
Abstract
Renal I/R injury is a severe medical condition contributing to acute kidney injury (AKI), leading to rapid kidney dysfunction and high mortality rates. It is generally observed during renal transplantation, shock, trauma, and urologic and cardiovascular surgery, for which there is no effective treatment. Cell death and damage are commonly linked to I/R. Cell death triggered by iron-dependent lipid peroxidation, such as ferroptosis, has been demonstrated to have a significant detrimental effect in renal IRI models, making it a new type of cell death currently being researched. Ferroptosis is a nonapoptotic type of cell death that occurs when free iron enters the cell and is a critical component of many biological processes. In ferroptosis-induced renal I/R injury, iron chelators such as Deferasirox, Deferiprone, and lipophilic antioxidants are currently suppressed lipid peroxidation Liproxstatin-1 (Lip-1), Ferrostatin-1 along with antioxidants like vitamin and quercetin. Ferroptosis has been considered a potential target for pharmaceutical intervention to alleviate renal IRI-associated cell damage. Thus, this review emphasized the role of ferroptosis and its inhibition in renal IRI. Also, Pharmacological modulation of ferroptosis mechanism in renal I/R injury has been conferred.

Graphical abstract
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Acute ischemic renal damage is a prevalent complication caused by various diseases that decrease the kidneys’ ability to receive adequate arterial blood flow. Ischemic renal damage is exacerbated by restoring blood flow (Linkermann et al. 2014a, b; Wang and Bellomo 2017). Apoptosis and necrosis are the only two cell death processes previously recognized to have a role in renal ischemia AKI pathogenesis (Thapa et al. 2022). In renal I/R models, ferroptosis emerged as a newly identified regulated mechanism of cell death with deleterious effects (Yan et al. 2020). Although cell death is a vital clinical sign of IRI, preventing cell death caused by IRI could be a new therapeutic strategy. However, it has previously been suggested that a comprehensive understanding of I/R-related cell death is essential in creating effective therapy options for IRI (Yarishkin et al. 2018; Sarkar et al. 2019). Oxidative stress-induced due to lipid peroxidation and elevated intracellular iron levels are hallmarks of IRI (Saklani et al. 2022a). In the most recent literature, iron-dependent ferroptosis has been found to cause cell damage and death (Thévenod and Lee 2013; Sharma et al. 2021). The cellular activity of iron-dependent ferroptosis can be blocked by iron chelation and antioxidants. In animal models of IRI, iron chelation is therapeutic (Rodríguez-Vargas et al. 2019; Jiang et al. 2021). Thus, ferroptosis has been recently found as a therapeutic target for renal I/R damage, according to recent findings.
Ferroptosis mechanism of cell death
Antioxidant mechanism
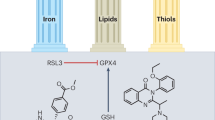
Selenium is an endogenous antioxidant required for the activity of glutathione peroxidase 4 (GPx4) (Ighodaro and Akinloye 2018). Asparagine, glutamine, tryptophan, and selenocysteine (Sec) or cysteine form hydrogen bonds with nitrogen atoms in GPx4’s catalytic core, forming a tetrad (Asn). One of GPx4’s unique properties is its structure, which makes it an excellent catalyst for cysteine residues’ rapid and specific oxidation (Tosatto et al. 2008). A wide range of substrates, comprising H2O2, tiny hydroperoxides, lipopeptides, and complex molecular lipids, such as phospholipid and cholesterol ester hydroperoxides, can be modified by GPx4 when injected into biomembranes or lipoproteins (Casañas-Sánchez et al. 2015; Saklani et al. 2022a, b). GPx4 is a crucial regulator of anti-lipid peroxidation and provides resistance to ferroptosis. Inhibition of GPx4 activity by ferroptosis agonists is accomplished through either direct (RSL3) or indirect (Erastin or Fin56) inhibition (Jiang et al. 2021). For GPx4 to work correctly, it needs GSH and selenium (Ursini and Maiorino 2020). It had been reported in 2003 that Erastin and RSL3 cause RAS mutation-dependent cytotoxicity, and ferroptosis had been linked to glutathione-metabolizing proteins (Zhou et al. 2020a, b). Neuronal death caused by glutamine has features comparable to ferroptosis, which implies that glutathione synthesis is involved in the method (Hayashima et al. 2021). Enzymatically generated glutathione (GSH) is an essential antioxidant involved in GPx4 renewal (Nehring et al. 2020; He et al. 2021; Tang et al.2021). The cytosolic enzymes glutathione synthetase (GSS) and glutamate-cysteine ligase (GCL) catalyze the GSH synthesis in two steps using glutamate, cysteine, and glycine (Aoyama et al. 2008; Franklin et al. 2009). Cysteine is required for cell survival, according to several studies. Due to low glutathione levels, human fibroblasts cannot be cultured in cystine-free media (Conrad and Sato 2012; Scindia et al. 2015). Disulfide-linked heterodimers of cystine/glutamate antiporter systems (Xct) are two subunits linked with the disulfide bond that allows glutamate and cysteine exchange across plasma membranes (Lewerenz et al. 2013; Liang et al. 2019). Solute carrier 3 member 2 (SLC3A2) is the first of these components, and then comes solute carrier 7 member 11 (SLC7A11). Cystine is quickly converted to cysteine when injected into the cell. Cystine/cystathionine and glutamate are the primary targets of erastin in ferroptosis, which is why this exchange is critical. Cysteine can be synthesized from methionine via the transsulfuration pathway in some cell types, and this mechanism may be resistant to erastin (Dixon et al. 2012). When selenium levels within cells are low, cysteine is positioned in the GPx4 active site (Ingold et al. 2018). For example, the enzyme’s activity is 1000 times lower than that of the naturally occurring GPx4 recombinant Cys mutant (Ingold et al. 2018). Na2SeO3 supplementation restored GPx4 function in SH-SY5Y cells exposed to methamphetamine (Yu et al. 2017). To prevent irreversible hyperoxidation suppression by Sec deprotonation, GPx4 requires selenium. Sec has an advantage over Cys in the central nervous system with its thiol groups because Sec can deprotonate fast (Barayuga et al. 2013; Brigelius-Flohé and Maiorino 2013; Song et al. 2014; Cardoso et al. 2017; Doll et al. 2017). GPx4 was assumed to be the only regulator of ferroptosis until now. No matter whether acyl-CoA synthetase long-chain family member 4 (ACSL4) is expressed or not, blocking GPx4 does not cause ferroptosis, contrary to the common belief (Wu et al. 2018; Doll et al. 2019; Bersuker et al. 2019). This demonstrates the possibility of other mechanisms of resistance. GPx4 and glutathione may work together with ferroptosis suppressor protein 1 (FSP1) and coenzyme Q10 (CoQ10) to reduce phospholipid peroxidation and ferroptosis (Wu et al. 2002; Han et al. 2020). Because of its molecular similarities to AIFM1, the apoptosis-inducing factor mitochondrial 2 (AIFM2) was anticipated to initiate apoptosis via a caspase-1-independent pathway (Chen et al. 2020a, b; Khan et al. 2022b). FSP1 functions as an oxidoreductase after being drawn to the cellular membranes by myristoylation to promote the regeneration of coenzyme Q10 (CoQ10) utilizing NADPH. Ubiquinol, a lipophilic radical-trapping antioxidant (RTA) (Frei et al.1990; Ng et al. 2019; Kalra et al. 2022), is a reduced form of CoQ10 that inhibits the formation of lipid peroxides, hence regulating ferroptosis. The mevalonate process uses acetyl-CoA to create ubiquinol. The mechanism of ferroptosis has been reported to be blocked by small molecules. Endogenous CoQ10 levels are depleted when FIN56 attaches to and activates squalene synthase (Mullen et al. 2016; Shimada et al. 2016) (Fig. 1).

Pathogenic factors trigger a Fenton reaction that oxidizes membrane lipids to lipid peroxides and regulates ferroptosis by increasing H2O2 or Fe2+ abnormally
Oxidation mechanisms
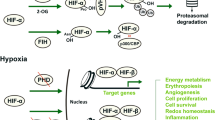
Lipid metabolism is essential for both pathological and physiological processes in the human body. For example, in cell signaling and energy metabolism, fatty acids are a critical component of membranes in biology (Olzmann and Carvalho 2019). Polyunsaturated fatty acids (PUFAs) contain two or more double bonds and are crucial for plasma and membrane formation (Kihara 2012; Zárate et al. 2017). Whereas cis-double bonds of methylene groups of fatty acids are quickly oxidized, making fatty acids more susceptible to autoxidation (Carvalho et al. 2010; Dixon and Stockwell 2019). Lipid hydroperoxides are a significant element in ferroptosis. A ferroptosis cell death signal is PE (phosphatidylethanolamine) that contains arachidonic acid (AA) (AA-PE). It is possible to lengthen adrenoyl (AdA) using an enzyme called elongase (Yang and Stockwell 2016; Chen et al. 2021). It has recently been discovered that AA-OOHPE is the most potent inducer of iron apoptotic phenotype in comparison to other phospholipids (PL)-OOH forms, and it catalyzes the conversion of acyl-CoA to acyl-PE via the acyl-CoA synthetase family 4 (ACSL4) (Müller et al. 2017; Lin et al. 2021). LPCAT3 is required to complete the esterification process, whereas LOXs and reactive oxidizing radicals accelerate the transformation of AA-PE to AA-OOH-PE (Hirschhorn and Stockwell 2019; Capelletti et al. 2020; Wang et al. 2020). Cytochrome P450 oxidoreductase (POR), a stress-induced enzyme, increases lipid peroxidation by providing electrons to downstream effectors. Ferroptosis occurs when AA-OOH-PE exceeds the reduction mechanism’s competence (Zou et al. 2020; Bagayoko and Meunier 2021). Lipid peroxidation can be caused by iron because of its ability of redox activation. The first step in the process is to release iron bound for Fenton reaction products from the labile iron pool (LIP), promoting ROS buildup (Jomova and Valko, 2011; Bertrand 2017; Li et al. 2020; Fujii et al. 2020). For example, iron’s role in enzyme reactions, such as lipoxygenases (LOXs) and NADPH oxidases, directly impacts the rate and extent of lipid peroxidation. According to these studies, iron homeostasis proteins also can regulate a condition known as ferroptosis. It has also been shown that silencing the iron response element-binding protein 2 (IREB2) via sh-RNA can reduce ferroptosis sensitivity (Tao et al. 2020; Chen et al. 2020a, b). One of the iron-sulfur cluster production enzymes, NFS1 (cysteine desulfurase) and Prominin2, a ferroptosis stress response protein, have also been discovered to suppress the formation of ferroptosis in lung cancer (Adam et al. 2006). In addition, ferroptosis can be triggered by ferritin autophagy in the lysosomes, which increases the decreased iron content (Hou et al. 2016; Liu et al. 2020a, b). To prevent cellular harm from iron-mediated degradation, cells undergo ferritinophagy, a mechanism regulated by the nuclear receptor coactivator 4 (NCOA4) cargo receptor (Cicenas et al. 2017; Gryzik et al. 2021) (Fig. 1).
Targeting ferroptosis in renal I/R injury
Thrombotic or embolic events, surgical procedures, and renal transplantation have been associated with acute kidney failure induced by IRI and have played a substantial role in patient morbidity and mortality (Malek and Nematbakhsh 2015). Necrosis and ferroptosis pathways exist simultaneously in renal IRI, suggesting a “synergistic effect” in recent studies on renal I/R (Haase et al. 2010; Linkermann et al. 2014a, b; Ni et al. 2019). Mixed lineage kinase domain-like protein (MLKL), a molecular switch in renal IRI, is activated by PUFA depletion and activates necroptosis. In contrast, ACSL4 is activated by necroptosis and makes the biomembrane less susceptible to MLKL-induced membrane permeabilization (Casserly and Dember 2003; Choi et al. 2019; Li et al. 2020). Iron chelators have already been reported to prevent renal tubular cell mortality in several studies of renal IRI, suggesting the significant role of iron in renal IRI (Paller and Hedlund 1994). Renal tubular cell death can be prevented by active in-transforming growth factor (TGF) receptor ALK4/5, which has been related to stress-induced renal injury (Sharma and Leaf 2019). There may be a correlation between ferroptosis and different cell death systems in the renal IRI model. Ferroptosis inhibitors are effective in a few preclinical studies for renal IRI, although discovering specific pathways regulating ferroptosis may yield better outcomes. As a result, future research efforts must focus on identifying novel mechanisms or pathways for preclinical and clinical evaluation of ferroptosis inhibitors to treat IRI (Fig. 2).

Esterification of arachidonic acid (AA) by lipoxygenase (LOX) produces phosphatidylethanolamine-AA species, which is then oxidized by ferroptosis-inducing mechanisms. Ferroptosis is initiated by ACSL4 and LPCAT3, which make it easier to generate AA-PE species. This is in contrast to GPX4, which adversely regulates ferroptosis by reducing the formation of lipid hydroperoxides (L-OOH). Lipid peroxidation inhibitors, iron chelators, antioxidants, and LOX inhibitors can be used to prevent ferroptosis
Lipid peroxidation inhibitors
Ferrostatin-1 (Fer-1) decreases membrane lipid breakdown and cell death via a reductive mechanism (Skouta et al. 2014). Lipid peroxides can be converted to alcohols (R-OOHR-OH), or lipid groups can be intercepted and scavenged by direct reduction (R-OR-OH) or hydrogen atom transfer (Hu et al. 2019; Gupta et al. 2021). SRS16-86 (a third-generation ferrostatin) was tested on an in vitro model of renal IRI, which offered better plasma, metabolic stability, improved kidney function, and prolonged life (Pefanis et al. 2019). In fact, ferrostatin 16–86, a more stable homolog, was more successful in experimental kidney IRI than Fer-1 (Angeli et al. 2014). The ferroptosis inhibitor Liproxstatin-1 was also found to be effective in inhibiting human proximal tubule epithelium, Gpx4/kidney, and a model of IRI-induced tissue injury (Angeli et al. 2014; Ran et al. 2015; Kajarabille and Latunde-Dada 2019). Ferroptosis in RSL3-stimulated mouse striatal cells was suppressed by vitamin E and its metabolites, such as quinone/hydroquinone, even though this research was conducted before ferroptosis was defined. Still, the influence on ferroptosis was not examined (Zhou et al. 2020a, b). Vitamin E also significantly reduced lipid peroxidation of renal cells induced by renal I/R in rats (Hinman et al. 2018). I/R-induced tubular epithelial cell injury and inflammation were also decreased by XJB-5–131 in mice by particularly inhibiting ferroptosis (Zhao et al. 2020). Quercetin (QCT) is a natural flavonoid-containing compound that has been proven to reduce ferroptosis in acute renal injury in patients with IRI via reducing levels of malondialdehyde (MDA) and ROS in renal proximal tubular epithelial cells while raising levels of glutathione (GSH) (Hatcher et al. 2009; Wang et al. 2021a, b, c, d, e).
Ferroptosis can also be prevented by lipid peroxidase inhibitors, such as lipoxygenase (LOX) inhibitors. Humans have several lipoxygenase isoforms (12/15-LOX, 5-LOX) isoform, and 5-LOX is the only human lipoxygenase with 3D structural data that exacerbates the ferroptosis process (Yang et al. 2016). Despite this, scientists have worked hard to identify inhibitors that specifically target particular isoforms of the protein. A definitive association between ferroptosis and a specific lipoxygenase isoform has yet to be established. For example, 15-LOX, which encodes lipoxygenase, can oxidize esterified FA and is assumed to be responsible for cell death, did not repair Gpx4 loss in genetic tests (Mao et al. 2019). Although ablation of 15-LOX in heterozygous Gpx4 mice can treat male subfertility, this suggests that Gpx4 and lipoxygenase have a complicated tissue-specific interplay (Friedmann Angeli et al. 2014). As it turns out, blocking more than one lipoxygenase had a more significant protective effect (Wang et al. 2021a, b, c, d, e) than inhibiting just one. As a result, it is necessary to determine whether a particular lipoxygenase or pan-lipoxygenase inhibitor may be used to develop new therapies that can successfully reduce ferroptosis in renal I/R injury. Cell death in human fibrosarcoma cells and GPX/mouse embryonic fibroblasts exposed to erastin was reduced by PD146176, a 15-LOX inhibitor (Doll et al. 2017).
Iron chelators
Experimental AKI has been effectively treated with iron chelation, preventing ferroptosis (Paller et al. 1998). Iron deficiency can lead to cell death in the proximal tubular cells; hence, none of these are currently used in clinical practice. However, the effect of Deferoxamine was evaluated on iron-mediated postischemic renal injury in rats; Deferoxamine infused during the first 60 min of reperfusion resulted in a marked improvement in renal function and reduction in lipid peroxidation (Paller et al. 1998). A dose-adapted form of Deferiprone can, on the other hand, be put to the test (Finazzi and Arosio 2014). H-ferritin (FtH) is expressed in the kidney’s proximal tubules to sequester iron and decrease free iron-mediated toxicity efficiently. The ferritin-H (FTH) component’s ferroxidase activity transforms Fe2+ to Fe3+, which is then stored in the ferritin mineral core by ferroxidase activity. As an iron chelator, FtH is vital since each molecule can bind to up to 4500 Fe2+ ions. HO-1 increases the expression of FtH to store the ferrous iron produced throughout the process (Matzanke et al. 1997). This protection is also dependent on FtH being upregulated by HO-1. In addition, HO-1 must increase the expression of FtH for it to be preserved. A study used proximal tubule-specific FtH mutant mice to evaluate the role of FtH in rhabdomyolysis and cisplatin-induced AKI. The removal of FtH from proximal renal tubules exacerbated kidney injury and increased mortality, despite HO-1 expression being considerably higher (Swaminathan 2018). In AKI, Hepcidin induces H-ferritin and sequesters iron from macrophages to prevent ferroptosis (Ho et al. 2011). The iron chelators Deferasirox and Hepcidin must be studied in a preclinical model of renal I/R damage to establish their iron-chelating capacities and impact on ferroptosis inhibition (Table 1).
Emerging mechanism of ferroptosis in renal I/R injury
Pannexin signalling
An ATP-releasing pathway protein, pannexin, is found in every cell type. Pannexin is a membrane channel composed of three proteins (Panx1, Panx2, and Panx3). Panx1 regulates ATP release as a damage-associated molecular pattern (DAMP) molecule that activates autophagy signaling or apoptosis under oxidative conditions (Penuela and Gehi 2013). Pannexin has been reported to exhibit pro-apoptotic effects, inflammation, oxidative stress, and cell death during kidney injury. The mechanism of ferroptosis in Parkinson’s disease was triggered by ATP binding to the P2Y7 receptor, activating signaling pathways such as PKC and MAPK (Karatas et al. 2013). In particular, a decrease in ATP release-dependent signaling has been found to lower oxidative stress and protect kidneys during IRI (Xu et al. 2018). P2Y7R-binding Panx 1 stimulates cell death that can be reversed by inhibiting ferroptosis, according to studies (Xu et al. 2018). A recent study found that silencing Panx1 expression in cultured HK-2 cells treated with erastin significantly attenuated ferroptotic lipid peroxidation and iron accumulation.
Further, Panx1 knockout mice subjected to renal IRI displayed reduced malondialdehyde (MDA), plasma creatinine levels, and tubular cell death compared to wild-type mice. Moreover, knockdown of Panx1 in mice offered protection against renal IRI by inducing expression of heme oxygenase (HO-1) and attenuating ferroptinophagy via MAPK/ERK activation (Su et al. 2019). Therefore, Panx1 could be a potential therapeutic target for managing acute kidney injury (AKI) due to IRI.
miRNAs
Various genes regulate the state of lipid peroxidation and iron concentrations in cells. SLC7A11 decreases ferroptosis by transferring cystine moiety into the cytosol to enhance GSH production, while GPX4 reduces lipid peroxidation in cells (Ghini et al. 2018). To affect gene transcription, microRNAs (miRNAs) target the 3′UTR of mRNA22 (Diallo et al. 2021). Many biological processes rely on miRNA-mRNA interaction, including immune responses, cell growth, autophagy, and death. A growing number of research suggest that miRNAs may play functional roles by cooperating with other noncoding RNAs (Xia et al. 2008). For example, in pediatric T-ALL cells, the expression of tumor suppressor genes PTEN and BIM was regulated by miRNAs hsa-20b-5p and hsa-363-3p and modulated survival of T-ALL cells (Drobna et al. 2020). In I/R-induced renal damage, miRNAs can slow its progression by modifying the expression of genes associated with the injury (Zager et al. 2009). Dysregulation of miR-182-5p and miR-378a-3p resulted in ferroptosis in I/R-induced kidney damage due to reduced GPX4 and SLC7A11 expression (Zager et al. 2009). As GPX4 and HMOX-1 are the essential regulatory genes in the ferroptosis form of programmed cell death and also cause injury to renal epithelial cells during IR. A recent study investigated the miRNA-mRNA regulatory system involved in ferroptosis following renal IR in which the bioinformatics analysis revealed a significant upregulation of HMOX1 in the early stages of renal IR injury, and miRNA-3587 was found to be a regulator of HMOX-1. Inhibition of miR-3587 in tubular epithelial cells of hypoxia reoxygenation (HR) model system showed a significant increase in HO-1 protein (encoded by HMOX1) compared to the HR group, resulting in a simultaneous increase in GPX4 protein levels, decreased Fe2+ and malondialdehyde level, and restored standard mitochondrial membrane potential. Therefore, the results indicated that miR-3587 suppression promoted HO-1 upregulation and protected renal tissues from IR-induced ferroptosis (Tao et al. 2021).
Heme oxygenase-1
Heme oxygenase is a well-known ubiquitous enzyme with a wide range of potential therapeutic applications in various disorders. Three different isoforms of the enzyme heme oxygenase 1 (HO-1) have been acknowledged: HO-1, HO-2, and HO-3, with the latter being a splice variation of HO-2. Inflammation and oxidative stress cause cell damage, which can be reduced by regulating iron metabolism (Khan et al. 2022a). Intracellular iron absorption and heme breakdown are facilitated by heme oxygenase-1 (HO-1) (Khan et al. 2022a, b, c; Khan et al. 2020). The intracellular ferrous iron level decreases when HO-1 expression or enzymatic activity is decreased. Thus, HO-1 is usually regarded as an essential regulator of iron metabolism. Gene deletion and transgenic techniques have established that HO-1 protects against AKI. It has been found that selective overexpression of HO-1 mice in the proximal tubule level is protective, whereas HO-1 deletion results in ferroptosis (Schipper et al. 2009; Liu et al. 2020a, b). However, toxin-induced HO-1 overexpression damages the kidneys and results in mitochondrial dysfunction (Wang et al. 2021a, b, c, d, e). Iron levels in the blood are maintained by FPN, the only iron export protein in mammals. In AKI mice, the FPN gene was knocked out, and renal function improved, possibly due to reduced ferroptosis and FtH chelation of ferrous iron (Shiraishi et al. 2000; Nath 2014; Wang et al. 2018; Fang et al. 2020). It has been found that HO-1 regulates oxidative stress and autophagy in a range of animal models of kidney injury, including ischemia–reperfusion, lipopolysaccharide, and cisplatin. Inflammation is regulated by HO-1, a critical contributor in this process (Adedoyin et al. 2018; Khan et al. 2020). Global HO-1 deficit in renal IRI mice model has shown to promote myeloid cell trafficking and monocyte chemoattraction through increased MCP-1 and MCP-1 trafficking (Pittock et al. 2005).
On the other hand, macrophage’s overexpression with HO-1 in patients with renal IRI has shown anti-inflammatory properties and contributed to renal recovery (Zarjou et al. 2013). Other systems, such as proximal tubular ferritin, are required to protect cells against iron-induced damage and death. For example, iron generated by HO-1-catalytic activity can be stored more effectively when the heavy chain of ferritin is upregulated. The ability of HO-1 to upregulate H-ferritin is essential for protection (Zarjou et al. 2013). Researchers have studied the role of FtH in rhabdomyolysis and cisplatin-induced AKI in proximal tubule-specific mutant mice (Zarjou et al. 2013). Removing FtH from renal tubules has been shown to aggravate kidney damage and increase mortality, even though HO-1 expression is significantly higher in these tubules (Mohammad et al. 2021). Hepcidin has also modulated iron metabolism via FPN in AKI by restoring iron homeostasis and lowering inflammation. As the primary regulator of ferroportin-mediated iron export and intracellular H-ferritin levels, Hepcidin is essential for iron homeostasis. Hepcidin, a protective molecule, protects against acute kidney injury. Promising agents targeting Hepcidin, H-ferritin, and ferroptosis pathways could be an effective treatment strategy to prevent renal IRI (Mohammad et al. 2021). NRF2 activation is influenced by metabolic proteins, including ferritin and heme oxygenase 1 (HO-1), that control iron availability and ferroptosis (Nie et al. 2018). Current research revealed that HO-1 deletion in hepatocellular carcinoma and kidney cells enhanced erastin-induced ferroptosis (Nie et al. 2018; Kim et al. 2021). At the same time, NRF2 activation by ulinastatin upregulated HO-1 expression and reduced acetaminophen-induced liver I/R injury via alleviating ferroptosis, suggesting that acetaminophen induced ferroptosis via downregulation of the NRF2/HO-1 signaling pathway (Wang et al. 2021a, b, c, d, e). Therefore, NRF2 function needs to be evaluated for its beneficial effects against renal I/R damage via targeting ferroptosis.
Conclusion and perspective
Ferroptosis has emerged as a possible target for designing a novel treatment regimen for a wide range of disorders. Dysregulation in iron metabolism and ROS generation are the primary causes of ferroptosis. Several studies have shown that ferroptosis has a significant function in renal I/R models. Lipid peroxidase inhibitors and iron chelators have been shown to inhibit renal I/R damage ferroptosis. However, further therapeutic options for preventing ferroptosis in renal I/R conditions need to be developed. Renal ischemia/reperfusion (I/R) models have shown many ferroptosis regulators’ expressions, comprising LOX, SLC7A11, and FTH1. Preventing ferroptosis in renal I/R injury may be possible by using pharmaceutical drugs to alter these parameters. Humans have several lipoxygenase isoforms (12/15-LOX, 5-LOX), and blocking one of these enzymes may prevent ferroptosis. Therefore, lipid peroxidase inhibitors may be evaluated in renal I/R models that may successfully reduce ferroptosis. PD146176, a 15 LOX inhibitor, had reduced cell death in GPX/mouse embryonic fibroblasts and human fibrosarcoma cells exposed to erastin. Hence, preclinical evaluation of these inhibitors in the renal I/R model should also be done. More understanding and identification of molecular mechanisms of ferroptosis in renal I/R models is required to assess the effect of modulating these mechanisms with the desired therapeutic agent. In renal I/R models, specific indicators for ferroptosis, such as caspase activation in the event of apoptosis and the development of autophagy lysosomes in the case of autophagy, have yet to be developed. Therefore, ferroptosis inhibition could be a promising strategy to reduce renal I/R damage and should be evaluated in preclinical and clinical platforms.
Data availability
Not applicable.
References
Adam AC, Bornhövd C, Prokisch H, Neupert W (2006) Hell K The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO Rep 25(1):174–183. https://doi.org/10.1038/2Fsj.emboj.7600905
Adedoyin O, Boddu R, Traylor A et al (2018) Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol 314(5):F702–F714. https://doi.org/10.1152/ajprenal.00044.2017
Angeli JPF, Proneth B, Hammond VJ et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in a therapeutically relevant mechanism. Free Radic Biol Med 76:S77–S78. https://doi.org/10.1016/2Fj.freeradbiomed.2014.10.276
Aoyama K, Watabe M, Nakaki T (2008) Regulation of neuronal glutathione synthesis. J Pharmacol Sci 0811120169-. https://doi.org/10.1254/jphs.08R01CR.
Bagayoko S, Meunier E (2021) Emerging roles of ferroptosis in infectious diseases. FEBS Lett. https://doi.org/10.1111/febs.16244
Barayuga SM, Pang X, Andres MA, Panee J (2013) Bellinger FP (2013) Methamphetamine decreases levels of glutathione peroxidases 1 and 4 in SH-SY5Y neuronal cells: protective effects of selenium. Neurotoxicology 37:240–246. https://doi.org/10.1016/j.neuro.2013.05.009
Bersuker K, Hendricks JM, Li Z et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575(7784):688–692. https://doi.org/10.1038/s41586-019-1705-2
Bertrand RL (2017) Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med Hypotheses 101:69–74. https://doi.org/10.1016/j.mehy.2017.02.017
Brigelius-Flohé R, Maiorino M (2013) Glutathione peroxidases. Biochim Biophys Acta Gen Subj 1830(5):3289–3303. https://doi.org/10.1016/j.bbagen.2012.11.020
Capelletti MM, Manceau H, Puy H, Peoc’h K (2020) Ferroptosis in liver diseases: an overview. Int J Mol Sci 21(14):4908. https://www.mdpi.com/1422-0067/21/14/4908.
Cardoso BR, Hare DJ, Bush AI, Roberts BR (2017) Glutathione peroxidase 4: a new player in neurodegeneration? Mol Psychiatry 22(3):328–335. https://doi.org/10.1038/mp.2016.196
Carvalho EBTD, Melo ILPD, Mancini-Filho J (2010) Chemical and physiological aspects of isomers of conjugated fatty acids. Food Sci Tech 30:295–307. https://doi.org/10.1590/S0101-20612010000200002
Casañas-Sánchez V, Pérez JA, Fabelo N, Quinto-Alemany D, Diaz ML (2015) Docosahexaenoic (DHA) modulates phospholipid-hydroperoxide glutathione peroxidase (Gpx4) gene expression to ensure self-protection from oxidative damage in hippocampal cells. Front Physiol 6:203. https://doi.org/10.3389/fphys.2015.00203
Casserly LF, Dember LM (2003) Thrombosis in end-stage renal disease. In Semin Dial (Vol. 16, No. 3, pp. 245–256). Malden, USA: Blackwell Science Inc. https://doi.org/10.1046/j.1525-139X.2003.16048.x.
Chen X, Li J, Kang R, Klionsky DJ, Tang D (2002) Ferroptosis: machinery and regulation. Autophagy 17(9):2054–2081. https://doi.org/10.1080/15548627.2020.1810918
Chen X, Yu C, Kang R, Tang D (2020) Iron metabolism in ferroptosis. Front Cell Dev Biol 2020:1089. https://doi.org/10.3389/2Ffcell.2020.590226
Chen J, Yang L, Geng L et al (2021) Inhibition of Acyl-CoA synthetase long-chain family member 4 facilitates neurological recovery after stroke by regulation ferroptosis. Front Cell Neurosci 15:93. https://doi.org/10.3389/fncel.2021.632354
Chen Y, Liu S, Li J,et al (2020b)The latest view on the mechanism of ferroptosis and its research progress in spinal cord injury. Oxid Med Cell Longev. 2020b. https://doi.org/10.1155/2020/6375938
Choi N, Whitlock R, Klassen J et al (2019) Early intraoperative iron-binding proteins are associated with acute kidney injury after cardiac surgery. J Thorac Cardiovasc Surg 157(1):287–297. https://doi.org/10.1016/j.jtcvs.2018.06.091
Cicenas J, Tamosaitis L, Kvederaviciute K et al (2017) KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma. Med Oncol 34(2):1–1. https://doi.org/10.1007/s12032-016-0879-9
Conrad M, Sato H (2012) The oxidative stress-inducible cystine/glutamate antiporter, system xc−: cystine supplier and beyond. Amino Acids 42(1):231–246. https://doi.org/10.1007/s00726-011-0867-5
Diallo I, Ho J, Laffont B, Laugier J et al (2021) Altered microRNA transcriptome in cultured human liver cells upon infection with ebola virus. Int J Mol Sci 22(7):3792. https://doi.org/10.3390/cells9051137
Dixon SJ, Stockwell BR (2019) The hallmarks of ferroptosis. Annu Rev Cancer Biol 3:35–54. https://doi.org/10.1146/annurev-cancerbio-030518-055844
Dixon SJ, Lemberg KM, Lamprecht MR et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell J 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Doll S, Proneth B, Tyurina YY et al (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13(1):91–98. https://doi.org/10.1038/nchembio.2239
Doll S, Freitas FP, Shah R et al (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575(7784):693–698. https://doi.org/10.1038/s41586-019-1707-0
Drobna M, Szarzyńska B, Jaksik R, Sędek Ł, Kuchmiy A, Taghon T, Van Vlierberghe P, Szczepański T, Witt M, Dawidowska M (2020) hsa-miR-20b-5p and hsa-miR-363-3p affect expression of PTEN and BIM tumor suppressor genes and modulate survival of T-ALL cells in vitro. Cells 9(5):1137. https://doi.org/10.3390/cells9051137
Fang X, Cai Z, Wang H et al (2020) Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res 127(4):486–501. https://doi.org/10.1161/circresaha.120.316509
Finazzi D, Arosio P (2014) Biology of ferritin in mammals: an update on iron storage, oxidative damage and neurodegeneration. Arch Toxicol 88(10):1787–1802. https://doi.org/10.1007/s00204-014-1329-0
Franklin CC, Backos DS, Mohar I et al (2009) Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol Asp Med 30(1–2):86–98. https://doi.org/10.1016/j.mam.2008.08.009
Frei B, Kim MC, Ames BN (1990) Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc Natl Acad Sci USA 87(12):4879–4883. https://doi.org/10.1073/pnas.87.12.4879
Friedmann Angeli JP, Schneider M, Proneth B et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16(12):1180–1191. https://doi.org/10.1038/ncb3064
Fujii J, Homma T, Kobayashi S (2020) Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic Res 54(11–12):969–980. https://doi.org/10.1080/10715762.2019.1666983
Ghini F, Rubolino C, Climent M, Simeone I, Marzi MJ, Nicassio F (2018) Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat Commun 9(1):1–5. https://doi.org/10.1038/s41467-018-05182-9
Gryzik M, Asperti M, Denardo A, Arosio P, Poli M (2021) NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim Biophys Acta Mol Cell Res 1868(2):118913. https://doi.org/10.1016/j.bbamcr.2020.118913
Gupta A, Khan H, Kaur A, Singh TG (2021) Novel targets explored in the treatment of alcohol withdrawal syndrome. CNS Neurol Disord - Drug Targets 20(2):158–173. https://doi.org/10.2174/1871527319999201118155721
Haase M, Bellomo R, Haase-Fielitz A (2010) Novel biomarkers, oxidative stress, and the role of labile iron toxicity in cardiopulmonary bypass-associated acute kidney injury. J Am Coll Cardiol 55(19):2024–2033. https://doi.org/10.1016/j.jacc.2009.12.046
Han C, Liu Y, Dai R, Ismail N, Su W, Li B (2020) Ferroptosis and its potential role in human diseases. Front Pharmacol 11:239. https://doi.org/10.3389/fphar.2020.00239
Hatcher HC, Singh RN, Torti FM, Torti SV (2009) Synthetic and natural iron chelators: therapeutic potential and clinical use. Future Med Chem 1(9):1643–1670. https://doi.org/10.4155/fmc.09.121
Hayashima K, Kimura I, Katoh H (2021) Role of ferritinophagy in cystine deprivation-induced cell death in glioblastoma cells. Biochem Biophys Res Commun 539:56–63. https://doi.org/10.1016/j.bbrc.2020.12.075
He L, Liu YY, Wang K et al (2021) Tanshinone IIA protects human coronary artery endothelial cells from ferroptosis by activating the NRF2 pathway. Biochem Biophys Res Commun 575:1–7. https://doi.org/10.3389/fcell.2020.586578
Hinman A, Holst CR, Latham JC et al (2018) Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS One 13(8):e0201369. https://doi.org/10.1371/journal.pone.0201369
Hirschhorn T, Stockwell BR (2019) The development of the concept of ferroptosis. Free Radic Biol Med 133:130–43. https://doi.org/10.1016/2Fj.freeradbiomed.2018.09.043
Ho J, Reslerova M, Gali B et al (2011) Urinary hepcidin-25 and risk of acute kidney injury following cardiopulmonary bypass. Clin J Am Soc Nephrol 6(10):2340–2346. https://doi.org/10.2215/cjn.01000211
Hou W, Xie Y, Song X, et al (2016) Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12(8):1425–8. https://doi.org/10.1080/2F15548627.2016.1187366. https://doi.org/10.1007/s00204-013-1110-9
Hu Z, Zhang H, Yang SK, Wu X, He D, Cao K, Zhang W (2019) Emerging role of ferroptosis in acute kidney injury. Oxid Med Cell Longev. https://doi.org/10.1155/2F2019%2F8010614
Ighodaro OM, Akinloye OA (2018) First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defence grid. Alexandria J Med 54(4):287–293. https://doi.org/10.1016/j.ajme.2017.09.001
Ingold I, Berndt C, Schmitt S et al (2018) Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell J 172(3):409–422. https://doi.org/10.1016/j.cell.2017.11.048
Jiang X, Stockwell BR, Conrad M (2021) Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22(4):266–282. https://doi.org/10.1038/s41580-020-00324-8
Jomova K, Valko M (2011) Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr Pharm Des 17(31):3460–3473. https://doi.org/10.2174/138161211798072463
Kajarabille N, Latunde-Dada GO (2019) Programmed cell-death by ferroptosis: antioxidants as mitigators. Int J Mol Sci 20(19):4968. https://doi.org/10.3390/ijms20194968
Kalra P, Khan H, Kaur A, Singh TG (2022) Mechanistic insight on autophagy modulated molecular pathways in cerebral ischemic injury: from preclinical to clinical perspective. Neurochem Res 7:1–9. https://doi.org/10.1007/s11064-021-03500-0
Karatas H, Erdener SE, Gursoy-Ozdemir Y et al (2013) Spreading depression triggers headache by activating neuronal Panx1 channels. J Sci 339(6123):1092–1095. https://doi.org/10.1126/science.1231897
Khan H, Kashyap A, Kaur A, Singh TG (2020) Pharmacological postconditioning: a molecular aspect in ischemic injury. J Pharm Pharmacol 72(11):1513–1527. https://doi.org/10.1111/jphp.13336
Khan H, Bangar A, Grewal AK, Bansal P, Singh TG (2022) Caspase-mediated regulation of the distinct signaling pathways and mechanisms in neuronal survival. Int Immunopharmacol 110:108951. https://doi.org/10.1016/j.intimp.2022.108951
Khan H, Garg N, Singh TG, Kaur A, Thapa K (2022b) Calpain inhibitors as potential therapeutic modulators in neurodegenerative diseases. Neurochem Res 1-25. https://doi.org/10.1007/s11064-021-03521-9
Khan H, Sharma K, Kumar A, Kaur A, Singh TG (2022c) Therapeutic implications of cyclooxygenase (COX) inhibitors in ischemic injury. Inflamm Res 1-6. https://doi.org/10.1007/s00011-022-01546-6
Kihara A (2012) Very long-chain fatty acids: elongation, physiology and related disorders. J Biochem 152(5):387–395. https://doi.org/10.1093/jb/mvs105
Kim S, Kang SW, Joo J, Han SH et al (2021) Correction: characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis 12(4):1–1. https://doi.org/10.1038/s41419-021-03667-y
Lewerenz J, Hewett SJ, Huang Y et al (2013) The cystine/glutamate antiporter system xc− in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 18(5):522–55. https://doi.org/10.1089/2Fars.2011.4391
Li J, Cao F, Yin HL,et al (2020) Ferroptosis: past, present and future. Cell Death Dis 11(2). https://doi.org/10.1038/s41419-020-2298-2
Li D, Li Y (2020) The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther 5(1):1. https://doi.org/10.1038/s41392-020-00216-5
Liang C, Zhang X, Yang M et al (2019) Recent progress in ferroptosis inducers for cancer therapy. J Adv Mater 31(51):1904197. https://doi.org/10.1002/adma.201904197
Lin Z, Liu J, Kang R, Yang M, Tang D (2021) Lipid metabolism in ferroptosis. Front Biol (beijing) 5(8):2100396. https://doi.org/10.1002/adbi.202100396
Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z (2014) Regulated cell death in AKI. J Am Soc Nephrol 25(12):2689–2701
Linkermann A, Skouta R, Himmerkus N et al (2014) Synchronized renal tubular cell death involves ferroptosis. PNAS USA 111(47):16836–16841. https://doi.org/10.1073/pnas.1415518111
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D (2020) Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol 27(4):420–435. https://doi.org/10.1016/j.chembiol.2020.02.005
Liu J, Zhang C, Wang J, Hu W, Feng Z (2020) The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int J Mol Sci 21(21):8387. https://doi.org/10.3390/ijms21218387
Malek M, Nematbakhsh M (2015) Renal ischemia/reperfusion injury; from pathophysiology to treatment. J renal inj prev 4(2):20. https://doi.org/10.12861/jrip.2015.06
Mao XY, Jin MZ, Li Q et al (2019) Lysyl oxidase promotes neuronal ferroptosis exacerbating seizure-induced hippocampal damage. BioRxiv 1:839852. https://doi.org/10.1101/839852
Matzanke BF, Winkelmann G, Carrano C (1997) Iron storage in microorganisms. Transition metals in microbial metabolism. Harwood Acad Publishers GmbH Amst Neth 23:117–158
Mohammad G, Matakidou A, Robbins PA, Lakhal-Littleton S (2021) The kidney hepcidin/ferroportin axis controls iron reabsorption and determines the magnitude of kidney and systemic iron overload. Kidney Int 100(3):559–569. https://doi.org/10.1016/j.kint.2021.04.034
Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ (2016) The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer 16(11):718–731. https://doi.org/10.1038/nrc.2016.76
Müller T, Dewitz C, Schmitz J et al (2017) Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci 74(19):3631–3645. https://doi.org/10.1007/s00018-017-2547-4
Nath KA (2014) Heme oxygenase-1 and acute kidney injury. Curr Opin Nephrol Hypertens 23(1):17. https://doi.org/10.1097/2F01.mnh.0000437613.88158.d3
Nehring H, Meierjohann S, Friedmann Angeli JP (2020) Emerging aspects in the regulation of ferroptosis. Biochem Soc Trans 48(5):2253–2259. https://doi.org/10.1042/BST20200523
Ng SW, Norwitz SG, Norwitz ER (2019) The impact of iron overload and ferroptosis on reproductive disorders in humans: implications for preeclampsia. Int J Mol Sci 20(13):3283. https://doi.org/10.3390/ijms20133283
Ni HM, Chao X, Kaseff J et al (2019) Receptor-interacting serine/threonine-protein kinase 3 (RIPK3)–mixed lineage kinase domain-like protein (MLKL)–mediated necroptosis contributes to ischemia-reperfusion injury of steatotic livers. Am J Clin Pathol 189(7):1363–1374. https://doi.org/10.1016/j.ajpath.2019.03.010
Nie J, Lin B, Zhou M, Wu L, Zheng T (2018) Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol 144(12):2329–2337. https://doi.org/10.1007/s00432-018-2740-3
Olzmann JA, Carvalho P (2019) Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 20(3):137–155. https://doi.org/10.1038/s41580-018-0085-z
Paller MS, Hedlund BE (1994) Extracellular iron chelators protect kidney cells from hypoxia/reoxygenation. Free Radic Biol Med 17(6):597–603. https://doi.org/10.1016/0891-5849(94)90099-X
Paller MS, Hedlund BE, Sikora JJ, Faassen A, Waterfield R (1998) Role of iron in postischemic renal injury in the rat. Kidney Int 4(4):474–480. https://doi.org/10.1038/ki.1988.205
Pefanis A, Ierino FL, Murphy JM, Cowan PJ (2019) Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int 96(2):291–301. https://doi.org/10.1016/j.kint.2019.02.009
Penuela S, Gehi R, Laird DW (2013) The biochemistry and function of pannexin channels. Biochim Biophys Acta Biomembr 1828(1):15–22. https://doi.org/10.1016/j.bbamem.2012.01.017
Pittock ST, Norby SM, Grande JP et al (2005) MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: pathophysiologic correlates. Kidney Int 68:611–622. https://doi.org/10.1111/j.1523-1755.2005.00439.x
Ran Q, Chen L, Hambright WS, Na R (2015) Ablation of GPX4 in neurons results in rapid motor neuron degeneration and paralysis. Free Radic Biol Med 1(87):S34. https://doi.org/10.1016/2Fj.freeradbiomed.2015.10.093
Rodríguez-Vargas JM, Oliver-Pozo FJ, Dantzer F (2019) PARP1 and poly (ADP-ribosyl) ation signaling during autophagy in response to nutrient deprivation. Oxid Med Cell Longev. https://doi.org/10.1016/j.mam.2013.01.007
Saklani P, Khan H, Gupta S, Kaur A, Singh TG (2022) Neuropeptides: potential neuroprotective agents in ischemic injury. Life Sci 288:120186. https://doi.org/10.1016/j.lfs.2021.120186
Saklani P, Khan H, Singh TG, Gupta S, Grewal AK (2022) Demethyleneberberine, a potential therapeutic agent in neurodegenerative disorders: a proposed mechanistic insight. Mol Biol Rep 3:1–3. https://doi.org/10.1007/s11033-022-07594-9
Sarkar S, Chakraborty D, Bhowmik A, Ghosh MK (2019) Cerebral ischemic stroke: cellular fate and therapeutic opportunities. Front Biosci 24:435–450. https://doi.org/10.2741/4727
Schipper HM, Song W, Zukor H, Hascalovici JR, Zeligman D (2009) Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J Neurochem 110(2):469–485. https://doi.org/10.1111/j.1471-4159.2009.06160.x
Scindia Y, Dey P, Thirunagari A et al (2015) Hepcidin mitigates renal ischemia-reperfusion injury by modulating systemic iron homeostasis. J Am Soc Nephrol 26(11):2800–2814. https://doi.org/10.1681/asn.2014101037
Sharma S, Leaf DE (2019) Iron chelation as a potential therapeutic strategy for AKI prevention. J Am Soc Nephrol 30(11):2060–2071. https://doi.org/10.1681/ASN.2019060595
Sharma A, Khan H, Singh TG, Grewal AK, Najda A, Kawecka-Radomska M, Kamel M, Altyar AE, Del-Daim MM (2021) Pharmacological modulation of ubiquitin-proteasome pathways in oncogenic signaling. Int J Mol Sci 22(21):11971. https://doi.org/10.3390/ijms222111971
Shimada K, Skouta R, Kaplan A et al (2016) Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12(7):497–503. https://doi.org/10.1038/nchembio.2079
Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A (2000) Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 278:F726–F736. https://doi.org/10.1152/ajprenal.2000.278.5.f726
Skouta R, Dixon SJ, Wang J et al (2014) Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136(12):4551–4556. https://doi.org/10.1021/ja411006a
Song E, Su C, Fu J et al (2014) Selenium supplementation shows protective effects against patulin-induced brain damage in mice via increases in GSH-related enzyme activity and expression. Life Sci 109(1):37–43. https://doi.org/10.1016/j.lfs.2014.05.022
Su L, Jiang X, Yang C, Zhang J et al (2019) Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. Int J Biol Chem 294(50):19395–19404. https://doi.org/10.1074/jbc.ra119.010949
Swaminathan S (2018) Iron homeostasis pathways as therapeutic targets in acute kidney injury. Nephron 140(2):156–159. https://doi.org/10.1159/000490808
Tang D, Chen X, Kang R, Kroemer G (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31(2):107–125. https://doi.org/10.1038/s41422-020-00441-1
Tao N, Li K, Liu J (2020) Molecular mechanisms of ferroptosis and its role in pulmonary disease. Oxid Med Cell Longev 2020. https://doi.org/10.1155/2020/9547127
Tao W, Liu F, Zhang J, Fu S, Zhan H, Qian K (2021) miR-3587 Inhibitor attenuates ferroptosis following renal ischemia-reperfusion through HO-1. Front Mol Biosci 8. https://doi.org/10.3389/2Ffmolb.2021.789927
Thapa K, Khan H, Kanojia N, Singh TG, Kaur A, Kaur G (2022) Therapeutic insights on ferroptosis in Parkinson’s disease. Eur J Pharmacol 175133. https://doi.org/10.1016/j.ejphar.2022.175133
Thévenod F, Lee WK (2013) Cadmium and cellular signaling cascades: interactions between cell death and survival pathways. Arch Toxicol 87(10):1743–1786
Tosatto SC, Bosello V, Fogolari F et al (2008) The catalytic site of glutathione peroxidases. Antioxid Redox Signal 10(9):1515–1526. https://doi.org/10.1089/ars.2008.2055
Ursini F, Maiorino M (2020) Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med 152:175–185. https://doi.org/10.1016/j.freeradbiomed.2020.02.027
Wang Y, Bellomo R (2017) Cardiac surgery-associated acute kidney injury: risk factors, pathophysiology and treatment. Nat Rev Nephrol 13(11):697–711. https://doi.org/10.1038/nrneph.2017.119
Wang X, Zheng X, Zhang J, Zhao S, Wang Z, Wang F, Shang W, Barasch J, Qiu A (2018) Physiological functions of ferroportin in the regulation of renal iron recycling and ischemic acute kidney injury. Am J Physiol Renal Physiol 315(4):F1042–F1057. https://doi.org/10.1152/ajprenal.00072.2018
Wang C, Liu T, Tong Y, Cui R, Qu K, Liu C, Zhang J (2021) Ulinastatin protects against acetaminophen-induced liver injury by alleviating ferroptosis via the SIRT1/NRF2/HO-1 pathway. Am J Trans Res 13(6):6031
Wang H, Cheng Y, Mao C et al (2021) Emerging mechanisms and targeted therapy of ferroptosis in cancer. Mol Ther 29(7):2185–2208. https://doi.org/10.1016/j.ymthe.2021.03.022
Wang Q, Zhan S, Han F, Liu Y, Wu H, Huang Z (2021) The possible mechanism of physiological adaptation to the low-Se diet and its health risk in the traditional endemic areas of Keshan diseases. Biol Trace Elem Res 8:1–5. https://doi.org/10.1074/jbc.m116.738930
Wang Y, Quan F, Cao Q et al (2021) Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J Adv Res 28:231–243. https://doi.org/10.1016/j.jare.2020.07.007
Wang J, Liu Y, Wang Y, Sun L (2021c) The cross-link between ferroptosis and kidney diseases. Oxid Med Cell Longev 2021c https://doi.org/10.1155/2021/6654887
Wu M, Xu LG, Li X, Zhai Z, Shu HB (2002) AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. Int J Biol Chem 277(28):25617–25623. https://doi.org/10.1074/jbc.m202285200
Wu JR, Tuo QZ, Lei P (2018) Ferroptosis, a recent defined form of critical cell death in neurological disorders. Mol Neurobiol 66(2):197–206. https://doi.org/10.1007/s12031-018-1155-6
Xia L, Zhang D, Du R et al (2008) miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer 123(2):372–379. https://doi.org/10.1002/ijc.23501
Xu J, Chen L, Li L (2018) Pannexin hemichannels: a novel promising therapy target for oxidative stress related diseases. J Cell Physiol 233(3):2075–2090. https://doi.org/10.1002/jcp.25906
Yan HF, Tuo QZ, Yin QZ, Lei P (2020) The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool Res 41(3):220. https://doi.org/10.24272/j.issn.2095-8137.2020.042
Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26(3):165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. PNAS 113(34):E4966–E4975. https://doi.org/10.1073/pnas.1603244113
Yarishkin O, Phuong TT, Bretz CA et al (2018) TREK-1 channels regulate pressure sensitivity and calcium signaling in trabecular meshwork cells. J Gen Physiol 150(12):1660–1675. https://doi.org/10.1172/jci44927
Yu L, Huang B, Po SS et al (2017) Low-level tragus stimulation for the treatment of ischemia and reperfusion injury in patients with ST-segment elevation myocardial infarction: a proof-of-concept study. JACC Cardiovasc Interv 10(15):1511–1520. https://doi.org/10.1016/j.jcin.2017.04.036
Zager RA, Johnson AC, Lund S (2009) Uremia impacts renal inflammatory cytokine gene expression in the setting of experimental acute kidney injury. Am J Physiol 297(4):F961–F970. https://doi.org/10.1038/s41419-020-03135-z
Zárate R, el Jaber-Vazdekis N, Tejera N, Pérez JA, Rodríguez C (2017) Significance of long chain polyunsaturated fatty acids in human health. Clin Transl Med 6(1):1–9. https://doi.org/10.1186/s40169-017-0153-6
Zarjou A, Bolisetty S, Joseph R et al (2013) Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J Clin Investig 123(10):4423–4434. https://doi.org/10.1172/JCI67867/
Zhao Z, Wu J, Xu H et al (2020) XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia− reperfusion injury. Cell Death Dis 11(8):1–5. https://doi.org/10.1038/s41419-020-02871-6
Zhou RP, Chen Y, Wei X et al (2020) Novel insights into ferroptosis: Implications for age-related diseases. Theranostics 10(26):11976. https://doi.org/10.7150/thno.50663
Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D (2020a) Ferroptosis is a type of autophagy-dependent cell death. Annu Rev Cancer Biol (Vol. 66, pp. 89–100). Academic Press. https://doi.org/10.1016/j.semcancer.2019.03.002.
Zou Y, Li H, Graham ET et al (2020) Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16(3):302–9. https://doi.org/10.1038/2Fs41589-020-0472-6
Acknowledgements
The authors extend their appreciation to the Chitkara University Punjab for support.
Author information
Authors and Affiliations
Contributions
Conceptualization: conceived and designed the experiments: Thakur Gurjeet Singh. Analyzed the data: Amarjot Kaur. Wrote the manuscript: Komal Thapa. Visualization: Amarjot Kaur. Editing of the manuscript: Komal Thapa, Amarjot Kaur, and Thakur Gurjeet Singh. Critically reviewed the article: Thakur Gurjeet Singh. Supervision: Thakur Gurjeet Singh. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Compliance with ethical standards
Not applicable.
Consent to participate
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Thapa, K., Singh, T.G. & Kaur, A. Targeting ferroptosis in ischemia/reperfusion renal injury. Naunyn-Schmiedeberg's Arch Pharmacol 395, 1331–1341 (2022). https://doi.org/10.1007/s00210-022-02277-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-022-02277-5