:485–498
Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR (2001) Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology 156(2–3):117–154
Glassman AH (1993) Cigarette smoking: implications for psychiatric illness. Am J Psychiatry 150(4):546–553
Graham FK (1975) The more or less startling effects of weak prestimulation. Psychophysiology 12:238–248
Hazlett EA, Buchsbaum MS, Haznedar MM, Singer MB, Germans MK, Schnur DB, Jimenez EA, Buchsbaum BR, Troyer BT (1998) Prefrontal cortex glucose metabolism and startle eyeblink modification abnormalities in unmedicated schizophrenia patients. Psychophysiology 35:186–198
Hazlett EA, Buchsbaum MS, Tang CY, Fleischman MB, Wei TC, Byne W, Haznedar MM (2001) Thalamic activation during an attention-to-prepulse startle modification paradigm: a functional MRI study. Biol Psychiatry 50(4):281–291
Hughes JR, Hatsukami DK, Mitchell JE, Dahlgren LA (1986) Prevalence of smoking among psychiatric outpatients. Am J Psychiatry 143(8):993–997
Jacobsen LK, D’Souza DC, Mencl WE, Pugh KR, Skudlarski P, Krystal JH (2004) Nicotine effects on brain function and functional connectivity in schizophrenia. Biol Psychiatry 55(8):850–858
Javitt DC, Zukin SR (1991) Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148(10):1301–1318
Kay SR, Fiszbein PS, Opler LA (1987) The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13:261–276
Kodsi MH, Swerdlow NR (1995a) Ventral pallidal GABA-A receptors regulate prepulse inhibition of acoustic startle. Brain Res 684:26–35
Kodsi MH, Swerdlow NR (1995b) Prepulse inhibition in the rat is regulated by ventral and caudodorsal striato–pallidal circuitry. Behav Neurosci 109:912–928
Kumari V, Gray JA (1999) Smoking withdrawal, nicotine dependence and prepulse inhibition of the acoustic startle reflex. Psychopharmacology 141:11–15
Kumari V, Soni W, Mathew VM, Sharma T (2000) Prepulse inhibition of the startle response in men with schizophrenia: effects of age of onset of illness, symptoms, and medication. Arch Gen Psychiatry 57:609–614
Kumari V, Postma P (2005) Nicotine use in schizophrenia: the self-medication hypotheses. Neurosci Biobehav Rev 29(6):1021–1034
Kumari V, Checkley SA, Gray JA (1996) Effect of cigarette smoking on prepulse inhibition of the acoustic startle reflex in healthy male smokers. Psychopharmacology 128(1):54–60
Kumari V, Cotter PA, Checkley SA, Gray JA (1997) Effect of acute subcutaneous nicotine on prepulse inhibition of the acoustic startle reflex in healthy male non-smokers. Psychopharmacology 132(4):389–395
Kumari V, Soni W, Sharma T (2001) Influence of cigarette smoking on prepulse inhibition of the acoustic startle response in schizophrenia. Hum Psychopharmacol 16:321–326
Kumari V, Gray JA, Geyer MA, Mitterschiffthaler M, Ffytche D, Vythelingum GN, Soni W, Simmons A, Williams SCR, Sharma T (2003a) Neural correlates of prepulse inhibition in normal and schizophrenic subjects: a functional MRI study. Psychiatry Res Neuroimaging 122:99–113
Kumari V, Gray JA, ffytche D, Mitterschiffthaler MT, Das M, Zachariah E, Vythelingum GN, Williams SCR, Simmons A, Sharma T (2003b) Cognitive effects of nicotine in humans: a functional MRI investigation. NeuroImage 19:1002–1013
Kumari V, Aasen I, Sharma T (2004) Sex effects in prepulse modification deficits in schizophrenia. Schizophr Res 69(2–3):219–235
Kumari V, Antonova E, Zachariah E, Galea A, Aasen I, Mitterschiffthaler MT, Sharma T (2005) Structural brain correlates of prepulse inhibition of the acoustic startle response in healthy humans. NeuroImage 26(4):1052–1058
Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. (2001) Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25:455–467
Leonard S, Adams C, Breese CR, Adler LE, Bickford P, Byerley W, Coon H, Griffith JM, Miller C, Myles-Worsley M, Nagamoto HT, Rollins Y, Stevens KE, Waldo M, Freedman R (1996) Nicotinic receptor function in schizophrenia. Schizophr Bull 22(3):431–445
Leonard S, Gault J, Adams C, Breese CR, Rollins Y, Adler LE, Olincy A, Freedman R (1998) Nicotinic receptors, smoking and schizophrenia. Restor Neurol 12(2–3):195–201
Ludewig K, Geyer MA, Vollenweider FX (2002) Stability of the acoustic startle reflex, prepulse inhibition, and habituation in schizophrenia. Schizophr Res 55(1–2):129–137
Ludewig K, Geyer MA, Vollenweider FX (2003) Deficits in prepulse inhibition and habituation in never-medicated first-episode schizophrenia. Biol Psychiatry 54:121–128
Mazoyer B, Zago L, Mellet E, Bricogne S, Etard O, Houde O, Crivello F, Joliot M, Petit L, Tzourio-Mazoyer N (2001) Cortical networks for working memory and executive functions sustain the conscious resting state in man. Brain Res Bull 54:287–298
McNamara D, Larson DM, Rapoport SI, Soncrant TT (1990) Preferential metabolic activation of subcortical brain areas by acute administration of nicotine to rats. J Cereb Blood Flow Metab 10:48–56
Nagata K, Shinohara T, Kanno I, Hatzawa J, Domino E (1995) Effects of tobacco cigarette smoking on cerebral blood flow in normal adults. In: Domino EF (ed) Brain imaging of nicotine and tobacco smoking. NPP, Ann Arbor, pp 95–107
Nyback H, Nordberg A, Langstrom B, Halldin C, Hartvig P, Ahlin A, Swahn CG, Sedvall G (1989) Attempts to visualize nicotinic receptors in the brain of monkey and man by positron emission tomography. Prog Brain Res 79:313–319
Ogawa S, Lee TM, Kay AR, Tank DW (1990) Brain magnetic resonance imaging with contrast dependent blood oxygenation. Proc Natl Acad Sci U S A 8868–8872
Russell MAH, Jarvis MJ, Jones G, Feyerabend C (1990) Non-smokers show acute tolerance to subcutaneous nicotine. Pscyhopharmacology 102:56–58
Sacco KA, Bannon KL, George TP (2004) Nicotinic receptor mechanisms and cognition in normal states and neuropsychiatric disorders. J Psychopharmacol 18(4):457–474
Sacco KA, Termine A, Seyal A, Dudas MM, Vessicchio JC, Krishnan-Sarin S, Jatlow PI, Wexler BE, George TP (2005) Effects of cigarette smoking on spatial working memory and attentional deficits in schizophrenia: involvement of nicotinic receptor mechanisms. Arch Gen Psychiatry 62(6):649–659
Schwarzkopf SB, McCoy L, Smith DA, Boutros NN (1993) Test–retest reliability of prepulse inhibition of the acoustic startle response. Biol Psychiatry 33:815–828
Semenova S, Bespalov A, Markou A (2003) Decreased prepulse inhibition during nicotine withdrawal in DBA/2J mice is reversed by nicotine self-administration. Eur J Pharmacol 472(1–2):99–110
Simpson JR Jr, Snyder AZ, Gusnard DA, Raichle ME (2001) Emotion-induced changes in human medial prefrontal cortex: I. During cognitive task performance. Proc Natl Acad Sci U S A 98:683–687
Spielewoy C, Markou A (2004) Strain-specificity in nicotine attenuation of phencyclidine-induced disruption of prepulse inhibition in mice: relevance to smoking in schizophrenia patients. Behav Genet 34(3):343–354
Stein EA, Pankiewicz J, Harsch HH, Cho JK, Fuller SA, Hoffmann RG, Hawkins M, Rao SM, Bandettini PA, Bloom AS (1998) Nicotine-induced limbic cortical activation in the human brain: a functional MRI study. Am J Psychiatry 155:1009–10015
Stevens KE, Wear KD (1997) Normalizing effects of nicotine and a novel nicotinic agonist on hippocampal auditory gating in two animal models. Pharmacol Biochem Behav 57(4):869–874
Stevens KE, Kem WR, Mahnir VM, Freedman R (1998) Selective alpha-7-nicotinic agonists normalize inhibition of auditory response in DBA mice. Psychopharmacology 136:320–327
Stitt CL, Hoffman HS, Marsh RR, Schwartz GM (1976) Modification of the pigeon’s visual startle reaction by the sensory environment. J Comp Physiol Psychol 90:601–619
Suemaru K, Yasuda K, Umeda K, Araki H, Shibata K, Choshi T, Hibino S, Gomita Y (2004) Nicotine blocks apomorphine-induced disruption of prepulse inhibition of the acoustic startle in rats: possible involvement of central nicotinic alpha7 receptors. Br J Pharmacol 142(5):843–850
Swerdlow NR, Geyer MA (1998) Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr Bull 24:285–301
Swerdlow NR, Geyer MA, Braff DL (2001) Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology 156(2–3):194–215
Talairach J, Tournoux P (1988) Co-planar stereotaxic atlas of the human brain. Thieme, Stuttgart
Uck A, Polat A, Bozkurt O, Meteris H (2004) Cigarette smoking among patients with schizophrenia and bipolar disorders. Psychiatry Clin Neurosci 58(4):434–437
Warburton DM (1990) Nicotine as a cognitive enhancer. In: Yamashita I (ed) Clinical neuropharmacology. Raven, New York, pp 579–580
Acknowledgements
VK was supported by the Wellcome Trust, UK (067427/427/Z/02/Z). MAG was supported in part by the National Institute of Mental Health, US (MH42228) and the US Veteran’s Administration VISN 22 Mental Illness Research, Education, and Clinical Center. MAG has an equity interest in Sand Diego Instruments, Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jeffrey A. Gray is deceased.
Rights and permissions
About this article
Cite this article
Postma, P., Gray, J.A., Sharma, T. et al. A behavioural and functional neuroimaging investigation into the effects of nicotine on sensorimotor gating in healthy subjects and persons with schizophrenia. Psychopharmacology 184, 589–599 (2006). https://doi.org/10.1007/s00213-006-0307-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-006-0307-5