
Abstract
The pharmacological profile of ipragliflozin (ASP1941; (1S)-1,5-anhydro-1-C-{3-[(1-benzothiophen-2-yl)methyl]-4-fluorophenyl}-d-glucitol compound with l-proline (1:1)), a novel SGLT2 selective inhibitor, was investigated. In vitro, the potency of ipragliflozin to inhibit SGLT2 and SGLT1 and stability were assessed. In vivo, the pharmacokinetic and pharmacologic profiles of ipragliflozin were investigated in normal mice, streptozotocin-induced type 1 diabetic rats, and KK-Ay type 2 diabetic mice. Ipragliflozin potently and selectively inhibited human, rat, and mouse SGLT2 at nanomolar ranges and exhibited stability against intestinal glucosidases. Ipragliflozin showed good pharmacokinetic properties following oral dosing, and dose-dependently increased urinary glucose excretion, which lasted for over 12 h in normal mice. Single administration of ipragliflozin resulted in dose-dependent and sustained antihyperglycemic effects in both diabetic models. In addition, once-daily ipragliflozin treatment over 4 weeks improved hyperglycemia with a concomitant increase in urinary glucose excretion in both diabetic models. In contrast, ipragliflozin at pharmacological doses did not affect normoglycemia, as was the case with glibenclamide, and did not influence intestinal glucose absorption and electrolyte balance. These results suggest that ipragliflozin is an orally active SGLT2 selective inhibitor that induces sustained increases in urinary glucose excretion by inhibiting renal glucose reabsorption, with subsequent antihyperglycemic effect and a low risk of hypoglycemia. Ipragliflozin has, therefore, the therapeutic potential to treat hyperglycemia in diabetes by increasing glucose excretion into urine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes is a metabolic disorder that results in hyperglycemia caused by a deficiency in the secretion and/or action of insulin. According to its pathophysiology, diabetes is classified into two categories: type 1 and type 2. Impaired insulin secretion is the major cause of type 1 diabetes, while both impaired insulin secretion and insulin resistance play important roles in the pathophysiology of type 2 diabetes. Irrespective of the type of diabetes, chronic hyperglycemia leads to glucose toxicity, which further worsens diabetic symptoms (Andrews et al. 1984; Prentki and Nolan 2006). In addition, high incidences of cardiovascular diseases and complications (e.g., nephropathy, neuropathy, and retinopathy) are associated with chronic hyperglycemia (Crofford 1995). Therefore, therapeutic strategies for diabetes are currently focused on controlling blood glucose levels.
To date, several oral drugs that enhance insulin secretion and/or improve insulin sensitivity have been developed for the treatment of diabetes. However, due to limited efficacy and adverse side effects, it is difficult to maintain good glycemic control in most diabetic patients. Therefore, additional agents, especially those that work independently of insulin secretion, are needed for the successful management of diabetes. Among these, the inhibitors of sodium–glucose cotransporter (SGLT) are an attractive option because their modes of action are connected to blood glucose reduction independently from insulin secretion (Idris and Donnelly 2009).
SGLTs exist in at least six subtypes. Among these, physiological and pathophysiological roles of two major SGLT isoforms, SGLT1 and SGLT2, are well investigated, while the roles of an additional four isoforms (SGLT3−6) are presently poorly identified (Wright and Turk 2004; Wright et al. 2007). SGLT2 is a low-affinity, high-capacity transporter that is specifically expressed in the kidneys (Wells et al. 1992; Wright 2001). Mutations in the SGLT2 gene are associated with familial renal glucosuria, a benign syndrome in which glucose excretion occurs in the absence of hyperglycemia (van den Heuvel et al. 2002); thus, SGLT2 plays a critical role in renal glucose reabsorption. In contrast, SGLT1 is a high-affinity, low-capacity transporter that is highly expressed in the small intestine (Pajor and Wright 1992). Mutations in the SGLT1 gene cause glucose/galactose malabsorption; therefore, SGLT1 plays an important role in dietary glucose absorption in the small intestine (Turk et al. 1991).
Phlorizin, a naturally occurring phenol glucoside first isolated from the bark of an apple tree, has a competitive inhibitory activity for SGLT1 and SGLT2. Early animal studies showed that administration of phlorizin induces glucosuria by inhibiting renal glucose reabsorption and ameliorates hyperglycemia in diabetic models without a risk of hypoglycemia (Rossetti et al. 1987a; Ehrenkranz et al. 2005). Phlorizin has also been found to decrease insulin resistance at the peripheral tissue level due to the improvement in hyperglycemia (Abdul-Ghani and DeFronzo 2008). Therefore, the renal glucose reabsorption system could be an important target for improving hyperglycemia in diabetes. However, phlorizin was not developed as a drug for the treatment of diabetes due to its nonselectivity and low bioavailability (Oku et al. 2000). Nonetheless, various orally active and selective SGLT2 inhibitors have been subsequently developed (Oku et al. 1999; Han et al. 2008). One of the approaches to improve bioavailability is to replace the unstable anomeric oxygen atom of the exo-glycosidic bond by a stable carbon atom (C-glycoside), as reported by two independent institutes including us in 2001 (Washburn 2009). In recent years, it has been demonstrated that some SGLT2 inhibitors, such as dapagliflozin, improve hyperglycemia in type 2 diabetic patients (List et al. 2009).
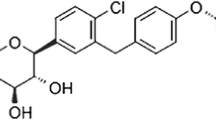
We have identified a novel orally active and selective SGLT2 inhibitor ipragliflozin (ASP1941; (1S)-1,5-anhydro-1-C-{3-[(1-benzothiophen-2-yl)methyl]-4-fluorophenyl}-d-glucitol compound with l-proline (1:1); Fig. 1a), which is a derivative of phlorizin (Fig. 1b). In this report, we describe the in vitro and in vivo pharmacology of ipragliflozin currently under clinical investigation for use as an antidiabetic agent.

Chemical structures of a ipragliflozin ((1S)-1,5-anhydro-1-C-{3-[(1-benzothiophen-2-yl)methyl]-4-fluorophenyl}-d-glucitol compound with l-proline (1:1)) and b phlorizin
Materials and methods
Materials
Ipragliflozin, dapagliflozin (Meng et al. 2008), and YM471 ([Z-4′-[4,4-difluoro-5-{2-(4-dimethylaminopiperidino)-2-oxoethylidene}-2,3,4,5-tetrahydro-1H-1-benzoazepine-1-carbonyl]-2-phenylbenzanilide monohydrochloride) (Tsukada et al. 2001) were synthesized by Astellas Pharma Inc. (Ibaraki, Japan). Phlorizin, phloretin, methyl-α-d-glucopyranoside (AMG), streptozotocin (STZ), glibenclamide, and furosemide were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Voglibose (BASEN®) was purchased from Takeda Pharmaceutical Company, Ltd. (Osaka, Japan). These drugs were dissolved or suspended in 0.5% methylcellulose solution and administered orally. The doses of ipragliflozin are expressed as the free base form. 14C-AMG (11.7 GBq/mmol) was obtained from Amersham Pharmacia Biotech (Little Chalfont, Buckinghamshire, UK).
Animals
Male Sprague–Dawley rats were purchased from Charles River Laboratories Japan (Kanagawa, Japan) at age 5–7 weeks. STZ was dissolved in 50 mM citric acid buffer before intravenous administration at 50 mg/kg. Normal control rats were given physiological saline. Blood glucose levels were measured in the diabetic rats 1 week later, after which the rats were grouped such that the blood glucose levels were similar in each group. Male Institute of Cancer Research normal and KK-Ay type 2 diabetic mice, which exhibit hyperglycemia, insulin resistance, hyperinsulinemia, hyperlipidemia, and obesity, were purchased from Japan SLC, Inc. (Shizuoka, Japan) and CLEA Japan (Kanagawa, Japan), respectively, at the age of 5–6 weeks. The diabetic mice were also grouped such that each group had similar blood glucose levels. All animals were housed under conventional conditions with controlled temperature, humidity, and light (12-h light–dark cycle) and were provided with a standard commercial diet and water (ad libitum). Animals were handled and cared for in accordance with the Guide for the Care and Use of Laboratory Animals, and all procedures were approved by the Animal Ethical Committee of Astellas Pharma Inc.
SGLT2 and SGLT1 inhibition assay
Human, rat, and mouse SGLT2 and SGLT1 full-length complementary deoxyribonucleic acid sequences were cloned and stably transfected into Chinese hamster ovary (CHO) cells using standard techniques as described previously (Katsuno et al. 2007). Cells were seeded into 96-well plates at a density of 3 × 104 cells/well in Ham’s F12 medium containing 10% fetal bovine serum. The cells were used 1 day after plating. Test compounds were initially dissolved in dimethyl sulfoxide and diluted to the desired concentration with sodium assay buffer (140 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 5 mM Tris–HCl, pH 7.4). After the medium was removed, the cells were preincubated in 100 μl choline assay buffer (NaCl in sodium assay buffer was replaced with the same concentration of choline chloride) at 37°C for 20 min. They were then incubated in the test compound solution (25 μl) containing 14C-AMG (2.2 μCi/ml) and nonlabeled AMG (final concentration 55 μM) at 37°C for 2 h. Cells were washed twice with 200 μl ice-cold wash buffer (choline assay buffer containing 10 mM AMG) and then solubilized in 0.5% sodium dodecyl sulfate (SDS) solution (25 μl). The cell lysate was mixed with 75 μl MicroScint MS-40 (Packard Instrument Co., Meriden, CT, USA), and radioactivity was measured using a Top Count Microplate Scintillation Counter (Packard Instrument Co.).
Specificity assay
Pharmacological screening assays for various receptors, ion channels, and transporters were performed according to the standard assay procedure of Sekisui Medical Co., Ltd. (Ibaraki, Japan).
Stability against mouse intestinal glucosidases
Under ether anesthesia, the small intestine was removed from overnight fasted normal mice, washed with cold saline, excised, and rinsed with phosphate buffer (48 mM NaCl, 5.4 mM KCl, 28 mM Na2HPO4, 43 mM NaH2PO4, 35 mM mannitol, 10 mM glucose, pH 6.5). The mucosal tissue was scraped off gently using a slide glass, homogenized with phosphate buffer, and used for the stability study. Test compounds were initially dissolved in acetonitrile at a concentration of 5 mM and then diluted to 100 μM with the phosphate buffer. Mucosal homogenates (5 mg/ml, 100 μl) were preincubated at 37°C in microtubes. Thereafter, each compound solution (100 μl, final concentration 50 μM) was added and incubated at 37°C for varying time periods. The reaction was stopped by the addition of ice-cold acetonitrile (200 μl), then 200 μl of methyl tert-butyl ether was added, and the mixture was centrifuged (15,000 rpm, 10 min). The supernatant was transferred into a tube and evaporated in a vacuum centrifugal concentrator. The residue was dissolved in mobile phase for use as the assay sample. Concentrations of compound in the assay sample were analyzed using a high-performance liquid chromatography (HPLC) with an ultraviolet detector (265 nm for ipragliflozin and 280 nm for phlorizin and phloretin) and a 4.6 × 250-mm reversed-phase ODS-80Ts column (Tosoh, Tokyo, Japan). The column temperature was maintained at 60°C, 20 mM ammonium acetate/acetonitrile [20:80 (v/v)] was used as the mobile phase, and the flow rate was 1.5 ml/min.
Pharmacokinetics
After oral administration of ipragliflozin (3 mg/kg) or phlorizin (100 mg/kg) to non-fasted normal mice, blood was withdrawn from the abdominal vena cava under ether anesthesia. The plasma concentrations of ipragliflozin or phlorizin were measured using HPLC. Acetonitrile (100 μl) and methyl tert-butyl ether (100 μl) were added to the plasma samples (100 μl), mixed, and then centrifuged (15,000 rpm, 10 min). The supernatant was transferred into a tube and evaporated in a vacuum centrifugal concentrator. The residue was dissolved in HPLC mobile phase, 0.1% formic acid solution/acetonitrile [55:45 (v/v)] for use as the assay sample. Concentrations of ipragliflozin or phlorizin in the assay samples were measured as described above.
Effect of ipragliflozin on urinary glucose excretion in normal mice
Ipragliflozin (0.01–10 mg/kg) was administered to non-fasted normal mice, and spontaneously voided urine was collected for 24 h after administration while the animals were kept in metabolic cages. After the urine volume had been measured, the glucose concentration in the urine was measured using the Glucose CII test reagent (Wako, Osaka, Japan).
Effect of single administration of ipragliflozin in diabetic animals
To investigate its antihyperglycemic effect, ipragliflozin (0.1–1 mg/kg) was administered to STZ-induced type 1 diabetic rats and KK-Ay type 2 diabetic mice in the fed condition. Blood glucose levels were then measured for 8 h under fasting conditions, in order to eliminate the influence of feeding during the experiment. To evaluate sustainability, ipragliflozin (0.1–1 mg/kg) was administered to both types of diabetic animals, which were then fasted for 12 h (overnight). A glucose solution (2 g/kg) was subsequently administered orally, and blood glucose levels were measured as described above.
Effect of repeated administration of ipragliflozin in STZ-induced type 1 diabetic rats
Ipragliflozin (0.3 and 1 mg/kg) was administered to STZ-induced type 1 diabetic rats once daily (at night) for 4 weeks. Body weight and food intake were measured every week. After drug administration on day 26, rats were transferred to metabolic cages and spontaneously voided urine was collected for 24 h. On the morning after the final drug administration on day 28, blood samples were collected under non-fasting conditions, and the pancreas was isolated under ether anesthesia. Blood and urinary glucose concentrations were measured as described above. The pancreas was homogenized by adding acid–ethanol solution (75% ethanol, 23.5% purified water, and 1.5% concentrated hydrochloric acid) and incubating at 4°C for 1 h to extract the insulin. Subsequently, the culture was centrifuged and the supernatant was used as a measurement sample. Plasma and pancreatic insulin concentrations were measured using an enzyme-linked immunosorbent assay (ELISA) kit (Morinaga Institute of Biological Science, Kanagawa, Japan). Hemoglobin A1c (HbA1c) levels were measured using a DCA2000 System (Bayer Medical, Tokyo, Japan).
Effect of repeated administration of ipragliflozin in KK-Ay type 2 diabetic mice
Ipragliflozin (0.3 and 1 mg/kg) was administered to KK-Ay type 2 diabetic mice once daily (at night) for 4 weeks. Body weight and food intake were measured every week. On the morning after the drug administration on day 28, blood samples were collected under non-fasting conditions. After the drug administration on day 30, mice were transferred to metabolic cages and spontaneously voided urine was collected for 24 h. Blood and urinary glucose concentrations, HbA1c, and plasma insulin levels were measured as described above. Urinary albumin concentration was measured using a mouse albumin ELISA.
Effect of ipragliflozin on fasting blood glucose levels in normal mice
In order to investigate their effects on postprandial hyperglycemia, ipragliflozin (0.03–100 mg/kg) or glibenclamide (0.3–300 mg/kg) was administered to normal mice that had been fasted overnight. After 30 min, glucose solution (2 g/kg) was administered orally, and blood glucose levels were measured. To investigate their effects on hypoglycemia, ipragliflozin (0.03–100 mg/kg) or glibenclamide (0.3–300 mg/kg) was administered to normal mice fasted overnight, and blood glucose levels were measured.
Effect of ipragliflozin on gastrointestinal carbohydrate contents in normal mice
After fasting for 24 h, mice received ipragliflozin (0.3–30 mg/kg) or voglibose (1 mg/kg). After 15 min, a liquid meal (ENSURE·H: carbohydrates 206 mg/ml, fats 53 mg/ml, proteins 53 mg/ml; Abbott, Osaka, Japan) was given orally at 20 ml/kg. Control mice were given purified water instead of a liquid meal. At 1 h after the liquid meal or water administration, blood glucose levels were measured, and gastrointestinal tracts (stomach, upper and lower small intestine, cecum, and lower large intestine) were isolated under ether anesthesia. Isolated gastrointestinal tracts were homogenized with purified water (5 ml) and centrifuged (3,000 rpm, 10 min) to retrieve the supernatant. Glucose concentration was measured as described above. Sucrose and maltose concentrations were measured by a previously described method with minor modifications (Dörner 1977). Fructose concentration was measured using a fructose assay kit (Sigma-Aldrich Co.).
Effect of ipragliflozin on plasma and urinary parameters in KK-Ay type 2 diabetic mice
Ipragliflozin (1 mg/kg), furosemide (10 mg/kg), or YM471 (3 mg/kg) was administered to non-fasted diabetic mice, which were then transferred to metabolic cages. The mice were allowed free access to food and water, and spontaneously voided urine samples were collected for 8 h. Thereafter, blood samples were collected from the tail vein for the determination of blood glucose level. Under ether anesthesia, urine was collected from the bladder, and blood samples were collected from the abdominal vena cava. The volume of spontaneously voided urine combined with the urine in the bladder was measured. Blood and urine samples were centrifuged (15,000 rpm, 10 min), after which the supernatants were used for the determination of several parameters. Plasma and urine osmolalities were measured using a freezing point depression osmometer (Advanced Instruments Inc., Norwood, MA, USA). Plasma and urine electrolyte (Na+, K+, and Cl−) concentrations were determined using a flame photometer (Hitachi, Tokyo, Japan), and the urinary electrolyte excretion was calculated as the product of the urine electrolyte concentration and the urine volume.
Statistical analysis
Experimental results are expressed as mean ± standard error of the mean (SEM), standard deviation (SD), or with 95% confidence limits. The median inhibition concentration (IC50) values were calculated using regression analysis. The areas under the blood glucose concentration–time curves were calculated from blood glucose concentrations measured over time. Student’s t test was used to analyze the differences between two groups; Dunnett’s multiple range test was used for comparisons among multiple groups. A value of p < 0.05 was taken to be significant. Statistical and data analyses were conducted using the SAS 8.2 software package (SAS Institute Japan, Ltd., Tokyo, Japan).
Results
SGLT2 and SGLT1 inhibition assay
Ipragliflozin concentration-dependently inhibited mouse, rat, and human SGLT2 activity at nanomolar concentrations (Table 1). For human SGLT2, the inhibitory effect of ipragliflozin was approximately five times greater than that of phlorizin, but for human SGLT1, it was only one ninth of that of phlorizin. Phloretin had very little potency to inhibit either SGLT. The ratios of selectivity (IC50 of human SGLT1/SGLT2) of ipragliflozin, phlorizin, and phloretin were 254, 6, and 3, respectively. In addition, ipragliflozin exhibited high selectivity for mouse and rat SGLT2. Dapagliflozin also potently and selectively inhibited mouse and human SGLT2 activity.
Specificity assay
To confirm the specificity, effects of ipragliflozin on several representative receptors, channels, and transporters were examined using radioligand binding and enzyme assays. Ipragliflozin did not interact with various receptors, ion channels, and transporters such as adrenergic (α1, α2, and β), muscarinic (M1, M2, and non-selective), angiotensin (AT1 and AT2), calcium channel (L-type and N-type), potassium channel (KATP and SKCa), sodium channel (site 2), cholecystokinin (CCKA and CCKB), dopamine (D1, D2, and transporter), endothelin (ETA and ETB), gamma-aminobutyric acid (GABAA and GABAB), glutamate (AMPA, kainate, and NMDA), serotonin (5-HT1, 5HT2B, and transporter), histamine (H1, H2, and H3), and neurokinin (NK1, NK2, and NK3), exhibiting IC50 values >3,000 nM.
Stability of ipragliflozin against mouse intestinal glucosidases
The in vitro biological stability of ipragliflozin and phlorizin against glucosidases was examined using mucosal homogenates of mouse small intestine. While ipragliflozin was not degraded at all (Fig. 2a), phlorizin was quickly degraded in mouse mucosal homogenates into its aglycon, phloretin (Fig. 2b).

Stability of a ipragliflozin and b phlorizin in mouse intestinal mucosal homogenates. Ipragliflozin (50 μM) or phlorizin (50 μM) was incubated with mouse intestinal mucosal homogenates (5 mg protein). Data are expressed as the mean ± SEM of three independent experiments
Pharmacokinetics
After oral administration of ipragliflozin (3 mg/kg) to normal mice, plasma concentrations of ipragliflozin reached a maximum at 1 h and then gradually decreased (Fig. 3). Obvious plasma concentrations were detected even 8 h after administration. In contrast, when phlorizin (100 mg/kg) was administered orally, plasma drug concentrations were low and rapidly eliminated.

Pharmacokinetics of ipragliflozin and phlorizin in normal mice. Ipragliflozin (3 mg/kg) or phlorizin (100 mg/kg) was administered orally to non-fasted normal mice. Data are expressed as the mean ± SD for three animals in each sampling time point
Effect of ipragliflozin on urinary glucose excretion in normal mice
In normal mice, ipragliflozin (0.01–10 mg/kg) dose-dependently and significantly increased urinary glucose excretion, and this effect was still apparent 12–18 h after administration at doses of ≥0.3 mg/kg (Fig. 4a). Urine volume was also significantly increased at doses of 3 and 10 mg/kg (Fig. 4b).

Effects of ipragliflozin on a urinary glucose excretion and b urine volume in normal mice. The values are the mean or mean ± SEM for four animals in each group. Each parameter was analyzed statistically over 24 h. *p < 0.05 vs. vehicle using Dunnett’s multiple range test
Effect of single administration of ipragliflozin in diabetic animals
In STZ-induced type 1 diabetic rats and KK-Ay type 2 diabetic mice, ipragliflozin (0.1−1 mg/kg) dose-dependently lowered blood glucose levels, and this effect was significant at all tested doses (Figs. 5a and 6a). During the oral glucose tolerance test (OGTT) 12 h after dosing, ipragliflozin (0.1–1 mg/kg) dose-dependently inhibited increases in blood glucose levels. In STZ-induced type 1 diabetic rats, this effect was significant at doses of 0.3 and 1 mg/kg (Fig. 5b), and in KK-Ay type 2 diabetic mice, the effect was significant at all tested doses (Fig. 6b).

Effects of ipragliflozin on blood glucose levels in streptozotocin-induced type 1 diabetic rats. a Ipragliflozin was administered to non-fasted diabetic rats, and blood glucose levels were measured for 8 h in a fasted state. b Ipragliflozin was administered, the rats were fasted for 12 h, then glucose solution was orally administered, and blood glucose levels measured for 2 h. The values are expressed as mean ± SEM for four animals in each group. *p < 0.05 vs. vehicle using Dunnett’s multiple range test

Effects of ipragliflozin on blood glucose levels in KK-Ay type 2 diabetic mice. a Ipragliflozin was administered to non-fasted diabetic mice, and blood glucose levels were measured for 8 h in a fasted state. b Ipragliflozin was administered, the mice were fasted for 12 h, then glucose solution was orally administered, and blood glucose levels were measured for 2 h. The values are expressed as mean ± SEM for four animals in each group. *p < 0.05 vs. vehicle using Dunnett’s multiple range test
Effects of repeated administration of ipragliflozin in STZ-induced type 1 diabetic rats
Compared to the normal control rats, STZ-induced type 1 diabetic rats had significantly higher mean levels of HbA1c, blood glucose, and urinary glucose excretion and significantly lower plasma insulin levels and pancreatic insulin content under non-fasting conditions (Table 2). Repeated administration of ipragliflozin (0.3 and 1 mg/kg) for 4 weeks significantly reduced the levels of HbA1c and blood glucose. Plasma insulin level was not significantly changed, but pancreatic insulin content was significantly increased at a dose of 1 mg/kg. Urinary glucose excretion was increased dose-dependently, and this was significant at the 1 mg/kg dose. Ipragliflozin did not affect body weight or food intake (data not shown) throughout the study.
Effects of repeated administration of ipragliflozin in KK-Ay type 2 diabetic mice
Repeated administration of ipragliflozin (0.3 and 1 mg/kg) for 4 weeks reduced HbA1c and blood glucose levels, with concomitant increases in urinary glucose excretion (Table 3). In addition, urinary albumin excretion was significantly decreased. Ipragliflozin treatment did not affect body weight or food intake (data not shown) throughout the study.
Effect of ipragliflozin on fasting blood glucose levels in normal mice
In normal mice, ipragliflozin (0.03–100 mg/kg) dose-dependently inhibited the increase in blood glucose level after glucose loading, and this effect was significant at doses ≥0.1 mg/kg (Fig. 7a). Glibenclamide (0.3–300 mg/kg) also dose-dependently inhibited the increase in blood glucose levels; this effect was significant at doses ≥3 mg/kg (Fig. 7c). In overnight-fasted mice, ipragliflozin (0.03–100 mg/kg) dose-dependently reduced blood glucose levels, but this effect was only significant at doses ≥10 mg/kg, which are 100-fold higher than that in the OGTT (Fig. 7b). Glibenclamide (0.3–300 mg/kg) also dose-dependently reduced fasting blood glucose levels at the same doses as in the OGTT (Fig. 7d). Ipragliflozin did not alter plasma insulin levels under fasting conditions but significantly reduced the increase in plasma insulin levels under glucose loading conditions. In contrast, glibenclamide significantly increased plasma insulin levels under both conditions (data not shown).

Effects of ipragliflozin and glibenclamide on fasting blood glucose levels in normal mice. a Ipragliflozin or c glibenclamide was administered to mice fasted overnight, glucose solution was orally administered 30 min after dosing, and blood glucose levels were measured for 6 h. b Ipragliflozin or d glibenclamide was administered to mice fasted overnight and blood glucose levels were measured for 6 h. The values are expressed as mean ± SEM for four animals in each group. *p < 0.05 vs. vehicle using Dunnett’s multiple range test
Effect of ipragliflozin on gastrointestinal carbohydrate contents in normal mice
Following liquid-meal loading in normal mice, gastrointestinal disaccharide (sucrose and maltose) and monosaccharide (glucose and fructose) contents increased significantly (Fig. 8). Voglibose (1 mg/kg) significantly increased gastrointestinal disaccharide content (Fig. 8a, b), decreased monosaccharide content (Fig. 8c, d), and significantly inhibited the increase in blood glucose levels (data not shown). In contrast, ipragliflozin (0.3–30 mg/kg) did not significantly affect gastrointestinal disaccharide content, even at the highest dose. In addition, ipragliflozin did not significantly affect gastrointestinal fructose content, and although it did dose-dependently increased glucose content, this effect was only significant at the maximum dose of 30 mg/kg. Ipragliflozin dose-dependently inhibited the increase in blood glucose levels, and this effect was significant at all tested doses (data not shown).

Effects of ipragliflozin and voglibose on gastrointestinal carbohydrate contents during the liquid meal tolerance test in normal mice. Effect of ipragliflozin and voglibose on gastrointestinal: a sucrose, b maltose, c glucose, and d fructose contents 1 h after liquid meal loading. The values are the mean or mean ± SEM for four animals in each group. a p < 0.05 vs. control group, b p < 0.05 vs. vehicle group using Student’s t test; c p < 0.05 vs. vehicle group using Dunnett’s multiple range test
Effect of ipragliflozin on plasma and urinary parameters in KK-Ay type 2 diabetic mice
In type 2 diabetic mice, the loop diuretic, furosemide (10 mg/kg), significantly increased urinary electrolyte (Na+, K+, and Cl−) excretion and urine volume, with a concomitant decrease in urinary osmolality (Table 4). This marked diuresis induced by electrolyte excretion also induced a significant decrease in plasma electrolyte concentrations and a significant increase in plasma osmolality. The vasopressin V1A/V2 receptor antagonist, YM471 (3 mg/kg), significantly increased urine volume without increasing electrolyte excretion and significantly decreased urinary osmolality. This marked water diuresis induced significant increases in plasma electrolyte concentrations and osmolality. In contrast, ipragliflozin (1 mg/kg) markedly increased urinary glucose excretion, with a concomitant slight increase in urine volume, and significantly decreased blood glucose levels, but did not significantly affect plasma or urinary electrolyte balance.
Discussion
Therapeutic strategies for diabetes currently focus on controlling elevated blood glucose levels. Chronic hyperglycemia has been shown to reduce insulin sensitivity and impair β-cell function in animal models (Harmon et al. 2001; Donath et al. 1999); therefore, the correction of hyperglycemia is predicted to improve these important physiological defects in patients with type 2 diabetes (Rossetti et al. 1987a, b). In recent studies, stimulating the excretion of excess glucose in the urine has been proposed as a new approach for the treatment of type 2 diabetes (Isaji 2007; Komoroski et al. 2009). SGLT2 is responsible for most of the glucose reabsorption in the kidneys and has been highlighted as a novel therapeutic target for diabetes. Ipragliflozin is a novel SGLT2 inhibitor, and we have explored its in vitro and in vivo characteristics in this study.
In CHO cells stably expressing either SGLT2 or SGLT1, ipragliflozin potently inhibited human, rat, and mouse SGLT2 with IC50 values in the low nanomolar range. However, its potency for SGLT1 inhibition was much weaker among all species studied, with IC50 values in the micromolar range. The inhibitory activities and selectivities of ipragliflozin were almost identical to those of dapagliflozin, and there was no appreciable species difference. Furthermore, ipragliflozin did not potently inhibit human SGLT4 and SGLT5 isoforms (IC50 > 1,000 nM) (unpublished data). In the specificity assays, ipragliflozin did not interact with various receptors, ion channels, and transporters. In addition, ipragliflozin did not inhibit several glucose transporter (GLUT) isoforms, including GLUT1 and GLUT4, in mouse 3T3-L1, rat L6, human Caco-2, and HepG2 cells (IC50 > 1,000 nM) (unpublished data). These in vitro studies suggest that ipragliflozin is a potent selective SGLT2 inhibitor. In the pharmacokinetic studies in mice, ipragliflozin showed good oral bioavailability and exhibited high drug concentrations for long periods. The absolute bioavailabilities of ipragliflozin were 71.7–90.7% and 74.5–75.3% in rats and monkeys, respectively (unpublished data). In contrast, phlorizin exhibited very low oral bioavailability. In the stability experiment using mucosal homogenates of mouse small intestine, phlorizin was quickly hydrolyzed to phloretin and glucose by intestinal glucosidases such as lactase-phlorizin hydrolase (Leese and Semenza 1973). This suggests that phlorizin administered orally would be easily degraded and poorly absorbed in the small intestine. The low oral bioavailability of phlorizin may be explained by its rapid enzymatic degradation in the gastrointestinal tract. In contrast, ipragliflozin exhibited stable properties against mouse intestinal glucosidases. In the same experiments using intestinal mucosal homogenates or microsomes of another species (rat, canine, monkey, and human), ipragliflozin also showed stable properties (data not shown). This intestinal stability of ipragliflozin is thought to contribute to its good oral bioavailability.
In normal mice, single administration of ipragliflozin induced marked and sustained increases in urinary glucose excretion which lasted beyond 12 h after administration, with concomitant increase in urine volume. The pharmacokinetic properties of ipragliflozin support these long-lasting glucosuric effects. In STZ-induced type 1 diabetic rats and KK-Ay type 2 diabetic mice, single oral doses ranging from 0.1 to 1 mg/kg of ipragliflozin produced dose-dependent and sustained reductions in hyperglycemia and improvements in glucose tolerance. These results suggest that ipragliflozin exhibits antihyperglycemic effects through the sustained stimulation of urinary glucose excretion via the inhibition of renal SGLT2. In addition, in both insulin-deficient and obese insulin-resistant diabetic models, once-daily ipragliflozin treatment at doses of 0.3 and 1 mg/kg for 4 weeks significantly lowered blood glucose and HbA1c levels. The concomitant increase in urinary glucose excretion indicates that the antihyperglycemic action of ipragliflozin is derived from the enhancement of urinary glucose disposal. Thus, ipragliflozin exhibits an antihyperglycemic effect, irrespective of the presence of impaired insulin secretion or insulin resistance. In STZ-induced type 1 diabetic rats, ipragliflozin increased pancreatic insulin content, suggesting that amelioration of hyperglycemia with ipragliflozin could induce the economization of pancreatic insulin content. In KK-Ay type 2 diabetic mice, ipragliflozin decreased urinary albumin excretion, the established parameter of early stage of diabetic nephropathy. Hyperglycemia is the principal factor for diabetic microvascular complications including nephropathy (UK Prospective Diabetes Study Group 1998); therefore, this would be mainly due to its antihyperglycemic effect. In addition, it has been reported the important role of enhanced glucose reabsorption in the proximal tubule in altering renal hemodynamics and the development of diabetic nephropathy (Vallon et al. 1999). Inhibition of renal glucose reabsorption induced by SGLT2 inhibitor might be expected to have an additional renoprotective effect; further studies are warranted to elucidate its mechanism of action in detail. In this study, chronic treatment of ipragliflozin slightly reduced body weights of obese diabetic mice, but this was not statistically significant. It has been reported that dapagliflozin reduced body weight in type 2 diabetes (List et al. 2009). This weight reduction was considered to be mainly due to steady caloric loss but might account at least in part for its water loss, through glucosuria. In the another experiment, we have confirmed that ipragliflozin reduced epididymal adipose tissue weights in KK-Ay type 2 diabetic mice (unpublished data), which suggests that SGLT2 inhibitors including ipragliflozin could improve obesity and further studies are warranted to elucidate their effects.
In this study, glibenclamide inhibited postprandial hyperglycemia and decreased fasting blood glucose levels. Both effects were induced by increases in plasma insulin levels at the same pharmacological doses. In clinical practice, when using sulfonylureas, the optimum dose should have a risk of hypoglycemia while maintaining sufficient therapeutic efficacy (Stahl and Berger 1999). The reduction in the fasting blood glucose level that occurred at the effective doses for glibenclamide is thought to be related to the risk of hypoglycemia. In contrast, by increasing urinary glucose excretion, ipragliflozin inhibited the increase in blood glucose levels after glucose loading in the absence of insulin secretion. Furthermore, as ipragliflozin had no effect on fasting blood glucose levels at pharmacological doses, there should be a low risk of hypoglycemia, making it safe for use as an antihyperglycemic agent. Furthermore, during the chronic treatment studies, significant correction of ambient hyperglycemia was achieved by ipragliflozin in type 1 and type 2 diabetic models, but no evidence of hypoglycemia was observed. The ability to induce an antihyperglycemic effect independently of insulin secretion is a property of ipragliflozin that represents a considerable advantage as a potential antidiabetic medicine for clinical use, and it could be efficacious in a wide variety of diabetic patients.
SGLT2, which is specifically expressed in the kidneys, plays an important role in renal glucose reabsorption (Jabbour and Goldstein 2008). In contrast, SGLT1 is highly expressed in the small intestine and mediates dietary glucose absorption (Pajor and Wright 1992). In order to elucidate the in vivo effect of ipragliflozin against intestinal SGLT1, we examined its effect on gastrointestinal carbohydrate absorption. α-Glucosidase inhibitors, such as voglibose, inhibit the small intestinal disaccharidases, sucrase and maltase (Matsuo et al. 1992). This slows the absorption of carbohydrates from the small intestine, thereby lowering postprandial hyperglycemia (Baron 1998). In this study, voglibose induced a significant increase in intestinal disaccharide content (by delaying disaccharide digestion) that inhibited the increase in blood glucose levels. Thus, α-glucosidase inhibitors are effective at preventing postprandial hyperglycemia by this mechanism. However, osmotic water retention induced by accumulation of intestinal disaccharide content can cause gastrointestinal symptoms such as soft feces or diarrhea (Vichayanrat et al. 2002). Ipragliflozin, however, did not affect intestinal disaccharide or fructose content, but at the maximum dose of 30 mg/kg, it did significantly increase intestinal glucose content. It is known that gastrointestinal expression of SGLTs and absorption of glucose depend mainly on SGLT1 (Turk et al. 1991) and that gastrointestinal drug concentrations immediately after oral administration are at a very high level (Masaoka et al. 2006). Thus, although ipragliflozin shows a 245-fold higher selectivity for mouse SGLT2 versus SGLT1, the significant increase in gastrointestinal glucose content with the highest dose of ipragliflozin is thought to be due to the inhibition of glucose absorption via SGLT1 in the small intestine. Nevertheless, gastrointestinal glucose elevation was only significant at a dose which is 100 times higher than that which significantly decreased postprandial hyperglycemia. It is therefore considered that therapeutic doses of ipragliflozin would not inhibit intestinal SGLT1, nor affect intestinal carbohydrate digestion and absorption. As such, the risk of gastrointestinal symptoms should be low. During the course of both acute and chronic experiments, no gastrointestinal side effects indicative of significant intestinal SGLT1 inhibition were noted at any dose.
Higher doses of ipragliflozin slightly increased urine volume along with a significant increase in urinary glucose excretion. Since sodium-ion transportation accompanies the glucose transport promoted by SGLT2, we evaluated the effect of ipragliflozin on plasma and urinary electrolyte balance. In this experiment, the loop diuretic, furosemide, induced a marked diuresis with concomitant increase in urinary electrolyte excretion, and decreased plasma concentrations of electrolytes including sodium. This furosemide-induced hyponatremia may be a severe adverse reaction (Sonnenblick et al. 1993). The vasopressin V1A/V2 receptor antagonist, YM471, also exerted a potent aquaretic effect and increased plasma electrolyte concentrations. In contrast, ipragliflozin (1 mg/kg) markedly increased urinary glucose excretion with a concomitant slight increase in urine volume, but did not affect plasma or urinary electrolyte balance. These results suggest that ipragliflozin would increase urinary glucose excretion without inducing either a marked osmotic diuresis or an electrolyte imbalance at pharmacological doses.
Mutations in the SGLT2 gene lead to the rare disorder, familial renal glucosuria, where glucose reabsorption in the kidneys is severely impaired and large amounts of glucose are excreted in the urine (Francis et al. 2004; Magen et al. 2005). Despite the severe glucosuria, patients appear to have a benign condition, with no serious adverse events or problems in kidney function. Familial renal glucosuria is also not associated with hypoglycemia and generally has no significant clinical manifestations (Santer et al. 2003). In this study, chronic administration of pharmacological doses of ipragliflozin did not induce significant adverse effects in diabetic models. Based on these findings, ipragliflozin seems to be an effective and safe drug for the treatment of hyperglycemia.
In conclusion, the present study shows that ipragliflozin is a potent selective SGLT2 inhibitor, possesses good pharmacokinetic characteristics, and exhibits a sustained antihyperglycemic effect by increasing urinary glucose excretion, without inducing hypoglycemia or insulin secretion. These results indicate the potential usefulness of ipragliflozin for further development as a therapeutic agent for hyperglycemia in patients with diabetes. Clinical trials with ipragliflozin are currently in progress, and proof-of-concept studies should clarify the suitability of ipragliflozin for the treatment of diabetes (Kashiwagi et al. 2010).
References
Abdul-Ghani MA, DeFronzo RA (2008) Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract 14:782–790
Andrews WJ, Vasquez B, Nagulesparan M, Klimes I, Foley J, Unger R, Reaven GM (1984) Insulin therapy in obese, non-insulin-dependent diabetes induces improvements in insulin action and secretion that are maintained for two weeks after insulin withdrawal. Diabetes 33:634–642
Baron AD (1998) Postprandial hyperglycaemia and alpha-glucosidase inhibitors. Diabetes Res Clin Pract 40(Suppl):S51–S55
Crofford OB (1995) Diabetes control and complications. Annu Rev Med 46:267–279
Donath MY, Gross DJ, Cerasi E, Kaiser N (1999) Hyperglycemia-induced β-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 48:738–744
Dörner KM (1977) Quantitative determination of lactose, maltose, and sucrose in urine. Eur J Pediatr 126:45–52
Ehrenkranz JR, Lewis NG, Kahn CR, Roth J (2005) Phlorizin: a review. Diabetes Metab Res Rev 21:31–38
Francis J, Zhang J, Farhi A, Carey H, Geller DS (2004) A novel SGLT2 mutation in a patient with autosomal recessive renal glucosuria. Nephrol Dial Transplant 19:2893–2895
Han S, Hagan DL, Taylor JR, Xin L, Meng W, Biller SA, Wetterau JR, Washburn WN, Whaley JM (2008) Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats. Diabetes 57:1723–1729
Harmon JS, Gleason CE, Tanaka Y, Poitout V, Robertson RP (2001) Antecedent hyperglycemia, not hyperlipidemia, is associated with increased islet cell triacylglycerol content and decreased insulin gene mRNA level in Zucker Diabetic Fatty rats. Diabetes 50:2481–2486
Idris I, Donnelly R (2009) Sodium–glucose co-transporter-2 inhibitors: an emerging new class of oral antidiabetic drug. Diabetes Obes Metab 11:79–88
Isaji M (2007) Sodium–glucose cotransporter inhibitors for diabetes. Curr Opin Investig Drugs 8:285–292
Jabbour SA, Goldstein BJ (2008) Sodium glucose co-transporter 2 inhibitors: blocking renal tubular reabsorption of glucose to improve glycaemic control in patients with diabetes. Int J Clin Pract 62:1279–1284
Kashiwagi A, Utsuno A, Kazuta K, Yoshida S, Kageyama S (2010) ASP1941, a novel, selective SGLT2 inhibitor, was effective and safe in Japanese healthy volunteers and patients with type 2 diabetes mellitus. Diabetes 59(Suppl 1):A21 (abstract 75-OR)
Katsuno K, Fujimori Y, Takemura Y, Hiratochi M, Itoh F, Komatsu Y, Fujikura H, Isaji M (2007) Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J Pharmacol Exp Ther 320:323–330
Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M (2009) Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther 85:513–519
Leese HJ, Semenza G (1973) On the identity between the small intestinal enzymes phlorizin hydrolase and glycosylceramidase. J Biol Chem 248:8170–8173
List JF, Woo V, Morales E, Tang W, Fiedorek FT (2009) Sodium–glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care 32:650–657
Magen D, Sprecher E, Zelikovic I, Skorecki K (2005) A novel missense mutation in SLC5A2 encoding SGLT2 underlies autosomal-recessive renal glucosuria and aminoaciduria. Kidney Int 67:34–41
Masaoka Y, Tanaka Y, Kataoka M, Sakuma S, Yamasita S (2006) Site of drug absorption after oral administration: assessment of membrane permeability and luminal concentration of drugs in each segment of gastrointestinal tract. Eur J Pharm Sci 29:240–250
Matsuo T, Odaka H, Ikeda H (1992) Effect of an intestinal disaccharidase inhibitor (AO-128) on obesity and diabetes. Am J Clin Nutr 55(Suppl):314S–317S
Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN (2008) Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem 51:1145–1149
Oku A, Ueta K, Arakawa K, Ishihara T, Nawano M, Kuronuma Y, Matsumoto M, Saito A, Tsujihara K, Anai M, Asano T, Kanai Y, Endou H (1999) T-1095, an inhibitor of renal Na+−glucose cotransporters, may provide a novel approach to treating diabetes. Diabetes 48:1794–1800
Oku A, Ueta K, Arakawa K, Kano-Ishihara T, Matsumoto M, Adachi T, Yasuda K, Tsuda K, Saito A (2000) Antihyperglycemic effect of T-1095 via inhibition of renal Na+-glucose cotransporters in streptozotocin-induced diabetic rats. Biol Pharm Bull 23:1434–1437
Pajor AM, Wright EM (1992) Cloning and functional expression of a mammalian Na+/nucleoside cotransporter. A member of the SGLT family. J Biol Chem 267:3557–3560
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Investig 116:1802–1812
Rossetti L, Shulman GI, Zawalich W, DeFronzo RA (1987a) Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J Clin Investig 80:1037–1044
Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA (1987b) Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Investig 79:1510–1515
Santer R, Kinner M, Lassen CL, Schneppenheim R, Eggert P, Bald M, Brodehl J, Daschner M, Ehrich JH, Kemper M, Li Volti S, Neuhaus T, Skovby F, Swift PG, Schaub J, Klaerke D (2003) Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol 14:2873–2882
Sonnenblick M, Friedlander Y, Rosin AJ (1993) Diuretic-induced hyponatremia. Review and analysis of 129 reported patients. Chest 103:601–606
Stahl M, Berger W (1999) Higher incidence of severe hypoglycaemia leading to hospital admission in type 2 diabetic patients treated with long-acting versus short-acting sulphonylureas. Diabet Med 16:586–590
Tsukada J, Tahara A, Tomura Y, Ki W, Kusayama T, Ishii N, Yatsu T, Uchida W, Taniguchi N, Tanaka A (2001) Effects of YM471, a nonpeptide AVP V1A and V2 receptor antagonist, on human AVP receptor subtypes expressed in CHO cells and oxytocin receptors in human uterine smooth muscle cells. Br J Pharmacol 133:746–754
Turk E, Zabel B, Mundlos S, Dyer J, Wright EM (1991) Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature 350:354–356
UK Prospective Diabetes Study (UKPDS) Group (1998) Intensive blood–glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853
Vallon V, Richter K, Blantz RC, Thomson S, Osswald H (1999) Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10:2569–2576
van den Heuvel LP, Assink K, Willemsen M, Monnens L (2002) Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet 111:544–547
Vichayanrat A, Ploybutr S, Tunlakit M, Watanakejorn P (2002) Efficacy and safety of voglibose in comparison with acarbose in type 2 diabetic patients. Diabetes Res Clin Pract 55:99–103
Washburn WN (2009) Evolution of sodium glucose co-transporter 2 inhibitors as anti-diabetic agents. Expert Opin Ther Patents 19:1485–1499
Wells RG, Pajor AM, Kanai Y, Turk E, Wright EM, Hediger MA (1992) Cloning of a human kidney cDNA with similarity to the sodium–glucose cotransporter. Am J Physiol 263:F459–F465
Wright EM (2001) Renal Na+-glucose cotransporters. Am J Physiol Renal Physiol 280:F10–F18
Wright EM, Turk E (2004) The sodium/glucose cotransporter family SLC5. Pflugers Arch 447:510–518
Wright EM, Hirayama BA, Loo DF (2007) Active sugar transport in health and disease. J Intern Med 261:32–43
Acknowledgments
The authors acknowledge Drs. Isao Yanagisawa, Seitaro Mutoh, Yasuaki Shimizu, Wataru Uchida, and Shinichi Tsukamoto (Astellas Pharma Inc.) for their valuable comments and continuing encouragement.
Conflict of interest
A. Tahara, E. Kurosaki, M. Yokono, D. Yamajuku, R. Kihara, Y. Hayashizaki, T. Takasu, M. Imamura, L. Qun, M. Sasamata, and M. Shibasaki are employees of Astellas Pharma Inc. H. Tomiyama, Y. Kobayashi, and A. Noda are employees of Kotobuki Pharmaceutical Co. Ltd. Ipragliflozin is in clinical development by Astellas Pharma Inc. and Kotobuki Pharmaceutical Co. Ltd.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tahara, A., Kurosaki, E., Yokono, M. et al. Pharmacological profile of ipragliflozin (ASP1941), a novel selective SGLT2 inhibitor, in vitro and in vivo. Naunyn-Schmiedeberg's Arch Pharmacol 385, 423–436 (2012). https://doi.org/10.1007/s00210-011-0713-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-011-0713-z