
Abstract
When blood flows through the renal capillaries, glucose is one of the many substances filtered by the kidney. However, glucose is subsequently recovered primarily by the sodium-dependent glucose transporter 2 (SGLT2) as the glomerular filtrate flows down the renal tubules. SGLT2 inhibitors inhibit this transporter leading to the loss of a significant fraction of the filtered glucose. The resulting glucosuria is of sufficient magnitude to reduce diabetes-related hyperglycemia and ameliorate-associated complications of diabetes. A systematic study was conducted to identify superior SGLT2 inhibitors based on a β- 1C-arylglucoside with substituted diarylmethane moieties. Such compounds are potent and selective SGLT2 inhibitors with metabolic stability that promote glucosuria when administered in vivo. Through this investigation, the β- 1C-arylglucoside dapagliflozin was identified as a potent and selective hSGLT2 inhibitor with an EC50 for hSGLT2 of 1.0 nM and 1,200-fold selectivity over hSGLT1. Dapagliflozin produced glucosuria in normal Sprague Dawley rats in a dose-dependent fashion. Moreover, a 0.1 mg/kg oral dose reduced blood glucose levels by as much as 55% in rats that had been made hyperglycemic by streptozotocin, a pancreatic toxin. These findings, combined with a favorable ADME profile and vivo data, led to nomination of dapagliflozin as a drug for the treatment of type 2 diabetes. The structural architecture of β- 1C-arylglucosides and their amphiphilic nature presented significant obstacles to the synthesis of dapagliflozin and similar candidates for toxicological and clinical testing, prompting the development of a new, safe, efficient, and economical process for the synthesis of C-4′ and C-4 substituted β- 1C-arylglucosides. A key element of the process was a remarkable discovery of novel crystalline complexes that enabled isolation and quality control.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The incidence of type 2 diabetes has become an increasing worldwide concern, as the number of patients suffering from type 2 diabetes is projected to increase from approximately 371 million people currently to 552 million by 2030 ([1], www.idf.org/diabetesatlas/5e/Update2012). Type 2 diabetes is characterized by hyperglycemia due to excessive hepatic glucose production, a deficiency in insulin secretion and/or peripheral insulin resistance. As the disease advances, hyperglycemia becomes the major risk factor for diabetes complications, including retinopathy, neuropathy, nephropathy, and macrovascular diseases [2–4]. Due to the progressive nature of the disease, combination therapy is usually necessary to achieve the target glycemic level, thereby necessitating development of alternative agents acting by novel mechanisms to control hyperglycemia [5, 6]. This need is underscored by the United Kingdom Prospective Diabetes Study (UKPDS) findings that at present only 25–50% of type 2 diabetics are effectively treated by current therapies [7, 8].
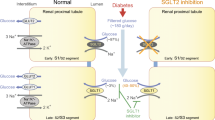
In healthy individuals, greater than 99% of the plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1% of the total filtered glucose being excreted in urine [9, 10]. This reabsorption process is mediated by two sodium-dependent glucose cotransporters: SGLT1, a low capacity, high affinity transporter expressed in gut, heart, and kidney [11, 12] and SGLT2, a high capacity, low affinity transporter that is expressed mainly in kidney [13, 14]. It is estimated that 90% of renal glucose reabsorption is facilitated by SGLT2 residing on the surface of the epithelial cells lining the S1 segment of the proximal tubule; the remaining 10% is likely mediated by SGLT1 localized on the more distal S3 segment of the proximal tubule [15–20]. Humans with SGLT1 gene mutations experience glucose-galactose malabsorption, resulting in frequent, watery diarrhea and dehydration when on a glucose diet, confirming that SGLT1 is the major glucose transporter in the small intestine. These individuals present with little or no glucosuria, suggesting that SGLT1 is not the major glucose transporter in the kidney [21, 22]. Persistent renal glucosuria is the sole reported phenotype of humans with SGLT2 gene mutations [23, 24].
Inhibition of SGLT2 has been proposed to aid in the normalization of plasma glucose levels in diabetics by preventing the glucose reabsorption process and promoting glucose excretion in urine [25]. Selective SGLT2 inhibitors would be desirable, since gastrointestinal side effects associated with SGLT1 inhibition would be minimized. This mechanism is expected to be associated with low risk of hypoglycemia, because there would be no interference with the normal counterregulatory mechanisms for glucose regulation.
A systematic study was conducted to identify superior SGLT2 inhibitors that are potent, selective over SGLT1, promote glucosuria when administered in vivo, and exhibit a pharmacokinetic profile compatible with once-daily administration.
2 Results and Discussion
2.1 Discovery
The natural product O-glucoside phlorizin (1, Fig. 1) is a well-documented, potent glucosuric agent that was subsequently shown to be a nonselective SGLT inhibitor [26]. The finding that chronic subcutaneous administration of 1 reduced plasma glucose levels of diabetic rodents supported this mechanistic approach [27–29]. However, phlorizin itself is not considered to be a suitable drug candidate because of its ability to inhibit SGLT1 and poor metabolic stability due to susceptibility to β-glucosidase-mediated cleavage resulting in release of the aglycone phloretin.

Structures of some known SGLT inhibitors
In a series of papers, researchers at Tanabe disclosed the structure–activity relationships (SAR) of phlorizin analogs resulting in the identification of selective, potent SGLT2 inhibitors. However, in order to achieve a significant reduction of hyperglycemia with a concurrent increase of glucosuria, the metabolic instability of the O-glucoside linkage necessitated oral administration of their lead compound T-1095A (2, Fig. 1) to KK-A y mice as the methyl carbonate pro-drug T1095 (3, Fig. 1) [25, 30–36]. Subsequently, Kissei disclosed two other series of O-glucosides containing SGLT2 inhibitors as potential treatment for type 2 diabetes which also required administration as pro-drugs [37–40].
In an attempt to increase the metabolic stability of the glucosyl linkage of O-arylglucoside 5a (Fig. 1), we synthesized C-benzylglucoside 5b (Fig. 1) [41]. This compound displayed a significant loss in SGLT2 activity (75-fold) as compared to compound 5a. Link et al., reported that a similar modification of 2 to generate the carbon analog 4 produced a >20-fold loss in potency [42]. Together, these findings imply that the isosteric replacement of the oxygen glucosidyl link with a methylene greatly attenuated previously favorable ligand–protein interactions. Possibly, the greater conformational freedom of 4 and 5b contributed to the reduction in SGLT2 affinity due to absence of the conformational constraints imposed by the exo-anomeric effect [43, 44].
Fortuitously, an alternative lead for C-glucoside-derived SGLT2 inhibitors surfaced upon characterization of a minor C-arylglucoside side-product that was generated during our SGLT2 program [45]. Of particular interest was the meta presentation of the glucosyl and benzyl appendages of 6 rather than the typical ortho presentation of O-glucoside-derived inhibitors. The activity (EC50 SGLT2 = 1,300 nM) and selectivity (>sixfold vs SGLT1) of 6 was unexpected especially since the SAR for all O-glucosides indicated that substitution of the aglycone with polar substituents markedly reduced SGLT2 activity. In hopes of achieving improved potency, 7a, the counterpart of a potent O-glucoside SGLT2 inhibitor 5a, was synthesized. The in vitro profile of 7a was extremely encouraging: SGLT2 EC50 = 22 nM; >600-fold selectivity vs SGLT1. The importance of a para substituent on the distal ring became readily apparent upon comparison of 7a to the parent 7b or meta isomer 7c, thereby underscoring the role of the substituent to properly orient the distal ring to achieve high affinity. A similar bias for para substitution of the distal ring had been observed for O-arylglucoside analogs of both 5b and dihydrochalcones reported by Hongu et al. [46].
These findings prompted a systematic study of meta-C-arylglucosides with varying linkers to evaluate proper placement of the distal aryl ring. The assumption was that high affinity SGLT2 inhibitors require not only the distal aryl ring to bear a lipophilic substituent but also that the distal ring assumes an orientation such that the lipophilic substituent can occupy a favorable binding pocket. Incorporation of a small lipophilic substituent at C-4 of the central aryl ring further augmented SGLT2 potency with little apparent impact on selectivity versus SGLT1, thereby maximizing potency while minimizing the potential for gastrointestinal (GI) side effects mediated by inhibition of intestinal SGLT1. A number of compounds with preferred C-4 substitutions were synthesized and evaluated to further understand the consequences of introduction of a zero, one, two, or three methylene spacer and impact of distal ring substitution with m-methyl or p-methyl groups.
Table 1 summarizes the structure–activity consequences upon alteration of the spacer moiety between the C-glucoside proximal and distal rings. Variation of the spacer from one (7) to zero (8), two (9), or three methylenes (10) reduced affinity ~ threefold for the unsubstituted (R = H) and m-methyl substituted examples. In contrast, changing the methylene spacer of 7a from one to zero (8c), two (9c), or to three methylenes (10c) reduced the binding affinity of the p-methyl substituted analogs 13-, 19-, and 29-fold, respectively. The unique advantage conferred by the single methylene of 7a is further confirmed by the respective threefold and 25-fold reduction in affinity upon replacement with a sulfur (12) or oxygen (11) bridging atom. Significant inhibition of human SGLT1 was not observed for any of the C-arylglucosides tested. In particular, the demonstrably high level of selectivity of 7a is expected to preclude gastrointestinal side effects.
Upon i.v. administration to rats and mice at 1 and 0.3 mg/kg, respectively, 7a produced maximum glucosuric levels of 230 and 600 mg/dL.Footnote 1 In contrast, upon administration of O-glucoside 5 under the same conditions, efficacy in rats was reduced ~ 50-fold relative to that obtained in mice. C-Arylglucoside 7a was found to be ~100-fold more stable in the presence of rat liver microsomes than the corresponding O-glucoside 5 [45]. We attribute the greater in vitro stability of 7a and the diminished variability in glucosuric response across species to 7a being impervious to glucosidase cleavage (unlike 5). This profile of the C-glucoside 7 to that of 5 reveals greater selectivity versus SGLT1, as well as enhanced metabolic stability. C-Arylglucosides show enhanced glucosuric activity in rats compared to O-arylglucosides that we attribute, in part, to the metabolic stability of the aryl-glucosyl C–C bond.
SAR exploration revealed meta-substituted diarylmethanes to be superior SGLT2 ligands to their biphenyl and 1,2-diarylethane counterparts [41]. Only small, hydrophobic para (C-4) substituents such as ethyl, methoxy, thiomethyl, and difluoromethoxy enhanced SGLT2 inhibitory activity resulting in EC50 values of ∼10 nM, whereas substitution at C-2 or C-3 was not beneficial. Compounds in this series exhibit >1,000-fold selectivity for SGLT2 than SGLT1. Further SAR exploration of the central aryl ring revealed that small lipophilic substituents at the C-4′ position of the central aryl ring further increased SGLT2 affinity such that EC50 values decreased to 1 nM. Although substitution at C-5′ or C-6′ modestly improved affinity, C-2′ substitution was deleterious (Fig. 2).

Structure–activity relationship for diarylmethane C-glucoside SGLT2 inhibitors
This SAR culminated in the discovery of several selective SGLT2 inhibitors such as compounds 13, 14, 15, 16, which exhibited properties warranting further progression as clinical candidates for the treatment of type 2 diabetes (Fig. 3).

Structures of advanced SGLT2 inhibitors
The in vitro SGLT inhibitory potential (EC50) of 16 and analogs were assessed by monitoring the inhibition of accumulation of radiolabeled α-methyl-d-glucopyranoside (AMG) by CHO cells stably expressing human or rat SGLT2 and SGLT1. As shown in Table 2, EC50 values of 1.0 nM for hSGLT2 and 1.6 μM for hSGLT1 were determined for 16 which corresponded to 1,200-fold selectivity for SGLT2 as compared with eightfold selectivity for phlorizin. The inhibitory potencies of 16 against rat and human SGLT2 were comparable, but the selectivity of 16 for rSGLT2 versus rSGLT1 decreased to 200-fold. At 20 μM, 16 was also found to weakly inhibit (8%) AMG uptake in human adipocyte mediated by GLUT1 and GLUT4 facilitative glucose transporter [47].
Statistically significant dose-dependent glucosuria occurred over a 24-hour period following oral administration of doses from 0.01 to 10 mg/kg of 16 to normal Sprague Dawley rats. This glucosuria was accompanied by dose-dependent increases in urine volume of 16–300% (Fig. 4). In this study, the loss of 700 and 2,400 mg of glucose per rat over 24 h following single oral dose of 0.1 and 10 mg/kg of 16, respectively, corresponded to a 1,000- and 10,000-fold elevation in glucose disposal relative to vehicle controls. In a separate experiment with streptozotocin-induced (STZ) diabetic rats (starting blood glucose levels of 480–530 mg/dL), a single 0.1 mg/kg oral dose of 16 followed by food restriction produced a 55% reduction in the elevated blood glucose level of treated versus control rats at 5 h after dosing (Fig. 5). This level of efficacy surpassed that of 13 (34% reduction in hyperglycemic blood glucose levels) and matched the efficacy observed for other analogs 14 and 15 (59%, and 62%) at this dose [48, 49]. The above correlation of SGLT2 inhibition, glucosuria, and blood glucose lowering effects suggested that selective SGLT2 inhibition with compounds 13–16 held promise as a viable approach to treat type 2 diabetes.

Mean total urine glucose excretion over 24 h following a single oral dose of dapagliflozin 16 in normal SD rats. n = 3. Mean total urine glucose excretion was statistically significant vs. vehicle at the 0.1, 1, and 10 mg/kg dose groups

Mean blood glucose values in streptozotocin-induced (STZ) diabetic SD rats following a single oral dose of 0.1 mg/kg dapagliflozin, 16. n = 6. *p < 0.05 vs. control group using a paired student t-test
Dapagliflozin 16 displayed a favorable ADME profile conducive to further development. At 10 μM in serum from rats and humans, the free fraction of 16 was 3 and 4%, respectively. Compound 16 is anticipated to be orally bioavailable in humans based on a high (>150 nm/s) caco-2 cell monolayer permeability value and 84% oral bioavailability in rats (Table 3). The steady-state volume of distribution value (1.6 L/kg) was greater than the total blood volume in rats, indicating that 16 distributed into the extravascular space. Low to intermediate in vitro metabolic rates were observed upon incubation of 16 with liver microsomes and hepatocytes from rats and humans. After oral administration of 1 mg/kg dose of 16 to rats, a C max of 0.6 μg/mL was obtained at 1.7 h with a low systemic clearance rate of 4.8 mL/min/kg. The elimination half-life for 16 following intra-arterial administration was 4.6, 7.4, and 3.0 h in rats, dogs, and monkeys, respectively. These pharmacokinetic parameters for dapagliflozin in preclinical species revealed a compound with adequate oral exposure, clearance, and elimination half-life, consistent with the potential for single daily dosing in humans [49].
In summary, dapagliflozin, compound 16, is a potent, selective SGLT2 inhibitor which stimulates glucosuria in normal rats. The promising significant reduction of blood glucose levels in diabetic STZ rats, combined with a favorable ADME profile, in vivo clinical data prompted nomination of dapagliflozin as new drug for the treatment of type 2 diabetes.
Clinical efficacy and safety of dapagliflozin were assessed in multiple Phase III placebo-controlled trials, and the results were recently reviewed [50]. Patients receiving dapagliflozin experienced improved glucose control as demonstrated by placebo-subtracted HbA1c reductions of >0.5%. Patients experienced additional benefits of weight loss (2–3%) and reductions in systolic blood pressure (4.4 mmHg) [50] that are attributed to caloric loss (glucosuria) and osmotic diuresis, respectively. Several studies have demonstrated the additional glucose-lowering effect of dapagliflozin on a background of insulin or oral glucose-lowering agents, indicating that the mechanism of action is complementary to other antidiabetic therapies [50].
2.2 Chemistry
To generate C-aryl glucosides, particularly with preferred C-4′ and C-4 substitutions, to support initial toxicological studies and Phase I clinical trials, a first generation synthesis was developed. This synthesis, based on the methodology developed by Kishi [51, 52] and Kraus [53], was used to gain rapid access to compounds 13, 15, and 16 starting with 2,3,4,5-tetra-O-benzyl-d-glucose 17 as outlined in Scheme 1. Lactone (18) [54–56] was prepared from compound 19 via TEMPO/bleach oxidation in >90% yield. On a large scale, purification of the syrupy product 20 was not practical; therefore, it was used directly in the next step without purification. Various m-bromo substituted aglycones [41] (19) employed in this route were lithiated and then added to 2,3,4,6-tetra-O-benzylgluconolactone (18). Reduction of the resultant lactols (20) with the sterically encumbered triisopropylsilane was preferred to generate predominantly β-linked glucosides [57] 21 that were deprotected by hydrogenolysis to give amorphous API (7–11, 13, 16) in which “A” is a methylene.

General route employed in the synthesis of C-arylglucosides
During process development of the first generation synthesis, we identified several disadvantages to using 2,3,4,6-tetra-O-benzyl-d-glucono-1,5-lactone 18 as a starting material. For instance, 18 was synthesized from commercially available, but expensive, 2,3,4,6-tetrabenzylglucose 17. Product 18 was difficult to isolate and purify on scale due to its syrupy nature. The crude product also gradually decomposed over several months, thereby limiting its usefulness in a commercial process. The reduction of intermediate lactol 20 required sterically hindered silanes to give favorable β-selectivity, which were expensive and difficult to obtain on commercial scale. More critically, the synthesis of C-arylglycosides via 2,3,4,6-tetra-O-benzyl gluconolactone 18 required a hydrogenolytic deprotection in the final step of the synthesis. Due to the amorphous nature of the active pharmaceutical ingredient (API) (7, 8, 9, 10, 11, 13, or 16) chromatography was required to remove palladium and process impurities. Although this route enabled rapid delivery of API to support early toxicology and preclinical studies, the inability to crystallize API was of paramount concern.
Despite extensive investigation, a neat crystalline form of the API has not been discovered, possibly due to the amphiphilic nature of the molecule. In addition, the amorphous, foamy API becomes a tacky or gummy solid on exposure to moisture. A syrupy, noncrystalline and hygroscopic form presented a formidable challenge to the development team. A lack of sufficient control over the isolation and purification of amorphous API, a high cost of goods, and unfavorable physical characteristics of several intermediates provided motivation for the development of a new process and a new final form for API, particularly to the nominated candidates 13, 14, 15, and 16, which required significant quantity for toxicology and clinical studies.
While some derivatives of glucose (such as 17) can be expensive and as such not realistic starting materials, we did realize the advantages of employing materials from the naturally occurring chiral pool that eliminated the need to control four of the five stereocenters present in the API. This strategy suggested that we should retain the glycoside core of 17 or 18 as a starting material and explore protecting group modifications to circumvent the adverse physical characteristics of the intermediates in the original synthesis. In this manner, proper protecting group selection could offer cost advantages and improvements to the physical form of certain intermediates for ease of isolation. A final desired change would entail the discovery of a simple deprotection method to avoid the use of the hydrogenolysis that was employed in the first generation synthesis.
Avoidance of chromatographic step for the final purification of the amorphous API was critical. We discovered that per-acetylation of API, particularly for compounds 13, 14, 15, and 16 yielded the corresponding tetraacetate 25 as a stable and pure crystalline solid that was amenable to purification by crystalization. The subsequent deprotection of this material to API (13, 14, 15, and 16) was facile and the resulting purity was very high (>99.9 area percent purity). Although the advantages presented by this crystalline derivative were apparent, there was a concern that acetates would not be compatible with the organometallic chemistry used to generate aglycone 17. As an alternative, we employed the transient protection of gluconolactone as the per-trimethylsilyl derivative [58–60], since the trimethylsilyl groups would resist lithioaromatics at low temperature but could also be cleaved during the work-up under mildly acidic conditions. Starting with d-gluconolactone 21, silyl protection was achieved using TMSCl/N-methyl morpholine in THF to give 21a. The work-up of this intermediate required a carefully controlled process as its oily nature and sensitivity to moisture precluded easy isolation. Therefore, the reaction mixture was diluted with toluene prior to aqueous washes; the per-silylated lactone 21a was isolated as a solution. The selection of toluene as solvent was critical as it efficiently extracted the product and enabled azeotropic drying, required in the following step.
Bromo-aglycone 17 was transmetallated with nBuLi to form the lithiated species 17a which was reacted at −78°C with the solution of 21a prepared above to give lactol 22. In this transformation, the toluene/THF solvent combination (3.5:1) proved ideal for yield and product purity; in THF solvent, a competing silyl transfer reaction dominates, forming trimethylsilyl-aglycone as a major by-product [61].
In all cases, lactol 22 was noncrystalline and problematic to isolate, therefore the toluene solution of 22 was quenched into methanol/methanesulfonic acid. This acidic quench served to: (1) neutralize any excess organometallic species, (2) promote in situ trans-ketalization with methanol, and (3) expedite concomitant removal of the TMS protecting groups to produce crude solutions of methyl glycosides 23. Intermediates 23 were typically amorphous, foamy solids. Extensive investigation failed to produce crystalline forms of 23. Thus, after an aqueous work-up, the solutions were dried via toluene azeotrope and further telescoped into the following step. Toluene solutions of 23 were typically isolated in >83% yield.
Based on our previous investigations [58–60], acetate groups were chosen to protect the hydroxyls in 23 since the reduction would provide advantageous crystalline per-acetylated API (13, 15 or 16). Toluene solutions of 23 were exhaustively acetylated with Ac2O/TEA, catalyzed by DMAP, to generate solutions of tetraacetate 24. Following quench of the remaining anhydride by aqueous phosphoric acid and aqueous work-up, the partially concentrated dry toluene solutions were ready for the key transformation: reduction.
We screened a wide variety of anomeric reduction conditions, starting with acid-catalyzed silyl hydride process. We investigated both Lewis and Bronsted acids as potential alternatives to BF3•Et2O along with triethylsilane as an alternative to the tri-isopropyl derivative. While several alternative catalysts gave encouraging results, we decided to retain BF3•Et2O for a combination of cost, product purity, and process robustness. The less expensive and more readily available triethylsilane (Et3SiH) replaced triisopropylsilane (iPr3SiH) since the resulting isomer ratio (>20:1, β:α) was similar.
The optimized process entailed dilution of toluene solutions of 24 with acetonitrile followed by reduction with Et3SiH, BF3•Et2O, and one equivalent of water. Water is critical for the reaction to progress to completion; and we believe that water and BF3•OEt2 generate a strong Bronsted acid, such as H+BF3OH−, which accelerates [62, 63] the formation of an oxa-carbenium ion intermediate. These strongly acidic conditions are essential to compensate for the deactivating effect of the carbohydrate acetoxy-substituents. Based on the literature [64] and our own studies [58–60], we believe that predominant axial hydride attack from the α-face on the oxacarbenium ion intermediate “A” is preferred resulting in β- C-arylglucosides 25 Footnote 2 as the major products (>20:1 β:α). Intramolecular stabilization via the C-2-acetate group (B) which would block the α-face does not seem to result in β-hydride attack (Fig. 6).

Proposed stereochemistry of hydride attack
The use of Et3SiH on scale presented safety concerns. Unreacted silane can generate hydrogen gas in spent reaction streams, which would render the waste streams more hazardous to storage and transport. To mitigate this risk, 2,2-dimethoxypropane was charged to the mature reaction mixture to quench excess Et3SiH. After complete disappearance of triethylsilane by GC analysis, aqueous work-up and crystallization of the product from ethanol produced a ~74% yield of the final intermediates 25 (>98.6 HPLC AP purity). Saponification of tetraacetates 25 (aq. NaOH/EtOH) produced API (13, 14, 15, and 16) as amorphous powders (Scheme 2).

Synthesis of tetraacetate 25
To solve the isolation and final crystal-form issues of the amorphous, amphiphatic drug substances 13, 14, 15, and 16, we first attempted traditional high-throughput crystallization screening studies, testing different solvents, solvent/antisolvent combinations and temperatures. These studies failed to produce a crystalline form. Subsequently, we focused on a co-crystallization approach [65] based on understanding of the structure of the drug substances. Although in principle virtually any pair of molecules may co-crystallize [66], we reasoned that another molecule containing structural elements that could favorably interact with both components of the drug substances should enhance the probability of crystallization. An amphiphilic co-crystallization component might interact with both the polar hydroxyls and the hydrophobic hydrocarbon regions of β- C-arylglucosides (13, 14, 15, and 16). The ideal co-crystallization component would be a GRAS (Generally Regarded as Safe) list compound, mitigating safety concerns. We identified several natural amino acids as possible partners for co-crystallization with 13, 14, 15, and 16 but concentrated on l-Phenylalanine. These efforts provided crystal forms (complexes) 26, 27, and 28, each composed of 1 mol of drug substance + 1 mol l-phenylalanine + 1 mol of water. In case of dapagliflozin, 16, the l-Phenylalanine complex was isolated but its physical properties were deemed not suitable for development. An S-propylene Glycol (S-PG), 29, complex was also identified. Crystals of 29 are fine needles that are stable under ambient conditions.
The structures of these hydrogen bonded complexes could only be solved from synchrotron X-ray diffraction data of the very fine, hair-like crystals (see footnote 2). The S-PG complex, comprised of 1 mol of drug + 1 mol S-PG + 1 mol of water, was found to be stable and suitable for development; in addition, it provided a significant weight advantage over the other complexes. Thus, the S-PG form was chosen for development (Fig. 7).

X-ray structure of complexes 26, 27, 28, and 29
Stable crystalline complexes with l-proline and d-phenylalanine were also prepared and their structures (see footnote 2) were determined via single X-ray analysis. The power of such “foreign” agents as co-crystallization “partners” in assembling crystal structures of pharmaceutical products that alone crystallize with difficulty (if at all) has been increasingly recognized in recent years (dedicated issue on “Pharmaceutical Co-crystal” please see: [67]).
The final step of the synthesis consists of deprotection of the acetyl protecting groups and formation of the crystalline complex. In contrast to the hydrogenation required to cleave the benzyl protecting groups of the final intermediate in the first generation synthesis (Scheme 1), the final step now required only aqueous base to remove the acetates. The saponification is rapid and irreversible and can be performed under a wide range of reagent concentration and temperature conditions.
There are no potential side reactions in this process which can impact the stereochemical integrity of the compound. Thus, in the final step the quality of the product was effectively preserved. Essentially no process changes were required to scale up the final intermediate 25. Deacetylation using EtOH or MeOH and aq. NaOH (Scheme 3) followed by neutralization with aq. HCl to pH 7 generated a solution of compounds 13, 14, 15, or 16 which was converted to the final complexes 26, 27, 28, or 29 respectively and crystallized as a monohydrate. After filtration and drying, the isolated product was obtained in 80–88% yield with 99.9 HPLC Area Percent purity.

Synthesis of co-crystalline complexes 26, 27, 28, and 29
In summary, a robust and practical synthesis of β- 1C-arylglucosides was developed that includes a co-crystalization approach with l-Phenylalanine or S-Propylene glycol to circumvent isolation and purification challenges.
3 Summary, Conclusions, Outlook
In summary, β- 1C-arylglucosides were identified as potent SGLT-inhibitors that are significantly more stable than their O-glycosides counterparts. Initial SAR studies of benzylaryl-O-glucosides led to discovery of C-arylglucosides that showed greater selectivity towards SGLT2 versus SGLT1 with increased metabolic stability. Further exploration of the in vivo SAR studies allowed us to identify superior SGLT2 inhibitors based on a β- 1C-arylglucoside with a diarylmethane moiety and C-4′ and C-4 substitutions. Through this investigation, β- 1C-arylglucoside dapagliflozin 16 was identified as a potent and selective hSGLT2 inhibitor with an EC50 for hSGLT2 of 1.0 nM and 1,600-fold selectivity over hSGLT. Initial toxicological studies, combined with a favorable ADME profile and vivo data, led to development of dapagliflozin 16 as a drug for the treatment of type 2 diabetes. Multiple placebo-controlled clinical trials demonstrated safety and efficacy of dapagliflozin. The results showed patients receiving dapagliflozin experienced improved glucose control, benefits of weight loss, and reductions in systolic blood pressure. The glucose-lowering effect of dapagliflozin occurs on a background of insulin or oral glucose-lowering agents, indicating that the mechanism of action is complementary to other antidiabetic therapies. Dapagliflozin was approved by the Australian TGA and the European Commission under the brand name Forxiga™ in a 10 mg daily dose (5 mg starting dose for severe hepatic impairment) as oral monotherapy for the treatment of diabetes. It was also approved by the US Food and Drug Administration (FDA) under the brand name Farxiga™, as a once-daily oral treatment to improve glycemic control in adults with type 2 diabetes. In addition, the European Commission granted Marketing Authorization for a fixed dose combination product of dapagliflozin and metformin hydrochloride, two anti-hyperglycemic products with complementary mechanisms of action to improve glycemic control, in a twice daily tablet (5 mg/850 mg and 5 mg/1,000 mg tablets) under the brand name Xigduo™. This is the first regulatory approval for a fixed dose combination of an SGLT2 inhibitor and metformin.
Notes
- 1.
Fasted male Sprague–Dawley rats or Swiss–Webster mice were anesthetized with I.P. Ketamine:Xylazine (0.001 mL/g), an abdominal incision made, and their bladders cannulated with a 16 gauge catheter. Drug was administered intravenously, and urine was collected over 60 min in 10 min intervals. Glucose concentration in urine samples were determined by COBAS MIRA.
- 2.
Crystallographic coordinates and unit cell data (excluding structure factors) for 25, 26, 27, 28, 29 and l-Proline forms 13-P1, 13-P2, 13-P3, 13-TP have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 756030–756038. These data can be obtained free of charge via www.ccdc.cam.ac.uk/const/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB21EZ, UK; fax + 44 1223 336033; e-mail deposit@ccdc.cam.ac.uk).
Abbreviations
- μg:
-
Microgram
- μM:
-
Micromolar
- Ac2O:
-
Acetic anhydride
- AcOH:
-
Acetic acid
- ADME:
-
Absorption, distribution, metabolism, and excretion
- AMG:
-
α-Methyl-d-glucopyranoside
- AP:
-
Area percent
- API:
-
Active pharmaceutical ingredient
- BF3•Et2O:
-
Boron trifluoride diethyl etherate
- Bn:
-
Benzyl
- CH3CN:
-
Acetonitrile
- CHO:
-
Chinese hamster ovary
- Cl:
-
Clearance
- C max :
-
Maximum concentration
- dL:
-
Deciliter
- DMAP:
-
4-(dimethylamino)pyridine
- EC50 :
-
Half maximal effective concentration
- EtOH:
-
Ethanol
- Et3SiH:
-
Triethylsilane
- F:
-
Fraction absorbed/bioavailability
- GC:
-
Gas chromatography
- GLUT:
-
Glucose transporter
- GRAS:
-
Generally regarded as safe
- h:
-
Hour(s)
- HbA1c:
-
Glycated hemoglobin
- HPLC:
-
High performance liquid chromatography
- iPr3SiH:
-
Triisopropylsilane
- iPrOAc:
-
Isopropyl acetate
- i.v.:
-
Intravenous
- Kg:
-
Kilogram
- KK-A y :
-
Mouse of the KK-strain carrying the Ay mutation
- L:
-
Liter(s)
- mg:
-
Milligram
- mg/kg:
-
Milligram per kilogram
- mL:
-
Milliliter
- mmHg:
-
Millimeters of mercury
- MeOH:
-
Methanol
- Me:
-
Methyl
- MSA:
-
Methanesulfonic acid
- n-BuLi:
-
n-Butyllithium
- nM:
-
Nanomolar
- nm/s:
-
Nanometer per second
- NaOH:
-
Sodium hydroxide
- NMM:
-
N-Methyl morpholine
- Pd–C:
-
Palladium on carbon
- PO:
-
Per os
- s:
-
Second(s)
- S-PG:
-
S-Propylene glycol
- SAR:
-
Structure–activity relationships
- SD:
-
Sprague Dawley
- SGLT:
-
Sodium-dependent glucose cotransporter
- STZ:
-
Streptozotocin, a pancreatic toxin used to induce diabetes
- TEA:
-
Triethylamine
- TEMPO:
-
(2,2,6,6-Tetramethylpiperidin-1-yl)oxy
- THF:
-
Tetrahydrofuran
- TMSCl:
-
Chlorotrimethylsilane
- T max :
-
Time to reach C max
- TMS:
-
Tetramethylsilane
- Tol:
-
Toluene
- TGA:
-
Therapeutic goods administration
- UKPDS:
-
United Kingdom Prospective Diabetes Study
- V ss :
-
Apparent volume of distribution at steady state
References
Skyler JS (2004) Diabetes mellitus: pathogenesis and treatment strategies. J Med Chem 47:4113–4117
Porte D (2001) Clinical importance of insulin secretion and its interaction with insulin resistance in the treatment of type 2 diabetes mellitus and its complications. Diabetes Metab Res Rev 17:181–188
Kikkawa R (2000) Chronic complications in diabetes mellitus. Br J Nutr 84:S183–S185
Edelman SV (1998) Importance of glucose control. Med Clin N Am 82:665–687
Rotella DP (2004) Novel “second-generation” approaches for the control of type 2 diabetes. J Med Chem 47:4111–4112
Mueller G (2002) Concepts and options for current insulin research and future anti-diabetic therapy. Recent Res Dev Endocrin 3:199–218
UK Prospective Diabetes Study (UKPDS) Group (1998) Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 352:854–865
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Deetjen P, von Baeyer H, Drexel H (1992) Renal glucose transport. In: Seldin DW, Giebisch G (eds) The kidney: physiology and pathophysiology, 2nd edn. Raven Press, New York, pp 2873–2888
Moe OW, Berry CA, Rector FC (2000) Renal transport of glucose, amino acids, sodium, chloride and water. In: Brenner BM, Rector FC (eds) The kidney, 5th edn. WB Saunders, Philadelphia, pp 375–415
Wright EM, Hirayama B, Hazama A, Loo DDF, Supplisson S, Turk E, Hager KM (1993) The sodium/glucose cotransporter (SGLT1). Soc Gen Physiol Ser 48:229–241
Wright EM, Turk E, Hager K, Lescale-Matys L, Hirayama B, Supplisson S, Loo DDF (1992) The sodium/glucose cotransporter (SGLT1). Acta Physiol Scand Suppl 146:201–207
Mackenzie B, Loo DDF, Panayotova-Heiermann M, Wright EM (1996) Biophysical characteristics of the Pig kidney Na+/glucose cotransporter SGLT2 reveal a common mechanism for SGLT1 and SGLT2. J Biol Chem 271:32678–32683
Kanai Y, Lee WS, You G, Brown D, Hediger MA (1994) The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J Clin Invest 93:397–404
Scheepers A, Joost H-G, Schuermann A (2004) The glucose transporter families SGLT and GLUT: molecular basis of normal and aberrant function. J Parent Enter Nutr 28:364–371
Wright EM (2001) Renal Na+-glucose cotransporters. Am J Physiol Renal Physiol 280:F10–F18
Wallner EI, Wada J, Tramonti G, Lin S, Kanwar YS (2001) Status of glucose transporters in the mammalian kidney and renal development. Ren Fail 23:301–310
You G, Lee WS, Barros EJG, Kanai Y, Huo TL, Khawaja S, Wells RG, Nigam SK, Hediger MA (1995) Molecular characteristics of Na+-coupled glucose transporters in adult and embryonic Rat kidney. J Biol Chem 270:29365–29371
Hediger MA, Rhoads DB (1994) Molecular physiology of sodium-glucose cotransporters. Physiol Rev 74:993–1026
Wells RG, Pajor AM, Kanai Y, Turk E, Wright EM, Hediger MA (1992) Cloning of a human kidney cDNA with similarity to the sodium-glucose cotransporter. Am J Physiol 263:F459–F465
Kasahara M, Maeda M, Hayashi S, Mori Y, Abe T (2001) A missense mutation in the Na+/glucose cotransporter gene SGLT1 in a patient with congenital glucose-galactose malabsorption: normal trafficking but inactivation of the mutant protein. Biochim Biophys Acta 1536:141–147
Turk E, Zabel B, Mundlos S, Dyer J, Wright EM (1991) Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature 350:354–356
van den Heuvel LP, Assink K, Willemsen M, Monnens L (2002) Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet 111:544–547
Santer R, Kinner M, Schneppenheim R, Hillebrand G, Kemper M, Ehrich J, Swift P, Skovby F, Schaub J (2000) The molecular basis of renal glucosuria: mutations in the gene for a renal glucose transporter (SGLT2). J Inherit Metab Dis 23(Suppl 1):178
Oku A, Ueta K, Arakawa K, Ishihara T, Nawano M, Kuronuma Y, Matsumoto M, Saito A, Tsujihara K, Anai M, Asano T, Kanai Y, Endou H (1999) T-1095, an inhibitor of renal Na+-glucose cotransporters, may provide a novel approach to treating diabetes. Diabetes 48:1794–1800
Ehrenkranz JRL, Lewis NG, Kahn CR, Roth J (2005) Phlorizin: a review. Diabetes Metab Res Rev 21:31–38
Jonas J-C, Sharma A, Hasenkamp W, Ilkova H, Patanè G, Laybutt R, Bonner-Weir S, Weir G (1999) Chronic hyperglycemia triggers loss of pancreatic β-cell differentiation in an animal model of diabetes. J Biol Chem 274:14112–14121
Dimitrakoudis D, Vranic M, Klip A (1992) Effects of hyperglycemia on glucose transporters of the muscle: use of the renal glucose reabsorption inhibitor phlorizin to control glycemia. J Am Soc Nephrol 3:1078–1091
Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA (1987) Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Invest 79:1510–1515
Doggrell SA, Castaner J (2001) T-1095 antidiabetic sodium-glucose cotransporter inhibitor. Drugs Future 26:750–753
Arakawa K, Ishihara T, Oku A, Nawano M, Ueta K, Kitamura K, Matsumoto M, Saito A (2001) Improved diabetic syndrome in C57BL/KsJ-db/db mice by oral administration of the Na+-glucose cotransporter inhibitor T-1095. Br J Pharmacol 132:578–586
Oku A, Ueta K, Arakawa K, Kano-Ishihara T, Matsumoto M, Adachi T, Yasuda K, Tsuda K, Saito A (2000) Antihyperglycemic effect of T-1095 via inhibition of renal Na+-glucose cotransporters in streptozotocin-induced diabetic rats. Biol Pharm Bull 23:1434–1437
Adachi T, Yasuda K, Okamoto Y, Shihara N, Oku A, Ueta K, Kitamura K, Saito A, Iwakura T, Yamada Y, Yano H, Seino Y, Tsuda K (2000) T-1095, a renal na+-glucose transporter inhibitor, improves hyperglycemia in streptozotocin-induced diabetic rats. Metab Clin Exp 49:990–995
Tsujihara K, Hongu M, Saito K, Kawanishi H, Kuriyama K, Matsumoto M, Oku A, Ueta K, Tsuda M, Saito A (1999) Na+-glucose cotransporter (SGLT) inhibitors as antidiabetic agents. 4. Synthesis and pharmacological properties of 4′-dehydroxyphlorizin derivatives substituted on the B ring. J Med Chem 42:5311–5324
Oku A, Ueta K, Nawano M, Arakawa K, Kano-Ishihara T, Matsumoto M, Saito A, Tsujihara K, Anai M, Asano T (2000) Antidiabetic effect of T-1095, an inhibitor of Na+-glucose cotransporter, in neonatally streptozotocin-treated rats. Eur J Pharmacol 391:183–192
Nawano M, Oku A, Ueta K, Umebayashi I, Ishirahara T, Arakawa K, Saito A, Anai M, Kikuchi M, Asano T (2000) Hyperglycemia contributes insulin resistance in hepatic and adipose tissue but not skeletal muscle of ZDF rats. Am J Physiol Endocrinol Metab 278:E535–E543
Kikuchi N, Fujikura H, Tazawa S, Yamato T, Isaji M (2004) Preparation of pyrazole glycoside compounds as SGLT inhibitors. PCT Int. Appl. WO2004113359; (2004) Chem Abstr 142:94061
Fushimi N, Yonekubo S, Muranaka H, Shiohara H, Teranishi H, Shimizu K, Ito F, Isaji M (2004) Preparation of glucopyranoside compounds having fused heterocycle as SGLT inhibitors. PCT Int. Appl. WO2004087727; (2004) Chem Abstr 141:332411
Fujikura H, Nishimura T, Katsuno K, Isaji M (2004) Preparation of D-glucose derivatives as human SGLT2 inhibitors. PCT Int. Appl. WO2004058790; (2004) Chem Abstr 141:123854
Fushimi N, Ito F, Isaji M (2003) Preparation of glucopyranosyloxybenzylbenzene derivatives as inhibitors of human SGLT2 (sodium-dependent glucose-transporter 2), medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof. PCT Int. Appl. WO2003011880; (2003) Chem Abstr 138:153771
Ellsworth BA, Meng W, Patel M, Girotra RN, Wu G, Sher PM, Hagan DL, Obermeier MT, Humphreys WG, Robertson JG, Wang A, Han S, Waldron TL, Morgan NN, Whaley JM, Washburn WN (2008) Aglycone exploration of C-arylglucoside inhibitors of renal sodium-dependent glucose transporter SGLT2. Bioorg Med Chem Lett 18:4770–4773
Link JT, Sorensen BK (2000) A method for preparing C-glycosides related to phlorizin. Tetrahedron Lett 41:9213–9217
Kishi Y (1993) Preferred solution conformation of marine natural product palytoxin and of C-glycosides and their parent glycosides. Pure Appl Chem 65:771–778
Wang Y, Goekjian PG, Ryckman DM, Miller WH, Babirad SA, Kishi Y (1992) Preferred conformation of C-glycosides 9. conformational analysis of 1,4-linked carbon disaccharides. J Org Chem 57:482–489
Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN (2008) Discovery of dapagliflozin: a potent, selective renalsodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2diabetes. J Med Chem 51:1145–1149
Hongu M, Tanaka T, Funami N, Saito K, Arakawa K, Matsumoto M, Tsujihara K (1998) Na + glucose cotransporter inhibitors as a antidiabetic agents. II. Synthesis and structure-activity relationships of 4′-dehydroxyphlorizin derevatives. Chem Pharm Bull 46:22–33
Wood IS, Trayhurn P (2003) Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr 89:3–9
Washburn WN, Meng W (2006) C-aryl glucoside SGLT2 inhibitors and method for the treatment of diabetes and related diseases. US. Pat. Appl. Publ. US 20060063722 A1 20060323
Obermeier MT, Yao M, Khanna A, Koplowitz B, Zhu M, Li W, Komoroski B, Kasichayanula S, Discenza L, Washburn W, Meng W, Ellsworth BA, Whaley JM, Humphreys WG (2009) In vitro characterization and pharmacokinetics of dapagliflozin (BMS-512148), a potent sodium-glucose cotransporter type II (SGLT2) inhibitor, in animals and humans. Drug Metab Dispos 38:405–414
Washburn WN, Poucher SM (2013) Differentiating sodium-glucose co-transporter-2 inhibitors in development for the treatment of type 2 diabetes mellitus. Expert Opin Investig Drugs 22:463–486
Lewis MD, Cha K, Kishi Y (1982) Highly stereoselective approaches to α- and β- C-glycopyranosides. J Am Chem Soc 104:4976–4978
Babirad SA, Wang Y, Kishi Y (1987) Synthesis of C-disaccharides. J Org Chem 52:1370–1372
Kraus GA, Molina MT (1988) A direct synthesis of C-glycosyl compound. J Org Chem 53:752–753
Rajanikanth B, Seshadri R (1989) A facile synthesis of nojirimycin. Tetrahedron Lett 30:755–758
Benhaddou R, Czernecki S, Farid W, Ville G, Xie J, Zegar A (1994) Tetra-n-propylammonium tetra-oxoruthenate(VII): a reagent of choice for the oxidation of diversely protected glycopyranoses and glycofuranoses to lactones. Carbohydr Res 260:243–250
Olah GA, Wang Q, Prakash GKS (1992) Synthetic methods and reactions 177. Ionic hydrogenation with triethylsilane-trifluoroacetic acid-ammonium fluoride or triethylsilane-pyridinium poly(hydrogen fluoride). Synlett 647–650
Ellsworth BA, Doyle AG, Patel M, Caceres-Cortes J, Meng W, Deshpande PP, Pullockaran A, Washburn WN (2003) C-Arylglucoside synthesis: triisopropylsilane as a selective reagent for the reduction of an anomeric C-phenyl ketal. Tetrahedron Asymmetry 14:3243–3247
Deshpande PP, Ellsworth BA, Buono FG, Pullockaran A, Singh J, Kissick TP, Huang M-H, Lobinger H, Denzel T, Mueller RH (2007) Remarkable β-1-C-arylglucosides: stereoselective reduction of acetyl-protected methyl 1- C-aryl-glucosides without acetoxy-group participation. J Org Chem 72(25):9746–9749
Horton D, Priebe W (1981) Synthetic routes to higher-carbon sugars. Reaction of lactones with 2-lithio-1,3-dithiane. Carbohydr Res 94(1):27–41
Deshpande PP, Singh J, Pullockaran A, Kissick T, Ellsworth BA, Gougoutas JZ, Dimarco J, Fakes M, Reyes M, Lai C, Lobinger H, Denzel T, Ermann P, Crispino G, Randazzo M, Gao Z, Randazzo R, Lindrud M, Rosso V, Buono F, Doubleday WW, Leung S, Richberg P, Hughes D, Washburn WN, Meng W, Volk KJ, Mueller RH (2012) A practical stereoselective synthesis and novel cocrystallization of an amphiphatic SGLT2 inhibitor. Org Process Res Dev 16:577–585
Barrett AGM, Pena M, Willardsen AJ (1996) Total synthesis and structural elucidation of the antifungal agent Papulacandin D. J Org Chem 61:1082–1100
Akiyama T, Takaya J, Kagoshima H (1999) Chemoselctive activation of aldimines in preference to aldehydes by the combination of BF3.OEt2 and water: novel catalyst for the Mannich-type reation. Chem Lett 28:947–948
Ribe S, Wipf P (2001) Water-accelerated organic tranformations. Chem Commun 299–307
Deslongchamps P (1993) Intramolecular strategies and stereoelectronic effects. Glycosides hydrolysis revisited. Pure Appl Chem 65:1161–1178
Gougoutas JZ (2002) Amino acid complexes of C-aryl glucosides for treatment of diabetes PCT Int. Appl, p 80, WO 2002083066
Mutig T, Kemnitz E, Troyanov SI (2010) Synthesis and molecular structures of heptafluoropropylated fullerenes: C70(n-C3F7)8, C70(n-C3F7)6O, and C70(C3F7). J Flour Chem 131:861–866
RodrÚguez-Hornedo N (2007) Molecular design of pharmaceutical materials. Mol Pharm 4:299–434
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Braem, A., Deshpande, P.P., Ellsworth, B.A., Washburn, W.N. (2014). Discovery and Development of Selective Renal Sodium-Dependent Glucose Cotransporter 2 (SGLT2) Dapagliflozin for the Treatment of Type 2 Diabetes. In: Seeberger, P., Rademacher, C. (eds) Carbohydrates as Drugs. Topics in Medicinal Chemistry, vol 12. Springer, Cham. https://doi.org/10.1007/7355_2014_41
Download citation
DOI: https://doi.org/10.1007/7355_2014_41
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-08674-3
Online ISBN: 978-3-319-08675-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)