
Abstract
The present study was carried out to investigate the effect of trimetazidine on the course of pentylenetetrazole (PTZ)-induced chemical kindling and oxidative stress markers in PTZ-kindled mice. Kindling was induced by repeated injections of a subconvulsive dose of PTZ (30 mg⁄kg, i.p.) on alternate days for 5 weeks or until stage 4 of the seizure score was evoked on three consecutive administrations. Trimetazidine was administered daily in three doses (5, 10 and 20 mg/kg) per orally (p.o.) along with alternate-day PTZ. Following PTZ kindling, oxidative stress parameters, i.e. levels of malondialdehyde (MDA) and reduced glutathione (GSH), were assessed in isolated homogenized whole brain tissue. The results showed that PTZ treatment progressively increased the seizure score in control mice. Biochemical analysis revealed a significant increase in MDA levels and decreased GSH levels in the brain homogenate of PTZ-kindled mice. Daily treatment with trimetazidine in doses of 10 and 20 mg/kg significantly decreased the PTZ-induced seizure score. However, a low dose of trimetazidine (5 mg/kg) failed to improve the seizure score. Pretreatment of trimetazidine in all doses showed an ameliorating effect on biochemical alteration induced by PTZ treatment. The results of the present study indicate the potential anticonvulsant activity of trimetazidine against PTZ-induced kindling in mice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epilepsy is one of the commonest neurological disorders of the brain, affecting approximately 1% of the population worldwide (Dhir et al. 2005; de Oliveira et al. 2008). The pathogenesis of neuronal damage in response to seizure has been widely studied. A number of observations indicate that a series of events occur in the process of epileptogenesis, leading to neuronal damage. Amongst these alterations in various ionic currents, oxidative damage via production of free radicals, calcium (Ca+2) influx into the cells, nitric oxide production and myocardial dysfunctions play an important role in epileptogenesis (Frantseva et al. 2000a; Waldbaum and Patel 2010). A growing body of evidence indicates the role of oxidative stress and mitochondrial dysfunctions both as consequence and as a cause of epileptic seizure (Patel 2004).
The generation of free radicals in the brain has been documented as a common pathway for cellular injury taking place in many neurological disorders including epilepsy (Patel 2002; Dhir et al. 2005; Akula et al. 2007). The role of oxidative stress has been implicated in the initial phases of seizure-induced pathology, as several studies have reported the production of reactive oxygen species in different brain regions following experimental seizures (Maertens et al. 1995; Frantseva et al. 2000b; Patel 2004; Devi et al. 2008; Waldbaum and Patel 2010). The role of oxidative stress in epilepsy is also supported by studies which suggest that exogenously administered antioxidants protected the brain against seizures (Nakao et al. 1996; Kabuto et al. 1998; Bashkatova et al. 2003; Gupta et al. 2003). Thus, drugs with antioxidant properties could be candidates for the prevention of oxidative damage and its consequences, like epilepsy.
Trimetazidine (1(2, 3, 4- trimethoxy-benzyl) piperazine, 2 HCl) is a well-known anti-ischemic drug. It has demonstrated protection against ischemic injury in a variety of tissues. In experimental studies, it has demonstrated gastroprotective, hepatoprotective, anti-inflammatory, anti-nociceptive and anti-apoptotic activities (Yin et al. 2004; Abdel-Salam and El-Batran 2005). Being lipid soluble in nature, it can cross the blood brain barrier (Harpey et al. 1989).
The mechanistic approach has shown its potent antioxidant activity on various tissues by reducing the excessive release of oxygen free radicals and inorganic phosphates (Maridonneau-Parini and Harpey 1985; Guarnieri and Muscari 1990). Moreover, trimetazidine has been reported to optimize energy metabolism in tissues by improving ATP production, reducing membrane lipid peroxidation, limiting the overproduction of free radicals, maintaining the activity of antioxidant enzymes and increasing the utilization of glucose (Aubert et al. 1989). Recently, some reports indicate the neuroprotective effects of trimetazidine in various experimental models. These effects have been suggested to be due to its antioxidant property (Mironovan et al. 2001; Dhote and Balaraman 2008; Serarslan et al. 2009).
Baltalarli et al. (2000) and Iqbal et al. (2002) showed that administration of trimetazidine during the period of ischemia–reperfusion injury reduced the number of damaged cells in cerebral tissue and preserved neurological function in transient global ischemia model in rats and gerbils, respectively. Another study demonstrated a neuroprotective effect of trimetazidine against excitotoxicity in vestibular ganglionic neurons of rats presumably mediated through the inhibition/modulation of AMPA/kainate receptors (Dayanithi et al. 2007). In rats, the protective effect of trimetazidine through its antioxidant activity has been shown against iron-induced epilepsy (Suzer et al. 2000). Trimetazidine protected the kidney and heart tissue from injury mediated by lipid peroxidation in experimental models of renal and cardiac damage (Guarnieri and Muscari 1990; Grekas et al. 1996). Recently, we have reported that trimetazidine is protective in an acute model of electroshock convulsions in mice, possibly through modulation of calcium channels (Jain et al. 2010). For further confirmation of the anticonvulsant activity of trimetazidine against epileptogenesis, the present study was carried out to assess its effect on pentylenetetrazole (PTZ)-induced kindling in mice.
Kindling is a widely used chronic model of epilepsy. Several studies describe its validity and application to study seizure, underlying the course of seizure and testing of anticonvulsant drugs. Kindling refers to a phenomenon in which generalised seizures are developed following repeated administration of subconvulsive electrical or chemical stimuli due to progressive intensification of seizure activity (Ali et al. 2005; de Oliveira et al. 2008). Kindling induced by PTZ is extensively used as a laboratory model of human partial complex epilepsy (Mason and Cooper 1972; Corda et al. 1990). Therefore, the present study was aimed to explore the possible effect of trimetazidine on a PTZ-induced kindling model in mice.
Materials and methods
Animals
Healthy, albino male mice weighing between 25 and 30 g, obtained from the central animal house of the University College of Medical Sciences (University of Delhi) and GTB Hospital, were used. The animals were housed in polypropylene cages (43 × 28.6 × 15.5 cm) in groups of ten mice per cage with free access to pellet diet and water and kept under controlled environmental conditions (temperature, 22 ± 2°C; humidity, 50–55%; natural light/day cycle). The animals were taken care of in accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals, India. The study was approved by the Institutional Animal Ethics Committee, University College of Medical Sciences, Delhi, India.
Drugs
Trimetazidine tablets (Trivedon, 20 mg tabs; Cipla Ltd, Mumbai, India) and PTZ powder (Sigma, MO, USA) were used in the study. The trimetazidine tablets were crushed and suspended with 1% carboxymethyl cellulose (CMC) in double-distilled water. PTZ was dissolved in normal saline. The control animals received a suspension of 1% CMC in double-distilled water, per orally daily. All drugs were given in a volume of 10 ml/kg.
Experimental design
Animals were randomly divided into five groups including 20 mice in each group. The first group of mice received vehicle only throughout the study and served as control group. Other groups were administered daily with vehicle (1% CMC in double-distilled water) or trimetazidine (5, 10 and 20 mg/kg) per orally in addition to alternate-day PTZ for 5 weeks. All animals were injected with a subconvulsant dose of PTZ (30 mg/kg, i.p.) on every alternate day till the kindling develops fully or to a maximum period of 5 weeks. Trimetazidine and the vehicle were given 1 hr before PTZ. After the study, the animals were sacrificed, and the whole brain was dissected for estimation of markers of oxidative stress.
The following groups were included:
-
Group 1:
vehicle (1% CMC) daily + saline, i.p. (on alternate days)
-
Group 2:
vehicle (1% CMC) daily + PTZ (30 mg/kg, i.p., on alternate days)
-
Group 3:
trimetazidine (5 mg/kg, p.o.) daily + PTZ (30 mg/kg, i.p., on alternate days)
-
Group 4:
trimetazidine (10 mg/kg, p.o.) daily + PTZ (30 mg/kg, i.p., on alternate days)
-
Group 5:
trimetazidine (20 mg/kg, p.o.) daily + PTZ (30 mg/kg, i.p., on alternate days)
PTZ-induced kindling
To induce kindling, a subconvulsant dose of PTZ (30 mg/kg, i.p.) was injected on every alternate day. Immediately after the PTZ injection, the animals were observed for the occurrence of convulsive behaviour for 30 min (Racine 1972).
The severity of seizure response was quantified using a five-point scoring system:
-
Stage 0:
no response
-
Stage 1:
ear and facial jerks
-
Stage 2:
myoclonic body jerks without upright position
-
Stage 3:
myoclonic jerks, upright position with bilateral forelimb clonus
-
Stage 4:
clonic–tonic seizures
-
Stage 5:
generalised clonic–tonic seizures, loss of postural control
The maximum response was recorded in each animal. When the animal had a seizure score of 4 on three consecutive administrations, it was defined as being kindled, and its treatment was discontinued. In all the groups, the animals that did not develop a seizure score of 4 were administered the vehicle and/or trimetazidine daily for 5 weeks. Cumulative kindling score (calculated by taking the average of all the individual behavioural scores and then dividing them with the number of animals) was plotted against duration of treatment.
Assessment of oxidative stress
At the end of the study, the animals were killed under ether anaesthesia. The brains were removed and rinsed with cold 0.9% saline, weighed and stored at −80°C until processing.
Tissue preparation
The brain tissues were homogenized with a volume of 10 times (w/v) ice-cold, 0.1 M sodium phosphate buffer (pH 7.4). The homogenate was centrifuged at 3,000 rpm for 15 min, and aliquots of homogenates were used for estimation of malondialdehyde (MDA) and reduced glutathione (GSH).
Measurement of lipid peroxidation
MDA, a measure of lipid peroxidation, was measured as described by Okhawa et al. (1979). Briefly, brain tissues were homogenized with 10 times (w/v) 0.1 M sodium phosphate buffer (pH 7.4). The reagents acetic acid 1.5 ml (20%), pH 3.5, 1.5 ml thiobarbituric acid (0.8%) and 0.2 ml sodium dodecyl sulphate (8.1%) were added to 0.1 ml of processed tissue sample. The mixture was then heated at 100°C for 60 min. The mixture was cooled with tap water, and 5 ml of n-butanol:pyridine (15:1 v/v) and 1 ml of distilled water were added. The mixture was vortexed vigorously. After centrifugation at 4,000 rpm for 10 min, the organic layer was withdrawn, and absorbance was measured at 532 nm using a spectrophotometer. The concentration of MDA was expressed as nanomoles per gram tissue.
Measurement of GSH
GSH was measured according to the method of Ellman (1959). Briefly, brain tissues were homogenized with 10 times (w/v) 0.1 M sodium phosphate buffer (pH 7.4). This homogenate was then centrifuged with 5% trichloroacetic acid to centrifuge out the proteins. To 0.1 ml of this homogenate, 2 ml of phosphate buffer (pH 8.4), 0.5 ml of 5, 5′-dithiobis (2-nitrobenzoic acid) and 0.4 ml of double-distilled water were added. The mixture was vortexed, and the absorbance read at 412 nm within 15 min. The concentration of GSH was expressed as micrograms per gram tissue.
Statistical analysis
All results are expressed as means ± standard error of the mean (S.E.M.). The data obtained were analysed using one-way analysis of variance (ANOVA) followed by post-hoc Tukey's test. A value of P < 0.05 was considered significant for comparison.
Results
Effect of trimetazidine on the development of PTZ kindling
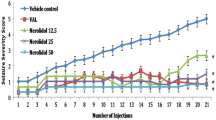
The repeated administration of subconvulsant dose of PTZ (30 mg/kg, i.p., on every alternate day) in the PTZ + vehicle-treated group resulted in increasing convulsive activity culminating into generalised clonic–tonic seizures, as revealed by a progressive increase in seizure score. Pretreatment with trimetazidine (5 mg/kg, p.o., daily, 1 hr before PTZ) did not modify the course of kindling induced by PTZ as compared to the PTZ-kindled group. In the PTZ + trimetazidine (5 mg/kg)-treated group, the administration of trimetazidine caused the appearance of stage 4 in six mice with five injections of PTZ (i.e. 9 doses of trimetazidine). The subsequent injections of PTZ in the trimetazidine (5 mg/kg)-treated group resulted in increased convulsive activity. However, the administration of trimetazidine in doses of 10 and 20 mg/kg suppressed the kindled seizure significantly, compared to the PTZ + vehicle-treated group (Fig. 1).

Effect of trimetazidine (5, 10 and 20 mg/kg, p.o.) administration on the PTZ-induced mean seizure score in mice (n = 20 in each group), assessed on weeks 1, 2, 3, 4 and 5 of the study. a P < 0.05, b P < 0.01, c P < 0.001 compared to the vehicle + PTZ-treated group. ** P < 0.01, *** P < 0.001 compared to the vehicle-treated group (ANOVA followed by Tukey's test)
Effect of trimetazidine on MDA and GSH levels
Repeated treatment with PTZ induced oxidative stress as indicated by a significant rise in brain MDA level. In the PTZ + vehicle-treated group, the MDA level was significantly higher than the saline + vehicle-treated group. However, trimetazidine (5, 10 and 20 mg/kg)-treated PTZ groups showed significantly lower levels of MDA as compared with the PTZ + vehicle-treated group (Fig. 2).There was a marked decrease in the GSH level in the PTZ-kindled group as compared with the saline + vehicle-treated group. The PTZ-treated mice administered with trimetazidine, 5, 10 and 20 mg/kg, showed higher GSH levels as compared to the PTZ-kindled mice (Fig. 3).

Effect of trimetazidine (5, 10 and 20 mg/kg, p.o.) administration on brain lipid peroxidation levels (MDA levels) in nanomoles per gram of wet tissue in the PTZ-treated mice (n = 20 for each group). Values are expressed as means ± SEM. ** P < 0.01, *** P < 0.001 compared to the vehicle + PTZ-treated group. **** P < 0.001 compared to the vehicle + saline-treated group (ANOVA followed by Tukey's test)

Effect of trimetazidine (5, 10 and 20 mg/kg, p.o.) administration on brain GSH levels in (micrograms per gram of wet tissue) in the PTZ-treated mice (n = 20 for each group). Values are expressed as means ± SEM. * P < 0.05 ** P < 0.01, compared to the vehicle + PTZ-treated group. *** P < 0.001 compared to the vehicle + saline-treated group (ANOVA followed by Tukey's test)
Discussion
The results of the present study indicate that administration of trimetazidine has a protective effect against PTZ-induced kindling in mice. It was observed that trimetazidine (10 and 20 mg/kg, p.o.) significantly reduced the seizure score in mice as compared to the PTZ + vehicle-treated group. However, in a dose of 5 mg/kg, trimetazidine failed to reduce the seizure score.
Results from previous studies have suggested that PTZ-induced kindling is associated with enhanced activity of a subpopulation of glutamatergic synapses using N-methyl-d-aspartate (NMDA) receptors, increase in extracellular glutamate levels and subsequent free radical species generation in neurons (Erakovic et al. 2003; Groves et al. 2006). Numerous studies have shown that oxidative stress plays an important role in brain tissue damage during seizure in the PTZ kindling model of epilepsy (Rauca et al. 1999; Frantseva et al. 2000b; Aldarmaa et al. 2010). Increased lipid peroxidation and a decrease in antioxidant enzymes have been reported following PTZ-induced kindling in both mice and rats (Erakovic et al. 2003; Singh et al. 2003). In line with previous studies, results of the present study also show an increase in the MDA and a decrease in GSH levels in brain tissue after PTZ administration. However, trimetazidine pretreatment in all the three doses reversed this increase in lipid peroxidation and increased the GSH levels. These results suggest that trimetazidine reduced oxidative brain damage after PTZ-induced kindling, enlightening its antioxidant property. Evidence from several studies indicated that administration of various antioxidants like lipoic acid, melatonin, N-acetylcysteine, resveratrol, tocopherol and vitamin C has protective effects against seizure manifestations and associated biochemical changes (Kabuto et al. 1998; Sudha et al. 2001; Sinha et al. 2002; Srivastava et al. 2002; Devi et al. 2008). Thus, in the present study, the observed protective effect of trimetazidine against PTZ-induced kindling could be due to the inhibition of oxidative injury, although other mechanisms such as effect on calcium channels, improvement of mitochondrial functions and effect on glutamate levels may also be playing a role in the anticonvulsant effect of trimetazidine.
Reports indicate that mitochondrial dysfunctions have an important role in the pathophysiology of neurological diseases such as epilepsy (Kudin et al. 2009). Neuropathological investigations indicate a similarity between ischemic and seizure-related alterations of neurons characterized by swollen and often disrupted mitochondria (Meldrum 1993). Impairment of mitochondrial functions lead to a reduction in ATP levels inside the cells, disturbances in neuronal calcium homeostasis, oxidative damage and the induction of apoptosis by opening the mitochondrial permeability transition pores (mPTP) that modulate neuronal excitability and synaptic transmission in epilepsy (Schinder et al. 1996; Javadov et al. 2009). Recently, mitochondria have emerged as promising targets for potential neuroprotective strategies in epilepsy (Bindokas et al. 1998; Kudin et al. 2009). Trimetazidine acts as a mitochondrial protective agent and has been shown to restore impaired mitochondrial functions (Guarnieri and Muscari 1993). Trimetazidine binding sites have been identified in purified brain mitochondria of rats (Morin et al. 2000). Several in vitro studies suggest that trimetazidine might modulate mitochondrial permeability transition. Argaud et al. (2005) reported trimetazidine, by inhibiting mPTP opening, protected the rabbit heart from prolonged ischemia–reperfusion injury. Topiramate, a well-known antiepileptic drug, also has been shown to be neuroprotective in the pilocarpine rat model of chronic epilepsy which is considered due to its inhibitory action on the mitochondrial permeability transition (Kudin et al. 2004). Another study showed that trimetazidine restored ATP synthesis in mitochondria of cyclosporine-exposed rat brain, indicating its mitochondria protective activity (Salducci et al. 1996).
Furthermore, the enhanced activity of glutamatergic neurotransmission system also plays an important role in the induction of convulsions. During epileptogenesis, increased production of free radicals can induce increased release of glutamate. Increased activity of glutamate transmitter causes neuronal damage and death by activation of membrane channels that induce massive Ca+2 influx in cells and generation of more free radicals which plays a crucial role in neuronal cell death during PTZ kindling in rats (Schroder et al. 1993; Rocha et al. 1996). Dayanithi et al. (2007) found that trimetazidine also modulates non-NMDA receptors. It blocked the increase in intracellular calcium by inhibiting the current through AMPA/kainate receptors in vestibulo-cochlear neurons and thus prevented neuronal depolarization and subsequent Ca+2 influx through voltage-dependent calcium channels. Thus, protection afforded by trimetazidine in the present study could be correlated to an indirect effect on the glutamatergic system and on the mitochondrial functions, and further studies could be carried out to establish these facts.
The anticonvulsant effect of various antiepileptic drugs like phenytoin, valproate, phenobarbitone and carbamazepine has been studied in the kindling model of epilepsy. Among these, valproate and phenobarbitone have been found to inhibit the development of kindling in experimental animals; however, phenytoin and carbamazepine failed to inhibit kindling development (Wada 1977; Albertson et al. 1984; Schmutz et al. 1988; Silver et al. 1991). Phenytoin, carbamazepine and phenobarbitone have been reported to cause an imbalance between the oxidative and antioxidant statuses. In experimental studies, treatments with phenytoin, carbamazepine and phenobarbitone have resulted in an increased level of MDA and decreased GSH level in the brain, while valproate has shown antioxidant action (Winn et al. 2003; Ilhan et al. 2005; Aycicek and Iscan 2007; Reeta et al. 2009; Arora et al. 2010). In the present study, trimetazidine is not only suppressing seizures but also reducing oxidative stress. In the background of the above-mentioned findings, it appears that trimetazidine has anticonvulsant and antioxidant effects against PTZ-induced kindled seizure similar to sodium valproate.
In conclusion, trimetazidine showed anticonvulsant effect against PTZ-kindled seizure in mice. Pretreatment with trimetazidine also significantly decreased MDA and increased GSH levels. These data support a role of trimetazidine in epilepsy in part at least by inhibition of oxidative injury. Our study is a preliminary study about the anticonvulsant and antioxidant effects of trimetazidine on PTZ-kindled seizure. Furthermore, behavioural and neurochemical studies are essential to determine the exact mode of action of trimetazidine and to identify its possible interaction with other neurotransmission systems involved in epilepsy.
References
Abdel-Salam OME, El-Batran S (2005) Pharmacological investigation of trimetazidine in models of inflammation, pain and gastric injury in rodents. Pharmacology 75:122–132
Akula KK, Dhir A, Kulkarni SK (2007) Systemic administration of adenosine ameliorates pentylenetetrazol-induced chemical kindling and secondary behavioral and biochemical changes in mice. Fundam Clin Pharmacol 21:583–594
Albertson TE, Joy R, Stark LG (1984) Carbamazepine: a pharmacological study in the kindling model of epilepsy. Neuropharmacology 23:1117–1123
Aldarmaa J, Liu Z, Long J, Mo X, Ma J, Liu J (2010) Anti-convulsant effect and mechanism of Astragalus mongholicus extract in vitro and in vivo; protection against oxidative damage and mitochondrial dysfunction. Neurochem Res 35:33–41
Ali A, Ahmad FJ, Pillai KK, Vohora D (2005) Amiloride protects against pentylenetetrazole-induced kindling in mice. Br J Pharmacol 145:880–884
Argaud L, Gomez L, Gateau-Roesch O, Couture-Lepetit E, Loufouat J, Robert D, Ovize M (2005) Trimetazidine inhibits mitochondrial permeability transition pore opening and prevents lethal ischemia–reperfusion injury. J Mol Cell Cardiol 39:893–899
Arora T, Mehta AK, Sharma KK, Mediratta PK, Banerjee BD, Garg GR, Sharma AK (2010) Effect of carbamazepine and lamotrigine on cognitive function and oxidative stress in brain during chemical epileptogenesis in rats. Basic Clin Pharmacol Toxicol 106:372–377
Aubert A, Bernard C, Clauser P, Harpey C, Vaudry H (1989) Effect of phenazine methosulfate on electrophysiological activity of the semicircular canal: antioxidant properties of trimetazidine. Eur J Pharmacol 174:215–225
Aycicek A, Iscan A (2007) The effects of carbamazepine, valproic acid and phenobarbital on the oxidative and antioxidative balance in epileptic children. Eur Neurol 57:65–69
Baltalarli A, Coskun E, Us MH, Rendeci O, Ortac R, Sirin BH (2000) Cerebral protection with trimetazidine in transient brain ischemia in rats. Int J Angiol 9:183–187
Bashkatova V, Narkevich V, Vitskova G et al (2003) The influence of anticonvulsant and antioxidant drugs on nitric oxide level and lipid peroxidation in the rat brain during pentylenetetrazole–induced epileptiform model seizures. Prog Neuropsychopharmacol Biol Psychiatry 27:487–492
Bindokas VP, Lee CC, Colmers WF, Miller RJ (1998) Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. J Neurosci 18:4570–4587
Corda MS, Giorgi O, Longoni M, Biggio G (1990) Decrease in the function of GABA-coupled chloride channel produced by the repeated administration of PTZ to rats. J Neurochem 55:1216–1221
Dayanithi G, Desmadryl G, Travo C, Chabbert C, Sans A (2007) Trimetazidine modulates AMPA/kainate receptors in rat vestibular ganglion neurons. Eur J Pharmacol 574:8–14
De Oliveira PA, Lino FL, Cappelari SE, da Silva Brum LF, Picada JN, Pereira P (2008) Effect of gamma-decanolactone on seizures induced by PTZ-kindling in mice. Exp Brain Res 187:161–166
Devi PU, Manocha A, Vohora D (2008) Seizures, antiepileptics, antioxidants and oxidative stress: an insight for researchers. Expert Opin Pharmacother 9:3169–3177
Dhir A, Naidu OS, Kulkarni SK (2005) Effect of naproxen, a nonselective cyclo-oxygenase inhibitor, on pentylenetetrazol-induced kindling in mice. Clin Exp Pharmacol Physiol 32:579–584
Dhote V, Balaraman R (2008) Anti-oxidant activity mediated neuroprotective potential of trimetazidine on focal cerebral ischaemia–reperfusion injury in rats. Clin Exp Pharmacol Physiol 35:630–637
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77
Erakovic V, Zupan G, Varljen J, Simonic A (2003) Pentylenetetrazol-induced seizures and kindling: changes in free fatty acids, superoxide dismutase and glutathione peroxidise activity. Neurochem Int 42:173–178
Frantseva MV, Velazquez JL, Hwang PA, Carlen PL (2000a) Free radical production correlates with cell death in an in vitro model of epilepsy. Eur J Neurosci 12:1431–1439
Frantseva MV, Perez Velazquez JL, Tsoraklidis G, Mendonca AJ, Adamchik Y, Mills LR, Carlen PL, Burnham MW (2000b) Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience 97:431–435
Grekas D, Dioudis C, Papageorgiou G, Iliadis S, Zilidis C, Alivanis P, Dimitradou A, Tourkantonis A (1996) Lipid peroxidation after renal ischemia and reperfusion in rats: the effect of trimetazidine. Ren Fail 18:545–552
Groves JO, Guscott MR, Hallett DJ, Rosahl TW, Pike A, Davies A, Wafford KA, Reynolds DS (2006) The role of GABA beta2 subunit-containing receptors in mediating the anticonvulsant and sedative effects of loreclezole. Eur J Neurosci 24:167–174
Guarnieri C, Muscari C (1990) Beneficial effects of trimetazidine on mitochondrial function and superoxide production in the cardiac muscle. Cardiovasc Drugs Ther 4:814–815
Guarnieri C, Muscari C (1993) Effect of trimetazidine on mitochondrial function and oxidative damage during reperfusion of ischemic hypertrophied rat myocardium. Pharmacology 46:324–331
Gupta YK, Veerendra Kumar MH, Srivastava AK (2003) Effect of Centella asiatica on pentylenetetrazole-induced kindling, cognition and oxidative stress in rats. Pharmacol Biochem Behav 74:579–585
Harpey C, Clauser P, Labrid C, Freyria JL, Poirier JP (1989) Trimetazidine, a cellular antiischemic agent. Cardiovasc Drug Rev 6:292–312
Ilhan A, Gurel A, Armutcu F, Kamisli S, Iraz M (2005) Antiepileptogenic and antioxidant effects of Nigella sativa oil against pentylenetetrazol-induced kindling in mice. Neuropharmacology 49:456–464
Iqbal S, Baziany A, Hussain M, James S, Wright S, Hemmings S, Shuaib A, Rajput A (2002) Trimetazidine as a potential neuroprotection in transient global ischemia in gerbils: a behavioral and histological study. Brain Res 928:1–7
Jain S, Bharal N, Mediratta PK, Sharma KK (2010) Trimetazidine exerts protection against increasing current electroshock seizure test in mice. Seizure 19:300–302
Javadov S, Karmazyn M, Escobales N (2009) Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther 330:670–678
Kabuto H, Yokoi I, Ogawa N (1998) Melatonin inhibits iron-induced epileptic discharges in rats by suppressing peroxidation. Epilepsia 39:237–243
Kudin AP, Debska-Vielhaber G, Vielhaber S, Elger CE, Kunz WS (2004) The mechanism of neuroprotection by topiramate in an animal model of epilepsy. Epilepsia 45:1478–1487
Kudin AP, Zsurka G, Elger CE, Kunz WS (2009) Mitochondrial involvement in temporal lobe epilepsy. Exp Neurol 218:326–332
Maertens P, Dyken P, Graf W, Pippenger C, Chronister R, Shah A (1995) Free radicals, anticonvulsants, and the neuronal ceroid-lipofuscinoses. Am J Med Genet 57:225–228
Maridonneau-Parini I, Harpey C (1985) Effects of trimetazidine on membrane damage induced by oxygen free radicals in human red cells. Br J Clin Pharmacol 20:148–151
Mason CR, Cooper RM (1972) A permanent change in convulsive threshold in normal and brain-damaged rats with repeated small doses of pentylenetetrazole. Epilepsia 13:663–674
Meldrum BS (1993) Excitotoxicity and selective neuronal loss in epilepsy. Brain Pathol 3:405–412
Mironovan OP, Zarubina IV, Krivoruchko BJ, Smirnov AV (2001) Antioxidant effects of amtizol and trimetazidine in brain ischemia. Patol Fiziol Èksp Ter 7:13–16
Morin D, Sapena R, Elimadi A, Testa B, Labidalle S, Ridant AL, Tillement JP (2000) (3H)- trimetazidine mitochondrial binding sites: regulation by cations, effect of trimetazidine derivatives and other agents and interaction with an endogenous substance. Br J Pharmacol 130:655–663
Nakao N, Grasbon FEM, Widner H, Brundin P (1996) Antioxidant treatment protects striatal neurons against excitotoxic insults. Neuroscience 73:185–200
Okhawa H, Ohishi N, Yagi K (1979) Assay of lipid peroxides in animal tissue by thiobarbituric acid reaction. Anal Biochem 95:351–358
Patel MN (2002) Oxidative stress, mitochondrial dysfunction, and epilepsy. Free Radic Res 36:1139–1146
Patel M (2004) Mitochondrial dysfuncions and oxidative stress: cause and consequence of epileptic seizures. Free Radic Biol Med 37:951–962
Racine RJ (1972) Modification of seizure activity by electrical stimulation: II. Motor seizures. Electroencephalogr Clin Neurophysiol 32:281–294
Rauca C, Zerbe R, Jantze H (1999) Formation of free hydroxyl radicals after pentylenetetrazol-induced seizure and kindling. Brain Res 847:347–351
Reeta KH, Mehla J, Gupta YK (2009) Curcumin is protective against phenytoin-induced cognitive impairment and oxidative stress in rats. Brain Res 1301:52–60
Rocha L, Briones M, Ackermann RF, Anton B, Maidment NT, Evans CJ, Engel J Jr (1996) Pentylenetetrazole-induced kindling: early involvement of excitatory and inhibitory systems. Epilepsy Res 26:105–113
Salducci MD, Chauvet-Monges AM, Tillement JP, Albengres E, Testa B, Carrupt P, Crevat A (1996) Trimetazidine reverses calcium accumulation and impairment of phosphorylation induced by cyclosporine A in isolated rat liver mitochondria. J Pharmacol Exp Ther 277:417–422
Schinder AF, Olson EC, SpitzerNC MontalM (1996) Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci 16:6125–6133
Schmutz M, Klebs K, Baltzer V (1988) Inhibition or enhancement of kindling evolution by antiepileptics. J Neural Transm 72:245–257
Schroder H, Becker A, Lossner B (1993) Glutamate binding to brain membranes is increased in pentylenetetrazole-kindled rats. J Neurochem 60:1007–1011
Serarslan Y, Bal R, Altug ME, Kontas T, Keles ON, Unal D, Unal B (2009) Effects of trimetazidine on crush injury of the sciatic nerve in rats: a biochemical and stereological study. Brain Res 1247:11–20
Silver JM, Shin C, Mc Namara JO (1991) Antiepileptogenic effects of conventional anticonvulsants in the kindling model of epilepsy. Ann Neurol 29:356–363
Singh A, Kumar G, Naidu PS, Kulkarni SK (2003) Protective effect of FK 506 (tacrolimus) in pentylenetetrazole-induced kindling in mice. Pharmacol Biochem Behav 75:853–860
Sinha K, Chaudhary G, Gupta YK (2002) Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci 71:655–665
Srivastava AK, Gupta SK, Jain S, Gupta YK (2002) Effect of melatonin and phenytoin on an intracortical ferric chloride model of posttraumatic seizures in rats. Meth Find Exp Clin Pharmacol 24:145–149
Sudha K, Rao AV, Rao A (2001) Oxidative stress and antioxidants in epilepsy. Clin Chim Acta 303:19–24
Suzer T, Coskun E, Demir S, Tahta K (2000) Lipid peroxidation and glutathione levels after cortical injection of ferric chloride in rats: effect of trimetazidine and deferoxamine. Res Exp Med 199:223–229
Wada JA (1977) Pharmacological prophylaxis in the kindling model of epilepsy. Arch Neurol 34:389–395
Waldbaum S, Patel M (2010) Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res 88:23–45
Winn LM, Kim PM, Nickoloff JA (2003) Oxidative stress-induced homologous recombination as a novel mechanism for phenytoin-initiated toxicity. J Pharmacol Exp Ther 306:523–537
Yin RX, Liang WW, Liu TW, Tao XZ, Zhu LG, Al-Ghazali R (2004) Inhibitory effect of trimetazidine on cardiac myocyte apoptosis in rabbit model of ischemia–reperfusion. Chin Med Sci J 19:242
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jain, S., Bharal, N., Khurana, S. et al. Anticonvulsant and antioxidant actions of trimetazidine in pentylenetetrazole-induced kindling model in mice. Naunyn-Schmied Arch Pharmacol 383, 385–392 (2011). https://doi.org/10.1007/s00210-011-0606-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-011-0606-1