
Abstract
Targeted therapies against cancer have improved both survival and quality of life of patients. However, metabolic rewiring evokes cellular mechanisms that reduce therapeutic mightiness. Resistant cells generate more glutathione, elicit nuclear factor erythroid 2-related factor 2 (NRF2) activation, and overexpress many anti-oxidative genes such as superoxide dismutase, catalase, glutathione peroxidase, and thioredoxin reductase, providing stronger antioxidant capacity to survive in a more oxidative environment due to the sharp rise in oxidative metabolism and reactive oxygen species generation. These changes dramatically alter tumour microenvironment and cellular metabolism itself. A rational design of therapeutic combination strategies is needed to flatten cellular homeostasis and accomplish a drop in cancer development. Context-dependent glutaminase isoenzymes show oncogenic and tumour suppressor properties, being mainly associated to MYC and p53, respectively. Glutaminases catalyze glutaminolysis in mitochondria, regulating oxidative phosphorylation, redox status and cell metabolism for tumour growth. In addition, the substrate and product of glutaminase reaction, glutamine and glutamate, respectively, can work as signalling molecules moderating redox and bioenergetic pathways in cancer. Novel synergistic approaches combining glutaminase inhibition and redox-dependent modulation are described in this review. Pharmacological or genetic glutaminase regulation along with oxidative chemotherapy can help to improve the design of combination strategies that escalate the rate of therapeutic success in cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
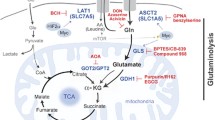
Organisms living in aerobic conditions and having an oxidative metabolism are constantly subjected to reactive oxygen species (ROS), which alter the signalling pathways and regulate carcinogenesis (Matés et al. 2012a). ROS refers to all molecular oxygen-derived free radicals like superoxide anion radicals (O2·–) and hydroxyl radicals (HO·), as well as the nonradical molecule hydrogen peroxide (H2O2) (Avolio et al. 2020). ROS play different roles on cellular homeostasis in vivo, being involved in energy production, cell growth and cellular interaction (Scialò et al. 2017). Further, ROS may be highly damaging, as they can attack biological macromolecules as lipids, polysaccharides, proteins and nucleic acids, leading to oxidation and membrane impairment, enzyme inactivation and DNA damage (Matés et al. 2008). It has been demonstrated that increased ROS levels contribute to genetic instability and cancer initiation and progression (Hu et al. 2010). l-Glutamine (Gln), a nonessential amino acid, is the most abundant amino acid in human plasma (0.5–0.8 mM), consistent with its versatile usage as a biosynthetic substrate (Jiang et al. 2019). Many tumours exhibit a remarkable preference for Gln as respiratory fuel, beyond its role as a proteinogenic amino acid (DeBerardinis and Cheng 2010). Different studies have confirmed that this preference correlates with increased glutaminase (GA) activity, which transform Gln into Glu (Benlloch et al. 2006; Lora et al. 2004). Glucose (Glc) and Gln are two nutrients that support biomass synthesis and energy production in the cell (Yuneva 2008). These two small molecules control macromolecular synthesis, adenosine triphosphate (ATP) generation, tricarboxylic acid (TCA) cycle, and oxidative stress (Fig. 1). Glc is the major source of carbon not only for ATP production via the TCA cycle, linked to oxidative phosphorylation (OXPHOS) in mitochondria, but likewise for the generation of precursors of nucleotides, amino acids, and fatty acids (FAs) (Kodama et al. 2020).

Schematic overview of the cellular functions of Gln and Gln-derived molecules. The first biochemical step for the transformation of glutamine (Gln) into glutamate (Glu) is mediated by glutaminase isoenzymes (GLS and GLS2). Carbon, nitrogen-α, and nitrogen-γ atoms derived from both amino acids are a source for the biosynthesis of many others such as alanine, asparagine, aspartate, cysteine, glycine, and serine. The Glu-derived intermediate α-ketoglutarate (αKG) enters to trycarboxylic acid (TCA) cycle and it is utilized to generate both nucleotides and fatty acids (FA). The Glu-derived tripeptide glutathione (GSH) is the major intracellular non-enzymatic antioxidant to combat reactive oxygen special (ROS), which are a by-product of glucose (Glc) and Gln metabolism. Myc is a key regulatory protein, which increases both Glc and Gln metabolism. This oncogenic factor is positively correlated with GLS expression. Likewise, Myc regulates the transcriptional function of E2F1 to control nucleotide metabolism. In addition, Gln is involved in mTORC1 signalling, another pathway essential for cancer growth. Color codes are defined as follows: blue = amino acids; orange = glutaminase isoenzymes; pink = transcription factors; yellow = Gln-derived metabolites (color figure online)
Tumour metabolism requires: (1) higher ATP and other energy equivalents needed for energy homeostasis; (2) large amounts of biochemical intermediates required for the synthesis of macromolecules for cell proliferation; and (3) reductive capacity to neutralize increased ROS (Lamonte et al. 2013). In cancer, the pleiotropic molecules Gln and Glu contribute to the supply of not only carbon but also nitrogen, which is required for biosynthesis of several key molecules such as purine and pyrimidine nucleotides, amino sugars (hexosamines and glucosamine 6-phosphate), polyamines, and nonessential amino acids (Márquez et al. 2017). On the other hand, Gln and Glu are necessary to synthethize glutathione (GSH), the most important intracellular antioxidant molecule (Matés et al. 2019). Glutamine generates Glu that is transformed into α-ketoglutarate (αKG) by glutamate dehydrogenase (GLUD1) (Márquez et al. 2016). Anyhow, Gln metabolism is more related with nucleotides, proteins, amino sugars or GSH biosynthesis than to energy production (Cetindis 2016). The nitrogen shift plays a key role in cancer progression and GA isoenzymes are decisive proteins to control the utilization of nitrogen derived from both Gln and Glu, affecting the efficiency of malignant transformation in vitro and in vivo (Kodama et al. 2020). Therapies targeting Glc and Gln metabolism are promising approaches to combat Glc/Gln-addicted tumours with great specificity. Unfortunately, these treatments usually elicit metabolic and oxidative stress, as well as resistance to therapy, a common characteristic of aggressive cancers (Matés and Campos-Sandoval 2018). Consequently, combination therapy is an actual requirement in most strategies to defeat cancer (Akins et al. 2018). Multiple approaches that include GA targeting have been reviewed in the last years (Masisi et al. 2020; Matés et al. 2020a, b; Wu et al. 2018). Although oxidative stress appears as an essential issue to be considered in GA-targeted therapy, so far scientific community has almost ignored it. Metabolic and redox reprogramming which follows GA overexpression in cancer will be analysed in this review. We will focus in the multiple roles of GA isoenzymes in both tumour growth and redox homeostasis, as well as in their clinical implications.
Mitochondria and redox homeostasis
Mitochondria have been recently defined as signalling organelles that regulate cell fate through both ROS and TCA intermediates, which can coordinate hypoxic response and post-translational protein modification (Martínez-Reyes and Chandel 2020). In mitochondria, the proteins of OXPHOS may or may not be elevated in some cancers when compared to the upregulation accomplished by the enzymes of glycolysis, glutaminolysis, pentose phosphate pathway (PPP), lipogenesis, and the antioxidant defence during induction of tumour proliferation (Torresano et al. 2020; Zhang et al. 2016a). Antioxidant enzymes can relieve oxidative stress, inhibit apoptosis, and facilitate the switch between metabolic pathways (Matés et al. 2006). Additionally, the induction of p53 in response to DNA damage or oncogenic stress is able to cause p53-dependent apoptosis associated with release of ROS from mitochondria (Eriksson et al. 2019). Thus, mitochondria are the fundamental bioenergetic organelles for maintaining redox homeostasis towards normal cell function (Zhu et al. 2019a, b). In many cancers, mitochondria have few alterations in tumour cells and do not suffer from extensive respiratory defects (Gentric et al. 2017). However, mitochondria of cancer cells are mainly characterized by a particular increase in the anaplerotic metabolic pathway named glutaminolysis, associated with elevated levels of Glu, diminished levels of Gln, and large GA expression (Matés et al. 2009). Mitochondria show increased Gln uptake by SLC25 transporter family system, and higher glutaminolysis to support OXPHOS and provide biosynthetic and bioenergetics requirements for growth (Sever et al. 2015). In addition to the positive correlation between OXPHOS and cancer proliferation, OXPHOS is related to others hallmarks of cancer as migration, invasion and stemness properties (Gentric et al. 2017). Although glycolysis generates less ATP than OXPHOS, it has been confirmed that aggressive tumours simultaneously use glycolysis and Glc oxidation by mitochondria through pyruvate dehydrogenase (PDH) (Marin-Valencia et al. 2012). Simultaneously, Gln is transformed into mitochondria by GA isoenzymes which start Gln metabolism to produce energy and raw material for cancer growth (Márquez et al. 2016). Glutamate is a key branch point in glutaminolysis that can be compelled toward mitochondrial oxidation to produce ATP and ROS, or toward de novo synthesis of GSH to control redox balance and abolish oxidative stress (Rashida et al. 2018). Of note, Glu is a limiting substrate for TCA cycle anaplerosis, rendering citrate to produce reduced nicotinamide adenine dinucleotide phosphate (NADPH) and lipids (Daemen et al. 2018). Besides, FAs beta-oxidation takes place in the mitochondria and makes acetyl-CoA, reduced nicotinamide adenine dinucleotide (NADH), and reduced flavin adenine dinucleotide (FADH2), cofactors used by the electron transport chain (ETC) (Gentric et al. 2017). On the other hand, induction of the apoptotic pathway in cancer cells is stimulated by ROS, which provoke shifts in mitochondrial membrane activity, and act as mediators between mitochondria and apoptosis (Matés and Campos-Sandoval 2018). The importance of mitochondria in tumour cell apoptosis and proliferation include provision of ATP, induction of Ca2+ signalling and generation of ROS, which favours cell proliferation, genetic instability, mutation acquisition, and drug resistance (Zhu et al. 2019a, b). In mitochondrial function, OXPHOS engenders electrons that produce superoxide anions (Matés et al. 2009). Antioxidant enzymes and high GSH levels are required to avoid mitochondrial bioenergetic collapse and cell death (Rashida et al. 2018).
Under hypoxia (another hallmark of human cancer), metabolic and mitochondrial disorders, or pharmacological treatment, homeostasis is crucial for the surviving of cancer cells (Dang et al. 2009). ETC conducts a sequence of redox reactions and engenders proton motive force, which is utilised by ATP synthase (complex V) to produce ATP through OXPHOS (Fig. 2). Being the most effective way of ATP production, OXPHOS must be tightly regulated (Gentric et al. 2017). The efficiency of OXPHOS is determined by the amount of inorganic phosphate (Pi) utilised for ATP production per amount of O2 consumed, and it can be adjusted by the level of uncoupling between inward mitochondrial H+ influx and ATP synthesis (Zhdanov et al. 2014). In cancer cells Glc incomes are fundamental, since glycolysis produces large amounts of ATP regardless of high availability of O2 (Warburg effect) (Lamonte et al. 2013). In turn, most of the pyruvate, instead of being transformed into acetyl-CoA and utilised in the TCA cycle, produces lactate, which is expelled from the cell (Zhdanov et al. 2014). Other intermediates of glycolysis are again re-directed to anabolic reactions generating intermediate metabolites for proliferating cancer cells (Sever et al. 2015). To optimize anaplerotic reactions and ATP production, cancer cells addictively utilise Gln, and more than half of ATP is produced through Gln-driven OXPHOS (Zhdanov et al. 2014). As a result, Gln-driven mitochondrial respiration in many cancer cells is active even at high Glc levels (DeBerardinis and Cheng 2010). Mitochondrial GLS and GLS2 essentially contribute to the synthesis of GSH and the scavenging of ROS, thereby maintaining functional integrity of mitochondria and defence against apoptosis (Okazaki et al. 2017; Zhdanov et al. 2014).

Oxidative mitochondrial metabolism. Glutamine (Gln) is converted by glutaminases (GLS and GLS2) into glutamate (Glu), which gives rise to α-ketoglutarate (αKG) in the matrix. The Gln pathway is reprogrammed in cancer to increase production of glutathione (GSH) and oxidized glutathione (GSSG), which ratio is regulated by glutathione reductase (GR) and glutathione peroxidase (GPx). Together with thioredoxin (Trx) redox system, they reduce mitochondrial oxidative stress. Reduced Trx (Trxred) catalyzes the reduction of disulfides within oxidized cellular proteins, such as peroxiredoxin (Prx). In this process Trx becomes oxidized (Trxox) which in return is reduced by thioredoxin reductase (TrxR) at the expense of NADPH. Catalase is another antioxidant enzyme, NADPH independent, which decreases ROS, converting hydrogen peroxide (H2O2) to water (H2O). Pyruvate (Pyr) is transported into the matrix by the mitochondrial pyruvate carrier (MPC). Cancer cells exhibit increased catabolism of Pyr and fatty acids (FA) to generate acetyl-CoA, which fuels TCA cycle. The oxidative phosphorylation, mediate by the electron transport chain (ETC), which is formed by complex I, complex II, ubiquinone (Q), complex III, cytochrome c and complex IV. ATP synthase captures protons (H+) from intermembrane space and uses their gradient to form ATP from ADP and inorganic phosphate (Pi). ETC uses the reducing equivalents (NADH and FADH2) produced by trycarboxylic acid (TCA) cycle and fatty acids oxidation (FAO), to form higher amounts of ATP. Color codes are defined as follows: blue = fuel molecules; lilac = coenzymes; orange = glutaminase isoenzymes; pink = enzymes involved in oxidative phosphorylation; yellow = antioxidant enzymes (color figure online)
Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) maintains mitochondrial bioenergetics and redox homeostasis by facilitation of GLS-mediated glutaminolysis (Zhu et al. 2019a, b). These authors demonstrated that MALT1 inhibitors MI-2 and VRPR elevated mitochondrial ROS and enhanced apoptosis by decreasing Glu, GSH and GLS gene expression in lymphoma cells. MALT1-mediated downstream signalling pathways modulate ROS and include nuclear factor kappa-B (NF-κB) as well as c-Jun (Hammouda 2020). Thus, MALT1 activates the nuclear translocation of c‐Jun, which is a transcription factor of GLS, directly bounding to the promoter region of the gene and promoting its expression (Zhu et al. 2019a, b). Further, aldehyde dehydrogenase (ALDH) expression is regulated by glutaminolysis because ALDH’s active site requires priming by Glu (Kamarajan et al. 2017). Glutaminolytic-ALDH driven mechanisms can determine carcinogenic transition of cancer stem cells (CSCs), a subset of cells within tumours that have the capacity to develop tumours (El-Hout et al. 2020). Hence, the induction of epithelial-mesenchymal transition (EMT) is associated with enhanced glycolysis and reduced glutaminolysis in mitochondria (Ramirez-Peña et al. 2019). Interestingly, ALDH expression follows activation of the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) that has a multifaceted role in mitochondria and controls numerous genes containing antioxidant response elements (AREs) (El-Hout et al. 2020). Of note, connection between ALDH and GA may help in the development of targeted therapies for tumour cell and its CSC compartment (Kamarajan et al. 2017).
Interestingly, GA inhibition evokes cell-cycle arrest in sensitive GLS-high expressing CSCs without inducing ROS or apoptosis. Therefore, susceptibility to GA inhibition using specific drugs correlates with exalted GLS expression (Koch et al. 2020). This is in line with similar observations in triple-negative breast cancer (TNBC) (Zhou et al. 2017). Furthermore, the observed anti-stemness effect of GLS inhibition has been reported by several studies highlighting how important is cell phenotype for an efficient pharmacological and or genetic GLS suppression (Koch et al. 2020).
Glutaminases
Human GLS gene (usually named in many articles with the incorrect terms GLS-1 or GLS-1: see HUGO Gene nomenclature at https://www.genenames.org/) is located in chromosome 2, spans 82 kb and splits into 19 exons (Márquez et al. 2016). Two different transcripts derive from this gene: the KGA transcript, first discovered in kidney, composed by 18 exons and assembled by joining exons 1–14 and 16–19, and the GAC transcript, which is built by alternative splicing and operates exons 1–15. The human GLS2 gene is located in chromosome 12, and has a length of approximately 18 kb and splits into 18 exons (Pérez-Gómez et al. 2003). Two transcripts have been characterized from the mammalian Gls2 gene: the longer transcript named GAB, completed by joining all 18 exons of the gene, and the shortest transcript termed LGA that lacks exon 1 and was first found in rat liver (Márquez et al. 2016).
Glutaminase isoforms KGA and GAC are usually referred as GLS, while isoforms GAB and LGA are collectively called GLS2 (Matés et al. 2013). Every isoenzyme produces glutaminolysis rendering Glu and providing biosynthetic precursors, energy and antioxidant power (Matés et al. 2020a, b). However, mammalian GLS and GLS2 isoenzymes show many kinetic differences such as the dependence of the activator inorganic phosphate (Pi), low for GLS2, high for GLS; the relative affinity for the substrate Gln, higher in GLS than in GLS2 isoforms; and the inhibitory effect of Glu, a unique characteristic only reported for GLS isoforms (Márquez et al. 2016). In their inactive states, KGA and GAC are dimeric species (Ferreira et al. 2013). In vitro, KGA or GAC can be activated utilizing Pi, which stimulates the formation of an active tetramer (Cassago et al. 2012). Meaningful, GAC is mainly expressed in several tumours with preference to KGA (Matés et al. 2013). For example, neuroblastoma cells in vitro exhibited much more GAC than KGA, as well as an unusually high GLS2 content (Xiao et al. 2015). It has been hypothesized that interspersed abundances of these isoenzymes could play a role in their diverse effects (Saha et al. 2019).
Mostly, GLS becomes upregulated while GLS2 is frequently repressed in many cancers (Matés et al. 2020a, b). Hence, GLS and GLS2 isoenzymes, depending on the context, can have opposite roles in cancer, including a nuclear assignment for GLS2 (Oliva et al. 2020). Some data point that GLS and GLS2 expression can be interconnected (Matés et al. 2020a, b). For example, using derivative MDA-MB-231 and TSE cell lines that ectopically express GLS2, the clones with higher GLS2 expression showed a moderate decrease in the level of GLS and greatly decreased sensitivity to specific GLS inhibition, thus mediating resistance to pharmacologic GLS inhibitors (Lukey et al. 2019). Additionally, the ratio GAC/KGA augmented in lung tumours compared with healthy lung tissue (Matés and Campos-Sandoval 2018). New investigations should be focused on the balance between GLS and GLS2 isoforms, which may be also involved in GLS2-mediating radioresistance (Xiang et al. 2013). Albeit much is known about GLS and GLS2 expression in many tumours (Matés et al. 2019), enlighten the mechanisms responsible for the GLS and GLS2 signalling pathway, as well as characterize the isoenzymes function, is required in every type of cancer. GLS/GLS2 signature is not an easy task. Any combined GLS and GLS2 signature has been correlated to the clinical outcomes of patients in some types of cancer (Saha et al. 2019). Accordingly, the molecular chaperone tumour necrosis factor receptor associated protein 1 (TRAP1), the most abundant heat shock protein 90 (HSP90) family member in mitochondria can act as an oncogene or a tumour suppressor, depending on the metabolic features of the individual tumour (Avolio et al. 2020). Chaperone HSP90 inhibition has been used as an anti-cancer strategy; but although cancer decreased growing when HSP90 inhibitors were used in clinical trials, the tumours spread after the drug treatment was stopped (Li et al. 2015). Conversely, heat shock protein 60 (HSP60), which silenced activated mitochondrial glutaminolysis, producing both oxaloacetate and aspartate, as well as isocitrate and acetyl-CoA, resulted in elevated de novo nucleotide and lipid biosynthesis (Daemen et al. 2018; Teng et al. 2019).
On the other hand, following GA inhibition there are other Gln to Glu converting enzymes that can compensate when GAs are inhibited, such as the enzymes catalyzing the synthesis of asparagine, nucleotides, NAD and glucosamine (Jiang et al. 2019). It has been recently stated the key role of the glutaminase II pathway (i.e., glutamine transaminases coupled to ω-amidase) in normal and malignant prostate cells as an essential source of anaplerotic αKG (Udupa et al. 2019). Eventually, it will be important to develop robust and selective inhibitors of Gln transaminase K (GTK), and ω-amidase as potential anti-cancer agents, possibly to be dispensed adjunctively with specific GA inhibitors (Dorai et al. 2020). Adaptive metabolic networks of tumours explain failure of single-targeted GLS inhibition in cancer and support the need for combination therapy (Udupa et al. 2019).
GLS
GLS isoforms have shown their oncogenic properties in many tumour cells types as breast, esophagus, head-and-neck, and blood cancers (Saha et al. 2019; Matés et al. 2020a, b). In addition, knocking down Gls induced apoptosis in Ehrlich ascitic tumour cells, caused oxidative stress and sensitized the cells to chemical agent methotrexate (Lora et al. 2004). Nonetheless, some recent studies have shown that no definitive conclusion in cancer can be established, neither for GLS isoforms. Neither genetic silencing nor pharmacological blockage of GLS altered the advancement of lung tumours in vivo (Kodama et al. 2020). GLS is differently regulated in many cancer types (Fig. 3).

GLS and GLS2 networks. GLS isoforms have oncogenic functions and are upregulated by regulatory pathways as mammalian target of rapamycin complex 1 (mTORC1), NOTCH1, c-Jun N-terminal kinase (JNK), nuclear factor kappa-B (NF-κB), and MYC, through microRNA (miR) miR-23a/b. Of note, oncoprotein MYC can be targeted by miR-145. Other miRs dowregulating GLS are miR-122, miR-137, miR-153, miR-203 and miR-513. Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES), CB-839 and compound 968 are chemicals that block GLS activity. GLS2 can work as a tumour suppressor in many types of cancer but, context dependent, also has potential oncogenic properties. GLS2 can be activated by (1) transcription factors: N-MYC, p53, GATA3, (2) drugs: phorbol 12-myristate 13-acetate (PMA), and (3) microRNAs (miR-16). GLS2 expression can be inhibited by miR-103a-3p, and by specific compounds AV-1 and 968. GLS2 can dowregulate to (1) other transcription factors as peroxisome proliferator-activated receptor gamma (PPAR-γ), (2) regulatory pathways as phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), and (3) GTPases as Rac1. Noteworthy, miR-34a is activated by ribonuclease DICER through a GLS2-dependent mechanism. Color codes are defined as follows: blue = transcription factors and regulatory proteins; green = chemicals; lilac = ribonucleases; orange = glutaminase isoenzymes; pink = GTPases; yellow = miRs (color figure online)
GLS regulation through microRNAs (miRNAs or miRs) appears to be a system by which tumour cells acquire concrete benefits for using alternative sources of carbon and nitrogen favouring their adjustment to new metabolic environments (Ortiz-Pedraza et al. 2020). First characterized miRNA controlling GA expression was miR-23a/b, which targets the GLS 3′ UTR through a mechanism mediated by oncoprotein MYC (Gao et al. 2009). Myc is an oncogene that produces c-Myc (hereafter referred to as MYC) protein that directly regulates Glc metabolism as well as other genes involved in mitochondrial network (Dang et al. 2009). MYC stimulates cell proliferation, and transcriptionally represses miR-23a and miR-23b, resulting in greater expression of GLS that affect energy and ROS homeostasis (Gao et al. 2009).
So far, GLS regulation by miRs has been characterized for miR-122, -137, -145, -153, -203, and -513c (Li et al. 2019b; Ortiz-Pedraza et al. 2020). Glutaminolysis is conversely related to miR-122 in primary human hepatocellular carcinoma (HCC) cells and in HCC patients. Hence, livers of miR-122 KO mice showed higher Gls expression whilst ectopic miR-122 supressed expression by direct targeting of Gls (Sengupta et al. 2020). MiR-137 was decreased in melanoma tissue. Besides, it works as a tumour suppressor in melanoma cells by targeting GLS (Luan et al. 2018). MiR-145 inhibited Gln consumption, α-KG production, and cellular ATP levels through MYC/GLS pathways in ovarian cancer cells (Li et al. 2019b). A reverse correlation was observed between GLS and miR-153 expression in human glioblastoma (GBM) tissues compared to matched non-tumour tissues. MiRNA-153 lowered the conversion of Gln–Glu by directly targeting GLS expression, promoting apoptosis and inhibiting tumour cell growth in GBM (Liu et al. 2017). GLS is also a downstream target gene of miR-203, which interacts directly with the 3′ untranslated region of GLS, limiting GLS expression and sensitization to drug temozolomide in vitro. Additionally, patients suffering from malignant melanoma showed downregulation of miR-203 and upregulation of GLS in their tissues (Chang et al. 2017). Finally, miR-513c, which supresses migration, invasion, and proliferation, is downregulated in human neuroblastoma tissues compared with their adjacent normal tissues. Molecularly, miR-513c bind to the 3′ untranslated region of GLS in human neuroblastoma cells (Xia et al. 2017).
On the other hand, GLS (GAC isoform) is dependent on NF-κB through a hyper-activated Rho GTPase mediated mechanism, that induces GLS phosphorylation, and it is needed for breast cancer and lymphoma B cells proliferation (Wang et al. 2010). In another metabolic adaptation required for tumorigenesis the transcription factor c-Jun N-terminal kinase (JNK), product of the proto-oncogene c-Jun, is sufficient to elevate GLS in breast cancer cells by direct binding of JNK to GLS promoter (Lukey et al. 2016). Similarly, JNK binds to GLS promoter to increase GLS in lymphoma cells (Zhu et al. 2019a, b).
GLS2
In contrast to GLS isoforms, much less is known with regard to the regulation of GLS2 isoforms (Oliva et al. 2020). GLS2 is activated by low phosphate levels and lightly inhibited by Glu (Matés et al. 2013). Although GAs are traditionally mitochondrial enzymes, GLS2 have been localized in nuclei, suggesting that GLS2 participate directly or as a co-regulator having essential impact upon the fine transcriptional regulation of Gln metabolism (Cardona et al. 2015; Oliva et al. 2020; Olalla et al. 2002). Interestingly, GLS2 contains an ankyrin-repeat domain in its C-terminal region, which may produce divergent properties and functions to GLS2 through protein–protein interactions (Márquez et al. 2017). GLS2 have been identified in many human cancers, so far: colorectal, gastric, bladder, lung, ovarian, thymoma, hepatoma cells, lymphocytic cells, myeloid cells, and breast cancer cells (Matés et al. 2019; Niu et al. 2019; Saha et al. 2019). Substantially, GLS2 was higher expressed in less aggressive cancers (Matés et al. 2020a, b; Ramirez-Peña et al. 2019). Oppositely, GLS2 expression was scant in colon, HCCs and GBMs, the most malignant brain tumours, both showing tremendous levels of GLS (Suzuki et al. 2010; Oliva et al. 2020).
GLS2 is also differently modulated in several cancers types (Fig. 3). Many studies have highlighted the role of GLS2 as a target gene of p53 in both non-tumour and tumour cells (Matés et al. 2020a, b). The human GLS2 gene contains a p53 consensus DNA-binding element, which is induced in response to DNA damage or oxidative stress, favouring the association of p53 with the GLS2 promoter (Suzuki et al. 2010). Hence, tumour suppressor p53 enhances GLS2 expression under both nonstressed and stressed conditions, targeting mitochondrial respiration, energy metabolism, ATP formation, and apoptosis, but produces larger GSH and NADH amounts to gain antioxidant power, therefore lowering intracellular ROS levels in cells (Hu et al. 2010). These results stated that GLS2 is a key enzyme in regulating the tumour-suppressive effects of p53, which has opposing roles in the regulation of ROS through transactivation of antioxidant and pro-oxidant genes (Suzuki et al. 2010). p53 induces the expression of sestrins (SESN), TP53-induced glycolysis and apoptosis regulator (TIGAR), glutathione peroxidase (GPx), and ALDH (Hu et al. 2010). The antioxidant role of p53 is essential to decrease oxidative stress-induced DNA damage and mutations, which induces the tumour suppression activity of p53 (Suzuki et al. 2010). Remarkably, antioxidant N-acetyl-l-cysteine (NAC) supplementation substantially boosted the survival of p53 null mice (Eriksson et al. 2019). Nevertheless, on the contrary to previous results of antioxidants being healthy supplements for cancer patients, other reports have found that ROS scavengers can accelerate cancer progression in mice and develop distant metastases in human tumour cells (Abu Aboud et al. 2017).
Following p53 regulation, GLS2 increases the levels of Glu, α-KG, OXPHOS and mitochondrial respiration rate (Hu et al. 2010), shortening cellular sensitivity to ROS-associated apoptosis conceivably through a GSH-dependent antioxidant defence system (Suzuki et al. 2010). GLS2 restraint in tumour cells might be explained by p53 mutations and lack of function frequently found in GBM and other aggressive cancers (Matés and Campos-Sandoval 2018). The GLS2 suppression mechanism identified in GBM, liver and colorectal cancer is promoter methylation, with independency of the p53 status (Obara-Michlewska and Szeliga 2020; Zhang et al. 2013). In addition, GLS2 binds to small Rac1 GTPase and impedes its interaction with Rac1 activators guanine-nucleotide exchange factors, which inhibits Rac1 to abolish migration, invasion and cancer metastasis in human HCC (Zhang et al. 2016b). Moreover, this research found GLS2 mediates p53′s function in metastasis suppression through inhibiting Rac1. Correspondingly, smaller and fewer lung tumours were detected in mice that had been injected with cells that provoked high levels of GLS2 (Zhang et al. 2016b). This function of GLS2 is independent of GLS2 enzymatic activity (Oliva et al. 2020).
Recent findings by Szeliga group show how transfection with GAB inhibits the growth of GBM cells (U87MG, T98G and LN229) and sensitizes them to oxidant agent H2O2. Additionally, increased sensitivity to H2O2 of all three GAB-transfected cell lines was mediated by downregulation of the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathway (Majewska et al. 2019). In other recent study, SH-SY5Y and HepG2 cells treated with the differentiation agent phorbol 12-myristate 13-acetate (PMA) increased GLS2 expression and nuclear accumulation, and it correlated with upregulation of tumour suppressors p53 and p21 (Oliva et al. 2020). A similar response was obtained by overexpression of GLS2 in T98G GBM cells, including reduction of MYC oncoprotein (Martín-Rufián et al. 2014). Upregulation of GLS2 in this cell model produced a decrease in proliferation, with cell-cycle arrest at the G2/M phase (Oliva et al. 2020). Experiments in vitro and in vivo, using HCC cells, exhibited that GLS2 stabilizes DICER protein to facilitate miR-34a maturation and subsequently represses SNAI1 expression in a GA activity independent manner, sharply inhibiting cell migration, invasion and metastasis (Kuo et al. 2016). On the other hand, the generation of ROS by treatment with oxidizing agents, like arsenic trioxide or H2O2, synergizes with GLS2 overexpression to eliminate aggressive properties of T98G glioma cells (Martín-Rufián et al. 2014). Accordingly, GLS2 provoked in these cells reduced MYC and BCL-2 protein levels, as well as an enhanced expression of the pro-apoptotic BID protein (Oliva et al. 2020). Hence, overexpression or derepression of GLS2 emerges as a potential therapeutic approach for some types of cancer (Matés et al. 2020a, b). Other evidences links GLS2 with cell differentiation, i.e., GLS2 and TAp63 expression increased during differentiation of primary human keratinocytes, whilst reduction of GLS2 suppressed skin differentiation (Giacobbe et al. 2013). We have hipothesized that nuclear GLS2 participates in the growth-arrest program modulating cell proliferation (Oliva et al. 2020). Strikingly, a nuclear location of GLS isoforms was also discovered in the prostate cancer cell line PC-3, associated with a diminished transcriptional activity of peroxisome proliferator-activated receptor-γ (PPAR-γ) (de Guzzi et al. 2018). This effect of GLS in transcriptional regulation was mediated by a direct interaction with PPAR-γ and independent of its catalytic activity, which supports the view of GAs as multifaceted proteins (Márquez et al. 2017).
Albeit lower expression of GLS2 is usually linked to malignant phenotypes, GLS2 is equally upregulated in some types of cancer. For example, the expression of GLS2 was significantly increased in resistant cervical carcinoma cells, HeLaR (Xiang et al. 2013). In this report, authors demonstrated that GLS2-silenced cells showed increased radiosensitivity with reduced colony survival and augmented apoptosis in response to radiation. At the molecular level, knockdown of GLS2 boosted ROS levels of HeLaR exposed to irradiation by reduction of the amounts of antioxidant GSH, NADH and NADPH, in both HeLaR cells and xenografts in vivo (Xiang et al. 2013). In other research supporting malignant properties of GLS2, specific inhibition of GLS2 by the alkyl benzoquinone AV-1 in A549 and HepG2 cancer cells led to autophagy via adenosine monophosphate-activated protein kinase (AMPK)-activation and mammalian target of rapamycin complex 1 (mTORC1) inhibition, eventually leading to growth arrest of cancer cells (Lee et al. 2014).
N-Myc, another important Myc family member, evokes conversion of Gln to Glu in N-MYC-amplified neuroblastoma cells, in vitro and in vivo, by specifically activating GLS2, but not GLS, transcription. In this study, GLS2 induced aggressive neuroblastoma progression, being a direct biomarker for unfavourable patient survival (Xiao et al. 2015). Besides, GLS2 knockout significantly reduced aerobic glycolysis as well as α-KG, ATP, and GSH, increasing ROS production, and improving radiosensitivities of resistant tumour cells. Moreover, GLS2 is essential for growth and proliferation of luminal-subtype breast tumours (Lukey et al. 2019). These authors found that GLS2 expression is driven by GATA3 and promoter methylation and its blockage can be achieved by chemical specific inhibitors. GATA3 and promoter hypermethylation also mediate GAB expression in GBM cell lines (Szeliga et al. 2016; Majewska et al. 2017). Furthermore, GLS2 is overexpressed in epidermal growth factor receptor (EGFR)-mutated non-small cell lung cancer (NSCLC) (Meijer et al. 2019). Surprisingly, treatment of TNBC cells with a specific GLS inhibitor evoked a drug-resistant population with increased endogenous GLS2, which sustained tumour proliferation (Dias et al. 2020). In this cell type, GLS2 expression was linked to higher in vitro cell migration and invasion, mesenchymal markers, and in vivo lung metastasis. In addition, miR-103a-3p interaction with GLS2 suppressed its expression and evoked an anti-tumour effect in gastric cancer cells by downregulating ROS generation, as well as cell proliferation, invasion and migration in vitro and in vivo (Niu et al. 2019). On the other hand, in renal cell carcinoma (RCC) MYC upregulated both GLS and GLS2, highlighting a regulation of Gln metabolism by proteasome inhibitors, which impacts the redox status and cytotoxicity of cancer cells by increasing ROS (Sourbier et al. 2019).
Consequently, GLS2 can show an opposite phenotype, or not, even though it elicits the similar metabolic effects as those observed in other cancers for GLS isoforms (Xiao et al. 2015). Context-dependent, GLS2 role as a tumour suppressor or as an oncogenic factor seems to be function of a complex interaction between oncogenic mutations and hypoxic tumour microenvironment (TME), as well as the histological subtype and aggressive tumour behaviour (Meijer et al. 2019).
Glutathione as an antioxidant and signalling molecule
Tripeptide GSH (l-γ-glutamyl-l-cysteinyl-glycine), chemically, (2S)-2-amino-5-[[(2R)-1- (carboxymethylamino)-1-oxo-3-sulfanylpropan-2-yl]amino]-5-oxopentanoic acid, is the most abundant low-molecular-mass thiol, essential in antioxidant defence (Matés et al. 2012b). GSH is not synthesized within mitochondria but is necessary for their normal function (Carretero et al. 2000). The ratio between the reduced and oxidized form (GSH/GSSG) is the primary redox couple that fixes the antioxidant capacity of cells (Lora et al. 2004). GSH and GSH/GSSG regulate many cellular events such as cell proliferation and apoptosis (Benlloch et al. 2006). GLS and GLS2 are essential for maintaining GSH homeostasis not only because the tripeptide consists in Glu, glycine and cysteine, but because Glu is equally necessary for the uptake of cystine, precursor of cysteine, which is the rate-limiting amino acid in the intracellular synthesis of GSH (Matés et al. 2013; Okazaki et al. 2017). Cysteine can be supplied through different circuits, i.e., from homocysteine and serine by the transsulfuration pathway (Hayano et al. 2016). Peroxiredoxins (Prxs) can also neutralize H2O2, reduce cystine and increase intracellular cysteine, which indirectly supports GSH synthesis to react with electrophilic and oxidizing species to work as a cofactor and electron donor of glutathione S-transferases (GST) and GPx (Eriksson et al. 2019).
Cellular GSH homeostasis is provided through (1) de novo synthesis from precursor sulfur amino acids methionine and cysteine, (2) regeneration from its oxidized form GSSG utilizing NADPH as a reductant, and (3) uptake of extracellular GSH via a Na+-dependent transport systems (Matés et al. 2012b). GSH and GSSG can be inter-converted by the enzymes GPX and glutathione reductase (GR), which reduces GSSG to GSH, constituting the major intracellular thiol-disulphide redox system in mammalian cells (Matés et al. 2013). Glutathione serves several vital functions, including (1) scavenging peroxides, (2) modulating key processes as cell proliferation, apoptosis, microtubular related events, and immunological function, (3) maintaining the essential thiol status of proteins by preventing oxidation of –SH groups or by reducing disulphide bonds induced by oxidative stress, (4) detoxifying electrophiles, and (5) affording a reservoir for cysteine (Matés et al. 2012a). Strikingly, diet supplemented with N-acetylcysteine that supplies extra cysteine for GSH biosynthesis, decreased the tumour growth (Erikson 2019). Similar results were found in RCC (Okazaki et al. 2017).
Glutathione is synthesized in two sequential ATP-dependent reactions catalyzed by γ-glutamylcysteine synthetase (GCL), the rate-limiting enzyme, and glutathione synthetase (GSS) (Fig. 4). Other factors in the regulation of the de novo GSH synthesis are the availability of cysteine and the concentration of GSH itself that inhibits, by a feedback mechanism, GCL activity (Matés et al. 2012b). In the presence of oxidative stress, GSH concentration drops while GSSG, potentially highly cytotoxic, soars because of the reduction of peroxides or a result of free radical scavenging (Matés et al. 2014). Consequently, the thiol redox status of the cell will shift and turn on some oxidant response transcriptional elements. Besides GSSG may be preferentially secreted from the cell and degraded extracellularly, increasing the cellular need for de novo GSH synthesis (Matés et al. 2012a). The high intracellular concentration of GSH (usually 5–10 mM) together with GPX and GR makes the molecule an effective redox buffer to scavenge free radicals (Matés et al. 2013). In mammalian cells, the cycling between GSH and GSSG serves to remove ROS such as H2O2 produced due to either mitochondrial respiration or metabolism of toxic molecules, and protect cells from oxidative damage (Matés et al. 2012a).

Glutathione equilibrium (GSH/GSSG) following Gln/Glu homeostasis. Glutamate (Glu), cysteine (Cys) and glycine (Gly) are required to synthetize glutathione. GCL gamma-glutamylcysteine synthetase, GSS glutathione synthetase, GSSG glutathione oxidized form, NADPH nicotinamide adenine dinucleotide phosphate, ROS reactive oxygen species, xCT solute carrier family 7 member 11 (SLC7A11), a cysteine (Cis)-glutamate antiporter. ASTC2, BOAT 1, LAT 1/2, and SNAT 1/2/3/5/7 are glutamine (Gln) transporters. Color codes are defined as follows: blue = amino acids; orange = glutaminase isoenzymes; pink = enzymes involved in GSH biosynthesis (color figure online)
GLS inhibition evoked lower GSH and GR levels in GLS-silenced cells (0.28AS-2) together with higher Gln levels, as well as a rise at intracellular ROS and a dramatic increase in apoptosis and a sensitization of cancer cells to chemotherapy by both H2O2 and methotrexate (Lora et al. 2004). In aggressive B16M-F10 melanoma cells, GSH depletion was achieved by BCL-2 AS (which hampers the Bcl-2-induced inhibition of GSH efflux) or by chemical inhibition of γ-glutamyl transpeptidase (which limits GSH synthesis by preventing cysteine generation), sensitizing malignant B16M-F10 cells to tumour necrosis factor alpha (TNF-α) (Benlloch et al. 2006). Additionally, oxidative stress-resistant metastatic cells displayed an adapted reply that comprised overexpression of detoxifying enzyme, as superoxide dismutases (SOD), but not the overall mitochondrial electron flow (Carretero et al. 2000). Interestingly, tumour cells in mice fed a Gln-enriched diet showed higher GLS and lower Gln synthetase (GS) activities than did cells isolated from mice fed with a standard diet. Subsequent increase in Glu content inhibited GSH uptake by tumour mitochondria and led to a selective depletion of mitochondrial GSH level, which may cause tumour cells to be more susceptible to oxidative stress-induced mediators as TNF-α (Benlloch et al. 2006). Tumour cell populations under active proliferative states and drug-resistant tumours have higher GSH levels to maintain increased cellular resistance. This is important because of the role of GSH in determining the sensitivity of cells to radiation and drug-induced cytotoxicity (Boysen et al. 2019; Carretero et al. 2000).
Glutathione depletion causes a JNK- and p38- mitogen activated protein kinase (MAPK)-mediated increase in expression of CGL (Matés et al. 2012a). Another example of signalling functions of GSH is its role in Glc transport (Lee et al. 2016b). Cancer cells can respond rapidly to oxidative stress by directing Glc flux through the oxidative PPP to form NADPH (Zhang et al. 2016a). Simultaneously, oxidized form of vitamin C, dehydroascorbate, is transferred into cells through the membrane Glc transporter GLUT1. Then, the cell imports dehydroascorbate, which is reduced back to vitamin C by GSH that becomes GSSG. Following, GSSG is transformed back to GSH by NADPH. Decrease of both GSH and NADPH augments ROS and provokes apoptosis (Zhang et al. 2016a). Importantly, by screening the effect of GLS and GCL inhibitors in a panel of 407 mesenchymal breast and lung tumour lines it has been found a strong co-dependency on GLS and de novo GSH synthesis. It was validated in vivo using four lung patient-derived xenograft models (Daemen et al. 2018). Intriguingly, GSH links to fumarate in fumarate hydratase (FH) deficient cancer cells to explain the boost of ROS, as well as ROS signalling activation of hypoxia-inducible factors (HIFs) and NRF2 (Sullivan et al. 2013).
Oxidative and metabolic adaptation in cancer
Redox homeostasis is an Achilles’s heel of cancer (Eriksson et al. 2019; Torresano et al. 2020). Metabolic and environmental factors reprogram tumour circuits to provide the distinct cellular biosynthetic and bioenergetic needs present during oncogenesis (Lamonte et al. 2013). For example, tumour cells compensate for Gln depletion by rerouting carbon from Glc and increasing pyruvate carboxylase (PC) expression (Bruntz et al. 2019). Notably, active glycolysis and glutaminolysis provide energy to cancer cells in physiological conditions (DeBerardinis and Cheng 2010). Cancer cells supplied with high amounts of both Glc and Gln are capable of producing a strong respiratory response and show an ATP-independent decrease in extracellular signal-regulated kinase (ERK) and AKT phosphorylation (Zhdanov et al. 2014). Besides, an increase in NAD(P)/NAD(P)H ratio and decrease in ROS, as well as higher levels of cytosolic Ca2+, affect phosphorylation of both kinases. Of note, these authors observed that a supply of Gln or pyruvate evoked a decrease in AMPKα phosphorylation, which is able to rise again following a ROS enhancement (Zhdanov et al. 2014). ROS generated in mitochondrial metabolism control KRAS-induced anchorage-independent growth through regulation of the ERK-MAPK signalling pathways (Weinberg et al. 2010). Major source of ROS generation required for anchorage-independent growth is the Q site of mitochondrial complex III, independently from OXPHOS (Fig. 2). KRAS- RAS-, MYC-, MAPK-, and AKT-dependent tumour cells require Gln metabolism through TCA cycle in the absence or presence of Glc (Anso et al. 2013; Son et al. 2013; Weinberg et al. 2010). Gln catabolism by the TCA cycle generates reducing equivalents (NADH and FADH2) and ROS by complex I, II, and III of the ETC (Fig. 2).
Aerobic glycolysis (Warburg effect), which is considered among the hallmarks of cancer, produces high amounts of lactic acid (Gentric et al. 2017). Acidosis leads to a decoupling of glutaminolysis and de novo GSH synthesis, repressing cellular proliferation but increasing sensitivity to ROS (Lamonte et al. 2013). In addition, acidic pH evoked a p53-mediated metabolic reprogramming towards an enhanced Gln metabolism (Schoonjans et al. 2020). Mechanically, acidosis increases GLS2 and redirects Glc away from lactate production and for the oxidative branch of the PPP, to enlarge NADPH production and counter the enhancement in ROS as consequence of acidosis (Lamonte et al. 2013). Thus, acidosis modulates cancer somatic evolution and progression to malignancy, by increasing invasion, metastasis and chemoresistance (Pillai et al. 2019).
ROS cause genomic instability but also induce tumorigenesis by activating several signalling pathways that modulate cellular proliferation, angiogenesis, and metastasis (Dang et al. 2009). ROS can activate transcription factors such as NF-κB and HIFs, which are fundamental for metabolic reprogramming and cancer progression (Anso et al. 2013). Hypoxia rewires cancer cell metabolism enhancing glycolysis and mitigating Glc oxidation in the mitochondria mainly through transcriptional factors HIF-1α and HIF-2α (Anso et al. 2013; Schoonjans et al. 2020). HIF-1α/2α switches on a Gln-maintained TCA cycle by the activation of reductive carboxylation of αKG, to circumvent Glc shortage and achieve de novo lipogenesis under hypoxia (Okazaki et al. 2017). Other cancer-linked mutations that deregulate cellular metabolic responses to hypoxia also rewire carbon metabolism to facilitate utilization of Gln (Dang et al. 2009). Elevated levels of MYC are a cellular signal for tumour growth that facilitates higher utilization of Gln for nucleotide production (Fig. 1). MYC induces the expression of enzymes in nucleotide biosynthetic pathways, including phosphoribosyl pyrophosphate amidotransferase, which shifts the γ-nitrogen of Gln to 5-phosphoribosyl pyrophosphate, a rate-limiting step in purine biosynthesis that was increased in patients suffering from lung adenocarcinoma (Kodama et al. 2020). Additionally, Gln stimulates mitochondrial respiration in MYC-dependent osteogenic sarcoma cells (Anso et al. 2013). MYC also induced a transcriptional program for hypoxic adaptation, through transcription factor E2F1 (Fig. 1), inducing genes involved in nucleotide metabolism, Glc metabolism, and DNA replication (Dang et al. 2009).
Complex mTORC1 integrates multiple signals from growth factors, nutrients, and cellular energy status to modulate autophagy as well as the biosynthesis of mRNA, proteins, nucleotides, and lipids (Li et al. 2015). Concordantly, mTORC1 controls GLS levels through a S6K1-dependent regulation of Myc, in pancreatic cancer cells, by improving its translation efficiency through phosphorylation of eukaryotic initiation factor 4 (eIF4) (Csibi et al. 2014). As GLS, GLUD1 can also activate mTORC1, upregulating glycolytic enzymes that promote tumour development, and modulates the resistance to chemotherapeutic drugs, e.g., cisplatin, 5-fluorouracil (5-FU) in gastric cancer cell lines, as well as radiation in oral squamous cell carcinoma (OSCC) cell lines (Cetindis et al. 2016). Thus, targeting glutaminolysis may synergistically reduce the glycolytic phenotype of cancer cells and the activation of mTORC1 as a positive regulator of cell growth (see “Glutathione as an antioxidant and signalling molecule”).
Several pancreatic tumour cells support redox and central carbon metabolism through an eIF4-dependent translation program (Csibi et al. 2014). Enzyme regulatory network includes proteins involved in GSH and NADPH synthesis in PDA cells (Chan et al. 2019). In this pancreatic cancer cells, inhibition of eIF4 using synthetic rocaglate CR-1-31-B (CR-31) induces Gln reductive carboxylation towards fatty acid synthesis. Besides, ALDH, cystathionine gamma-lyase (CTH), and GST, all proteins dealing with the oxidative stress response network, are upregulated following GLS inhibition (Hensley and DeBerardinis 2015). Of interest, renal cell carcinoma progression generates oxidative stress and is susceptible to GLS inhibition (Teng et al. 2019). In this situation, NRF2-mediated oxidative stress response activates enzymes involved in GSH and NADPH synthesis, namely, GLS, GCL, GSS, GST, thioredoxin reductase (TrxR), and GR (Fig. 5). Accordingly, blocking GLS evoked nucleoside reduction and ROS increase that drove to DNA replication stress and induction of an intra–S phase checkpoint, abolishing the survival of RCC cells (Okazaki et al. 2017). HSP60 silencing induced Gln addiction, by MEK/ERK/MYC axis, switching mitochondria from ATP production to biosynthesis for promoting tumour growth and metastasis (Teng et al. 2019). On the other hand, KRAS, which upregulates NRF2, is the most commonly mutated oncogenic driver in NSCLC, colorectal cancer, and other solid tumours (Chen et al. 2020; Mukhopadhyay et al. 2020; Son et al. 2013). Many KRAS-mutant cancers carry loss-of-function mutations in the Keap1 gene encoding Kelch-like ECH-associated protein 1 (KEAP1), a negative regulator of NRF2 (Fig. 5). KEAP1 has many redox-sensitive cysteine residues that can react with oxidants, which affects its conformation and disturbs its ability to inhibit NRF2 (Eriksson et al. 2019). Serine/threonine kinase STK11 (also called LKB1) is the second most commonly altered tumour suppressor in NSCLC (Galan-Cobo et al. 2019). LKB1 loss produces increased energetic and redox stress defined by decreased levels of ATP and increased levels of intracellular ROS as well as declined NADPH/NADP+ ratio, and GSH levels (Galán-Cobo et al. 2019). Gln deprivation impairs GSH synthesis and increase damaging intracellular ROS (Okazaki et al. 2017). Cancer cells frequently shift their metabolism to be more Gln/GA/GSH-dependent (Matés et al. 2020a, b). Thus, deregulation of the KEAP1/NRF2 axis alters metabolic requirements evoking tumour cells to be more sensitive to Gln metabolism (Galan-Cobo et al. 2019). Accordingly, NRF2 activation reprogrammed metabolism by elevating pathways involved in glutaminolysis, as well as inducing genes encoding PPP enzymes, providing chemoresistance in cancer (Kitano et al. 2018; Mukhopadhyay et al. 2020).

NRF2 controls cancer cells oxidative stress. The nuclear factor erythroid 2-related factor 2 (NRF2) pathway is frequently mutated in cancer, either the NRF2 gene itself or its regulator KEAP1, resulting in stabilized NRF2, increased expression of NRF2 target genes, and elevated capacity to cope with oxidative stress. Oxidation of Keap1 cysteine residues inhibits Keap1 ubiquitin ligase activity, leading to NRF2 release. NRF2 is also activated by NOTCH1 signalling, which equally promotes glutaminase (GLS) expression. Hence, Nrf2 regulates many genes involved in glutathione (GSH) synthesis such as GLS, γ-glutamylcysteine synthetase (GCL) and glutathione synthetase (GSS). NRF2 leads to transactivation of ARE genes, including glutathione reductase (GR) and thioredoxine reductase (TrxR). TrxR activates p53 antioxidant activity inducing targets genes such as superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx), which is regulated by the glutaminase isoenzyme GLS2. TP53-induced glycolysis and apoptosis regulator (TIGAR) generates NADPH that is used for positive activation of ARE genes. NRF2 increases hypoxia-inducible factor-1 (HIF-1) to control ROS levels and activate p53. NRF2 also induces mouse double minute 2 (MDM2) that targets p53 for degradation in the proteasome, thus antagonizing p53 activity. On the contrary, MDM2 activates the nuclear factor kappa-B (NF-κB) signalling pathway by enhancing the transcription factor NF-κB. NF-κB increases antioxidant capacity against reactive oxygen species (ROS) through induction of antioxidant enzymes SOD, CAT and GPx. In addition, NF-κB can rise GLS expression to provide higher amounts of GSH. Tumor suppressor p53 can activate expression of antioxidant proteins like sestrins (SESN) or oxidant enzymes as aldehyde dehydrogenase (ALDH). Color codes are defined as follows: blue = regulatory proteins; orange = glutaminase isoenzymes; pink = enzymes involved in GSH biosynthesis; red = oxidant enzymes; yellow = antioxidant enzymes (color figure online)
GLS2 is a key p53 transcriptional target gene involved in regulation of the cellular redox homeostasis (Hu et al. 2010). Mutant p53 has been shown to bind NRF2 and modulate its transactivation capacity, stimulating the production of GSH, taurine and S-adenolsylmethionine through several target genes and routes, i.e., TIGAR, GLS2, SESN1/2, and p21-dependent activation of NRF2 (Eriksson et al. 2019; Kitano et al. 2018). In this context, mutant p53 proteins can achieve gain-of-function activities and determine the cellular oxidative balance, regulating redox signalling systems. Figure 1 shows an overview of redox regulation and sources of ROS, including cellular circuits that produce NADPH. The cell employs enzymatic and non-enzymatic antioxidant systems to balance oxidative stress (Matés et al. 2013). While catalase (CAT) and SOD neutralize highly oxidant species (O2·–, HO·, and H2O2) without consuming significant amounts of NADPH, the two major antioxidant systems thioredoxin (Trx) and GSH utilize NADPH for their dithiol-disulfide exchange reactions (Matés et al. 2012b). Trx reduces many proteins as ribonucleotide reductase, methionine sulfoxide reductase and Prxs (Eriksson et al. 2019), besides, rescues non-enzymatic antioxidants such as cysteine (Lee et al. 2016b). Main source of cellular NADPH are metabolic enzymes such as malic enzymes (ME), isocitrate dehydrogenases (IDH), and the PPP where glucose-6-phosphate dehydrogenase (G6PD) catalyzes the rate-limiting step (Lee et al. 2016b; Matés et al. 2020b).
Tumour microenvironment
Tumour-associated mutations switch on multiple cellular networks that promote adaptation of tumour cells to its TME, where oxygen and nutrient availability are restricted (Dang et al. 2009; Olson et al. 2016). In return, TME also adapts to cancer needs (Ippolito et al. 2020; Wu et al. 2019). Cancer cells need to reprogram ROS detoxification as well as nutrient acquisition and utilization to supply their high requirements for biosynthesis, energy production, and antioxidant defence (Dang et al. 2009; Matés et al. 2013). However, most tumours are resistant to the enormous ROS accumulation within the TME (Galán-Cobo et al. 2019). A hallmark of cancer metabolic reprogramming is the elevated utilization of Gln (DeBerardinis and Cheng 2010). Glutamine metabolism is modulated by many factors, including tissue of origin, the underlying genetics, oncogene/tumour suppressor status, epigenetic alterations, and TME (Lee et al. 2020; Wu et al. 2019; Yuneva et al. 2012). Despite the evidences of Gln addiction for progression and survival in tumour cells, the level of Gln in the TME used to be scarce (Jiang et al. 2019). Since targeting Gln uptake and utilization is both a hallmark of tumour growth, and an emerging therapeutic strategy in cancer (Matés et al. 2020a, b), it is fundamental to understand how TME affects Gln metabolism and tumour progression (Ippolito et al. 2020). Glutamine demand is very heterogeneous among different cancer cell lines, ranging from those that are Gln auxotrophs, to those that can growth and proliferate without an exogenous Gln supply (Son et al. 2013). For in vitro experiments TME conditions can be modelled, as the components of the culture media can be customized and other imperative variables can be controlled (Cluntun et al. 2017; Martinez-Outschoorn et al. 2017). In vivo, tumour growth is accompanied by many metabolic changes to the surrounding cells that result in a heterogeneous and complex TME (Marín-Valencia et al. 2012; Wu et al. 2019). For instance, glutaminase II pathway induces the metabolic rewiring in the stromal cells, enriching the aggressiveness associated with advanced prostate cancer (Udupa et al. 2019).
On the other hand, tumour-associated myeloid cells are one of the preeminent constituents of solid cancers, working as main immune regulators for the TME (Engblom et al. 2016). Wu et al. (2019) have found that hematopoietic precursor cells abound in the tissues of many types of tumours and can differentiate into immature myeloid cells (IMC). These authors have found that tumour-infiltrating IMCs are eminently glycolytic, proliferative, and immunosuppressive (through the Glu-N-methyl-d-aspartate receptor axis). Importantly, cancer cells acidify the TME and lessen nutrients and oxygen, weakening the function of T cells and countering their cytotoxicity against tumour cells (DeBerardinis 2020). IMCs response is linked with Gln metabolism, regardless Glc availability. Glc deprivation upregulated a set of genes in IMCs that have been implicated in glutaminolysis, as GLS, glutamine synthetase (GLUL), branched chain amino acid transaminase 1 (BCAT1), and SCL transporters, SLC1A3 and SLC7A11 (Wu et al. 2019). In addition to inhibiting GAs, the control of Gln availability for reducing tumour growth has been implemented using other strategies: (1) blocking Gln uptake by cells by targeting transporter ASCT2 (SLC1A5) using V-9302 (Schulte et al. 2018), (2) inhibiting both ASCT2 by benzylserine and GLUL by L-methionine sulfoximine (Ye et al. 2018), (3) blocking ASCT2 by benzylserine, l‐γ‐glutamyl‐p‐nitroanilide or shRNA (Wu et al. 2015), (4) silencing Gln transporter SNAT2 (SLC38A2) by siRNA (Luo et al. 2020; Morotti et al. 2019), (5) targeting the cystine-Glu antiporter xCT (LeBoeuf et al. 2020), (6) dowregulating ferroptosis (including GPX, SLC transporters and glutaminase), using β-elemene and cetuximab (Chen et al. 2020).
Metabolic and oxidative therapy
Because of plasticity of adaptive metabolic reprogramming in tumours, successful single treatments against cancers are scarce (Vaziri-Gohar et al. 2018). In a variety of cancers, multi-targeted therapy always includes some specific inhibitor of glutaminolysis (Matés et al. 2019). After the first attempt and failure to use 6-diazo-5-oxo-l-norleucine (DON) as an anti-cancer drug, many small molecules have been assayed to block GLS and GLS2 isoenzymes (Matés et al. 2020a, b). Dibenzophenanthridine-968, is an allosteric inhibitor of both GLS and GLS2 isoforms (Katt et al. 2012). The specific inhibitors most frequently used are bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES), and a derived molecule known as CB-839, which is a more potent, selective, and orally bioavailable inhibitor of both GLS splice variants (GAC and KGA) than BPTES (Aboud et al. 2017). CB-839 is currently being administered to humans in phase 1 clinical trials for some types of cancers (Gregory et al. 2019; Lee et al. 2020; Matés et al. 2020a, b). These molecules prevent the formation of phosphate-induced oligomers, which are active for GA activity (Márquez et al. 2016). Additional specific chemicals inhibiting GLS (CB-839 selenadiazole-derivatives CPD-20 and CPD-23, UPGL00004, zaprinast, physapubescin K), GLS2 (alkyl benzoquinone AV-1), or both (thiazolidine-2,4-dione compounds), have been used to fight against the proliferation of several types of tumours (Matés et al. 2020a, b).
Cell viability in high-grade chondrosarcomas was significantly decreased using monotherapy with CB-839, metformin, phenformin, and chloroquine (CQ). Every drug disrupts glutaminolysis: (1) CB-839 specifically targets GLS, (2) metformin and phenformin indirectly inhibits GLS via c-Myc, inhibits complex I of ETC, and represses mTORC1 signalling, (3) CQ inhibits GLUD1 (Peterse et al. 2018). Although reduction in tumour growth in human pancreatic cancer xenografts was similarly obtained by single agent therapies using BPTES, autophagy inhibitor CQ, anti-diabetic agents phenformin and metformin, transaminase inhibitor aminooxyacetate, and pyruvate dehydrogenase kinase (PDK) inhibitor dichloroacetate (Rajeshkumar et al. 2017), combination therapy is the best choice for clinical cancer treatments. Unique therapy uses to generate metabolic rewiring and drug resistance (Avolio et al. 2020). For example, in human RCC cells, inhibition of GLS using BPTES selectively impairs the growth of tumours cells that adapt to GLS repression by stimulating Glc oxidation in the TCA cycle (Okazaki et al. 2017). Hence, glutaminase-resistant tumours can use different carbon sources to keep TCA cycle precursors, while GA-sensitive tumours are highly dependent on Gln for TCA cycle supply (Galan-Cobo et al. 2019). For instance, following GLS inhibition by CB-839, a decrease in oxaloacetate levels observed only in the CB-839-resistant patient-derived xenografts models represents an adaptive response leading to their maintenance of Glu and citrate levels despite GLS inhibition (Daemon et al. 2018). This adaptive metabolic reprogramming leading to maintain anaplerosis and GSH levels can be achieved by conversion of oxaloacetate to Glu by transamination reactions (Son et al. 2013) and/or by citrate synthesis through PC and PDH activity (Cheng et al. 2011).
Dual therapy is usually projected by a variety of genetic and pharmacologic approaches (Lee et al. 2016a; Matés et al. 2020b). In a recent discovery, injections of adenovirus expressing Cre-recombinase in mice made animals deficient in aryl hydrocarbon receptor nuclear translocator, the common binding partner for both HIF-1α and HIF-2α transcription factors. This strategy, together with suppressing GLS by CB-839, significantly reduces tumour growth in undifferentiated pleomorphic sarcomas in vivo (Lee et al. 2020). Lack of other targeted therapies for soft tissue sarcomas endows GLS inhibition as a highly promising therapy, offering a new approach towards treating these patients. Notably, GLS targeting is a potential objective to block either antioxidant pathways or metabolic progression (Matés et al. 2019). LKB1-deficient cells harbouring KEAP1/NRF2 pathway inactivation were fully sensitive to GLS inhibition using CB-839, suggesting that LKB1 deficiency together with the KEAP1/NRF2 axis provides a specific vulnerability to block glutaminolysis and cell proliferation, enhancing the metabolic and oxidative stress response (Galan-Cobo et al. 2019). KEAP1/NRF2-mutant lung adenocarcinoma cells are dependent on increased glutaminolysis, and this metabolic feature has been exploited through the pharmacological inhibition of GLS using CB-839 and BPTES (Romero et al. 2017). In a similar study, LeBoeuf et al. demonstrated that GLS inhibition, using CB-839, suppresses KEAP1 mutant tumour growth in vivo through activation of the NRF2 antioxidant response pathway by limiting Glu availability (LeBoeuf et al. 2020). Of note, many KRAS-dependent tumours became resistant to mTORC1 inhibition by upregulating Gln metabolism, and mTORC1 inhibition in combination with CB-839 was able to overcome such resistance (Galan-Cobo et al. 2019). Moreover, blocking GLS abrogates the generation of functional immune cells, enhancing the therapeutic efficacy of other treatments (Wu et al. 2019). On the other hand, standard monotherapy for acute myeloid leukaemia (AML) has very poor results. Activating mutations of FMS-like tyrosine kinase 3 (FLT3) are one of the most frequently detected genetic events in this haematological malignancy. AC220 (FLT3 inhibitor) represses Gln import. Its combination with CB-839 synergized to deplete GSH, increase ROS, and produced apoptotic cell death in vitro and in vivo, improving survival significantly in a patient-derived xenograft AML mouse model (Gregory et al. 2018). Similarly, a high-level of resistance has been found in the standard therapy for T-cell acute lymphocytic leukemia (T-ALL) with Notch1 inhibitors; however, the combination of Notch1 inhibition and blocking of GLS/glutaminolysis with BPTES yielded complete regression of T-ALL xenotransplanted in mouse models (Herranz et al. 2015).
Other drugs that have shown synergistic effects with CB-839 are: (1) THZ1, a covalent inhibitor of cyclin-dependent kinase 7, evoking cell-cycle arrest in human NSCLC cells (Cheng et al. 2019); (2) gemcitabine, a deoxycytidine analogue that inhibits DNA replication and tumour growth, in pancreatic ductal adenocarcinoma (PDA) (Mokhapadyahay et al. 2020); (3) erastin, an inhibitor of xCT that depletes GSH levels, in lung adenocarcinoma (LeBoeuf et al. 2020); (4) CR-31, an eIF4 inhibitor that targets OXPHOS and glycolysis, in PDA cells (Chan et al. 2019); (5) β-lapachone, a NADPH:quinone oxidoreductase that leads to NADPH depletion through ROS and poly(ADP ribose) polymerase (PARP) hyperactivation, in PDA cells (Chakrabarti et al. 2015); metformin or rotenone, targeting mitochondrial complex I, in osteosarcoma cells in vitro and in vivo, also reducing metastasis (Ren et al. 2020).
Furthermore, olaparib, PARP inhibitor, synergized with BPTES to suppress the growth of RCC cells in vitro and in vivo (Okazaki et al. 2017). Similarly, a synergistic effect inducing cell death in vitro and in vivo, was obtained in NSCLC using BPTES/GLS knockdown by siRNA plus 5-FU, a thymidylate synthase inhibitor that elicited cell-cycle arrest and ATP depletion (Lee et al. 2016a). Another combinatory therapy includes sensitization to radiation (Xiang et al. 2013). So, CB-839 treatments produce a 50% reduction of serum GSH concentrations in several lung tumour cell lines and human lung-derived tumour xenografts in mice, increasing response to radiotherapy by 30% (Boysen et al. 2019). An interesting example are IDH mutant glioma that generate (R)-2-hydroxyglutarate (2-HG), an inhibitor of BACATs, which complements GLS inhibition by CB-839 to deplete Glu content and GSH availability and, consequently, sensitizing mutant glioma cells to oxidative stress in vitro and to radiation in vitro and in vivo (McBrayer et al. 2018). These findings were confirmed using many cell lines, engineered isogenic cells and patient-derived non-isogenic GBM stem cell lines, as well as in both subcutaneous and orthotopic tumour models.
Eventually, current single or combined treatments are not still easy approaches to eradicate cancer because metabolic heterogeneity (Chen et al. 2019; Cluntun et al. 2017; Wang et al. 2016). For instance, treatment with 968 compound and CB-839 suppresses glioma carcinogenic cells in vitro and in vivo, but not in all tumour cells. Sensitivity to GLS inhibition correlates with how much elevated is GLS expression of cells (Koch et al. 2020).
Oxidative unbalance and glutaminase reprogramming
ROS can be damaging agents, but also essential for tumour growth working as fundamental signalling species regulating proliferative pathways, i.e., AKT, HIF, MAPK, NF-κB in several types of cancers (Matés et al. 2008; Sullivan et al. 2013). Oxidative levels and redox homeostasis determine a thin line of balance that resolves life or death for the cell (Aboud et al. 2017; Zhang et al. 2020). Because normal cells have lower basal levels of ROS, an increase in oxidative stress can selectively kill tumour cells, and this strategy has been used as a therapeutic tool against some cancers (Martín-Rufián et al. 2014). Singularly, MYC-dependent osteogenic sarcoma cells, which are dependent on Glc and Gln/GSH/GLS compared to osteocytes, are dependent on mitochondrial ROS to proliferate (Anso et al. 2013). Hence, these authors demonstrated that although depletion of GSH by oxidant agents such as beta-phenylethyl isothiocyanate or buthionine sulfoximine (BSO) did not cause apoptosis in MYC-dependent osteogenic sarcoma cells, reducing mitochondrial ROS levels by a mitochondrial-targeted vitamin E declined cell proliferation. Additional studies have confirmed the value of mitochondrial-targeted antioxidants against cancer in different human models, including HCC (Dilip et al. 2013), gastric, pancreatic, umbilical vein epithelial, and breast tumours cells (Du et al. 2019), and leukaemia (Gregory et al. 2019). It is tempting to speculate whether a dual therapy including mitochondrial-targeted antioxidants and GLS inhibitors would have synergistic effect as a valuable therapeutic strategy. Supporting that point, a high correlation between tumour sensitivity to GLS (using BPTES) and GCL inhibition (using BSO) has been demonstrated among a broad panel of > 400 tumour cell lines (Daemen et al. 2018). Besides, although inhibition of Gln pathway decreases pyrimidine synthesis (indispensable for DNA formation), it has been suggested that GLS inhibition might be a less damaging chemical or genetic therapy than the use of other more cytotoxic chemotherapeutic approaches that have stronger consequences on cells and organisms (Gregory et al. 2018; Matés and Campos-Sandoval 2018).
Mitochondrial antioxidant and pro-oxidant strategies have been described for anti-cancer therapy (Ippolito et al. 2020). Targeting glutaminolysis in combination with drugs that unbalance mitochondrial redox state are widely used for treating multiple types of cancers. For example, combinatorial strategy firstly disrupting redox homeostasis (inhibiting glutaminolysis and lowering GSH) followed by oxidant drugs might yield a better anti-tumour efficacy (Wang et al. 2016). Next, we will outline some of such therapeutic designs (Table 1). First, when HCC cells were treated with compound 968 they became more sensitive to dihydroartemisinin-mediated cytotoxicity in vitro and in vivo because combination therapy evoked excessive intracellular ROS that strongly induced apoptosis (Wang et al. 2016). Similarly, CB-839 evoked a synergistic effect increasing apoptosis when combined with H2O2 in RCC (Aboud et al. 2017). Two different FDA-approved pro-oxidant drugs that are strong inducers of mitochondrial ROS, arsenic trioxide (ATO) and homoharringtonine (HHT) showed great activity against AML when combined with the CB-839, both in vitro and in vivo (Table 1). Inhibition of GLS provoked accretion of ROS, activation of apoptosis, and made AML cells susceptible to those adjuvant drugs that further perturbed mitochondrial redox state (Gregory et al. 2019). Moreover, there are other hematologic targets, such as multiple myeloma, T-ALL cells and leukaemia stem cells, which have been found to be highly dependent on Gln metabolism for proliferate (Gregory et al. 2019).
Combination therapy produced synergistic effects in breast cancer cells, including TNBC. Thus, GLS was blocked using compound 968, but metabolic rewiring to maintain Glu levels activated both lipid metabolism and autophagy (Halama et al. 2018). Synergistic inhibition of cell proliferation was achieved by a dual treatment consisting in compound 968 and inhibitors of lipolysis (etomoxir or trimetazidine), or compound 968 and inhibitors of autophagy (CQ and bafilomycin A1). Hence, an interesting experiment would be a triple therapy targeting (1) GLS, (2) autophagy, and (3) fatty acid oxidation (FAO). In a similar strategy, compound 968 was used for repressing GLS, and CQ to suppress induced autophagy in NSCLC. This combined treatment inhibited cell growth and migration downregulating EGFR/ERK pathway and inducing G1/G0-phase cell-cycle arrest (Han et al. 2017). Almost coincident, BPTES was used for GLS blocking and synergistically annulling the metabolic reprogramming, in SiHa cervix cancer cells and HCT-116 colon cancer cells, after dichloroacetate treatment for PDK inhibition and subsequently abolishing HIF-1α activity and enhancing p53 activity to decrease Glc uptake and tumour growth (Schoonjans 2020). Again, dual inhibition of GLS (by BPTES) and HSP90 (using 17AAG drug) caused a synergistic regression of mTORC1-driven cell xenograft tumours through apoptosis induction due to a deregulated redox balance (Li et al. 2015). Compound 968 synergistically reduced tumour growth and enhanced apoptosis when combined with apigenin (4′,5,7,-trihydroxyflavone), a drug having multiple targets (including GLUT1, NF-κB, PI3K, EGFR, and HIF‑1α), that produced accumulation of intracellular ROS through the depletion of GSH and NADPH, sensitizing lung cancer cells to inhibition of Gln utilization (Lee et al. 2016b).
Another dual treatment, including ATO or H2O2 plus silencing GLS or overexpressing GLS2, synergistically inhibited survival of GBM cell lines through a MIY- BID- BCL-2-dependent mechanism (Martín-Rufián et al. 2014). Selenite (SeO32−) induced ROS-dependent apoptosis in both A549 lung cancer cells in vitro and patient lung cancer tissues ex vivo, as well as in a variety of cancer cells including prostate, glioma, and colorectal cancers (Bruntz et al. 2019). Strikingly, selenite exerts a dual function as ROS source and Gln metabolism disrupter. In this research, authors have shown how selenite inhibits glutaminolysis and GSH synthesis by suppressing GLS expression, and disrupting TCA cycle, inhibiting PC and malic enzyme (ME) activities, affecting multiple anaplerotic targets and minimizing the likelihood of resistance development in lung cancer cells. Markedly, CB-839 elicited coincident metabolic responses as selenite (Bruntz et al. 2019). Differently, because prostate cancer cells metastasizes preferentially to the skeleton, a cell model has been characterized to analyse how tumour progression led to an increase of the “bone cell-like” differentiation from the original LNCaP human prostate cells (Dorai et al. 2018). Increase in the interaction of membrane protein caveolin-1 with the cytoplasmic nicotinamide riboside: quinone oxidoreductase (NQO2), as well as an increment in the oxidative stress and metastatic potential was detected in cells showing the most osteomimetic properties. The associated extrusion of large extracellular vesicles, containing NQO2-caveolin-1, was increased in the bone metastatic cells, but this process was repressed using BPTES, which also decreases the cells’ osteomimetic properties (Dorai et al. 2018). Consequently, a dual therapy consisting on BPTES or CB-839, plus inhibitors of NQO2 (quercetin, resveratrol, melatonin, CQ and imatinib) might be useful for advanced prostate cancer patients to avoid bone metastatic complications.
Future prospects and troubles
Glutamine and glutaminases not only are fundamental in the control of redox homeostasis and intermediary metabolism but also as molecular tools for monitoring Gln-dependent cancers (Hensley et al. 2013). Because many cancers are Gln avid, and that the positron emission tomography (PET) radiotracer 18F-(2S,4R)4-fluoroglutamine (18F-Gln) is efficiently taken up by such Gln-dependent tumours cells both in vitro and in vivo, Gln-PET is being instrumented in clinical oncology to complement traditional 18F-fluorodeoxyglucose PET imaging (Zhu et al. 2019b). Additionally, Glc-PET is not effective in evaluating gliomas in vivo because of the high Glc metabolism in the normal brain derivatives in suboptimal tumour detection (Venneti et al. 2015). Besides, gliomas have so high Gln content, because its de novo synthesis, uptake from the blood supply, and reduced rate of export, that 18F-Gln can serve as an imaging biomarker (Marin-Valencia et al. 2012). Thus, the use of Gln-PET has been optimized in last years to quantify tumour cellular Gln pool size, whose change might likewise report the pharmacodynamic effect of GLS inhibitors targeting glutaminolysis (Zhou et al. 2017). Even better, an alternative strategy has described radiolabel Gln-derived trifluoroborate PET tracer (18F-Gln-BF3) for tumour imaging and in vivo evaluation by biodistribution (Li et al. 2018). Imaging and Gln distribution in vivo has proved to be a valuable tool for monitoring Gln reprogramming in antioxidant production in orthotopic mice models (Aboud et al. 2017). Consequently, Gln dependence can be used to obtain Gln-PET imaging, leading to stratification of patients into those whose tumours are more sensitized to GLS inhibition (Venneti et al. 2015). This strategy might be amenable to in vivo monitoring of the progress of tumours to GLS inhibition therapy by following Gln levels in the presence of specific GLS inhibitors (Aboud et al. 2017). Another strategy for monitoring cancer growth and metabolic rewiring, evoking tumour resistance, is the quantification of metabolites, mostly from the 13C enrichment of molecules from central carbon metabolism (Chen et al. 2019).
Omics data are a new source of valuable knowledge of metabolic reprogramming and its therapeutic consequences (Faubert et al. 2020). Together with genomics, proteomics, transcriptomics, and metabolomics material, the interactome of enzymes involved in Gln metabolism, as GLS and GLS2, is starting to be uncovered adding a new level of regulatory complexity with important functional ramifications, including their specific and regulated targeting (Márquez et al. 2017). For instance, Daemen et al. implemented a predictive pancancer metabolic signature for GLS/GCL co-dependency in vivo, identifying Glu and citrate as main predictive biomarkers. Among sensitive tumours, increased GAC levels associate with higher response to GLS repression by CB-839 (Daemen et al. 2018). Therefore, in the near future, extensive database compelling information of oncogenotype, gene expression, protein expression, and therapeutic sensitivity, as well as selected metabolites as significant biomarkers will enable the user to bring to light new relationships between metabolism and oncological processes (Chen et al. 2019). Ex vivo and in vivo applied fluxomics for cancer tracking is another key instrument to be flourished in the future years to better understand metabolic sensitivities and therapeutic requirements (Yuan et al. 2019). The hoarded data point out that the cancer rewiring of the diverse metabolic and energy provision networks will offer biomarkers (including tracing of labelled metabolites) that, alone or in combination, could be handled to arrest cancer in a near future successful personalized oncology (Torresano et al. 2020).
New research is necessary to better underpin whether patient’s omics patterns are origin or effect of individual cellular programs (Matés et al. 2020a, b). Another demanding obstacle in cancer therapy is the metabolic heterogeneity among cells in a single tumour of an individual patient (Matés and Campos-Sandoval 2018). Understanding the cellular and molecular basis of oxidative and metabolic reprogramming will improve integration of data through systems biology to illustrate the changeling networking between cellular metabolism and redox signalling in each heterogeneous lone tumour (Chen et al. 2019). Furthermore, compiled evidence suggests that the TME modifies cancer metabolism and symbiotic relationships within tumours might provoke a reset of both oxidative and metabolic networks to initiate resistance (Faubert et al 2020; Muir et al. 2018; Olson et al. 2016). Otherwise, neoplasms can secrete functional species (i.e., lactate, ROS, protons) to shape an unfavourable metabolic environment for immune cells, which trigger cancer progression (Schulze annnd Harris 2012).
Conclusions
In general, a favourable TME for cancer proliferation is characterized by high Glc utilization (glycolysis), and low ATP yield, while a harsh TME can be defined by lower Glc utilization (OXPHOS) and higher ATP yield (Vaziri-Goher et al. 2018). Although switch between aerobic glycolysis and OXPHOS is a major metabolic signature in a variety of cancers (Kreitz et al 2019), it seems that a unified model of altered tumour metabolism might not exist, neither unique rewiring metabolic pathways to support both energy/biosynthetic demands and redox homeostasis (Chen et al. 2019; Zhang et al. 2020).
Anyhow, increased antioxidant capacity of tumour cells is required for adaptation to hypoxia and ROS of rapidly proliferating cancer cells (Hlouschek et al. 2018). Metabolic or redox therapy against cancer has resulted in many interesting findings, using specific targeting against key enzymes, proteotoxic stress or oxidative stress, but tumour resistance by adaptive metabolic rewiring is a common response in several types of cancers (Daemon et al. 2018; Okazaki et al. 2017). Many tumours activate intrinsic antioxidant systems as a means to counteract excessive ROS levels during periods or locations of environmental stress, such as a highly metabolically aggressive malignancy (Martín-Rufián et al. 2014). On the contrary, upon GLS inhibition with CB-839 or BPTES, the RCC cell lines SN12PM-6-1 (SN12) and 786-O exhibited lower survival and higher apoptosis associated with a decreased GSH/GSSG ratio, increased NRF2, and structural damage within the cell, including enhanced DNA damage (Aboud et al. 2017). Albeit the oxidative stress generated by ROS can be protumorigenic by inducing mutations, it can operate in an antitumorigenic fashion too, by inducing apoptosis and reducing HIF-1α level (Schoonjans et al. 2020). So, the adjustment of pro- and antioxidant systems needs to be finely balanced for the proliferation of the tumour, although this characteristic also has the potential to be overturned for curative purposes (Aboud et al. 2017). In addition, reductive stress (excessive levels of GSH, NADPH and NADP) must be counteracted to avoid its related adverse negative consequences in OXPHOS and mitochondrial homeostasis (Xiao and Loscalzo 2020). A better understanding of the metabolic diversity leading to a flexible redox homeostasis will improve our ability to delimit their contribution to aggressive cancer progression, and to characterize more metabolic effectors to be adopted as therapeutic targets (Faubert et al. 2020; Kreitz et al 2019; Vaziri-Gohar et al. 2018). In aggregate, new research is needed to discover novel biomarkers for patient stratification and response to therapy.
Combination therapies inhibiting oncometabolism offer enormous benefits for inhibiting multiple targets and signalling pathways to synergistic kill cancer cells, avoiding or postponing the appearance of drug resistance (Li et al. 2015). Among regulatory proteins and signalling pathways MYC, p53, NRF2 and antioxidants enzymes play essential roles in the strict regulation of GLS and GLS2, and then of GSH content, which modulates apoptosis and survival of tumour cells (Galán-Cobo et al. 2019; Matés et al. 2013; Mukhopadhyay et al. 2020). Glutamine and Glu are essential fuels for cancer that can be drained inhibiting GLS (Wu et al. 2018) and GLUL activities (Ye et al. 2018), as well as blocking ASCT2 transporters (Jiang et al. 2020). Nevertheless, it is important to consider that although ASCT2 and LAT1 systems use to have an oncogenic function, coupling model of Gln transporters is not universal across different cancer types (Cormerais et al. 2018). Again, metabolic heterogeneity among types of cancers, patients, and even different cells inside the same tumour is a wall to wreck (Martinez-Outschoorn et al. 2017). As a hope, metabolic therapies arranged to target specific cancers are designed on the basis that some tumour cells are less metabolically plastic than their normal counterparts, and this may be a factor to selectively target malignancies while sparing healthy cells (Olson et al. 2016). Remarkably, a new Gln antagonist is JHU083, a prodrug that can release DON inside tumours, thereby annulling its toxic effects on brain, liver, heart, and other basic organs (DeBerardinis 2020). JHU083 abolishes tumour growth in mice by a dual function: (1) blocking cancer cells from using Gln, and (2) upgrading TME for effector T cells, which, in turn, rise their immune response against the tumour (Leone et al. 2019). Importantly, these effects conclude in unremitting anti-tumour immunity (DeBerardinis 2020).
In conclusion, multiple and synergistic targeting has reached better results in both blocking tumour proliferation, and sensitizing cancer cells to other treatments (Fatima et al. 2019). GA isoenzymes as well as their interacting biomolecules and signalling pathways are established targets for cancer therapy (Fig. 3). To analyse and customize results in cancer patients will be necessary to ensure a clinical use (Faubert et al. 2020). Importantly, proteomics and metabolomics studies, following GLS targeting, will anticipate responsiveness to combination therapy. Further work is required to investigate the metabolic and oxidative consequences of GLS silencing and GLS2 inhibition/overexpression to generate new anti-cancer strategies (Matés et al. 2019). In addition, most efficient therapeutic approaches will have to target the multiple mechanisms used by cancer cells to corrupt the basal TME, including metabolic and redox homeostasis (Zhang et al. 2020). Nonetheless, specific targeting will not be easy because metabolic circuits that activate the growth of most tumours are different from the metabolic processes that promote metastasis and therapy resistance (Faubert et al. 2020).
References
Aboud OA, Habib SL, Trott J et al (2017) Glutamine addiction in kidney cancer suppresses oxidative stress and can be exploited for real-time imaging. Cancer Res 77(23):6746–6758
Akins NS, Nielson TC, Le HV (2018) Inhibition of glycolysis and glutaminolysis: an emerging drug discovery approach to combat cancer. Curr Top Med Chem 18(6):494–504
Anso E, Mullen AR, Felsher DW, Matés JM, Deberardinis RJ, Chandel NS (2013) Metabolic changes in cancer cells upon suppression of MYC. Cancer Metab 1(1):7
Avolio R, Swann Matassa D, Criscuolo D, Landriscina M, Esposito F (2020) Modulation of mitochondrial metabolic reprogramming and oxidative stress to overcome chemoresistance in cancer. Biomolecules 10(1):135
Benlloch M, Mena S, Ferrer P et al (2006) Bcl-2 and Mn-SOD antisense oligodeoxynucleotides and a glutamine-enriched diet facilitate elimination of highly resistant B16 melanoma cells by tumor necrosis factor-alpha and chemotherapy. J Biol Chem 281(1):69–79
Boysen G, Jamshidi-Parsian DMA et al (2019) Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int J Radiat Biol 95:436–442
Bruntz RC, Belshoff AC, Zhang Y et al (2019) Inhibition of anaplerotic glutaminolysis underlies selenite toxicity in human lung cancer. Proteomics 19(21–22):e1800486
Cassago A, Ferreira AP, Ferreira IM et al (2012) Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci USA 109(4):1092–1097
Cardona C, Sánchez-Mejías E, Dávila JC et al (2015) Expression of Gls and Gls2 glutaminase isoforms in astrocytes. Glia 63:365–382
Carretero J, Obrador E, Pellicer JA, Pascual A, Estrela JM (2000) Mitochondrial glutathione depletion by glutamine in growing tumor cells. Free Radic Biol Med 29(9):913–923
Cetindis M, Biegner T, Munz A, Teriete P, Reinert S, Grimm M (2016) Glutaminolysis and carcinogenesis of oral squamous cell carcinoma. Eur Arch Otorhinolaryngol 273(2):495–503
Chakrabarti G, Moore ZR, Luo X et al (2015) Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ß-lapachone. Cancer Metab 3:12
Chan K, Robert F, Oertlin C et al (2019) eIF4A supports an oncogenic translation program in pancreatic ductal adenocarcinoma. Nat Commun 10(1):5151
Chang X, Zhu W, Zhang H, Lian S (2017) Sensitization of melanoma cells to temozolomide by overexpression of microRNA 203 through direct targeting of glutaminase-mediated glutamine metabolism. Clin Exp Dermatol 42:614–621
Chen PH, Cai L, Huffman K et al (2019) Metabolic diversity in human non-small cell lung cancer cells. Mol Cell 76(5):838–851.e5
Chen P, Li X, Zhang R, Liu S et al (2020) Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 10(11):5107–5119
Cheng ZJ, Miao DL, Su QY et al (2019) THZ1 suppresses human non-small-cell lung cancer cells in vitro through interference with cancer metabolism. Acta Pharmacol Sin 40(6):814–822
Cheng T, Sudderth J, Yang C et al (2011) Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA 108:8674–8679
Cluntun AA, Lukey MJ, Cerione RA, Locasale JW (2017) Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer 3(3):169–180
Cormerais Y, Massard PA, Vucetic M et al (2018) The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J Biol Chem 293(8):2877–2887
Csibi A, Lee G, Yoon SO et al (2014) The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol 24(19):2274–2280
Daemen A, Liu B, Song K et al (2018) Pan-cancer metabolic signature predicts co-dependency on glutaminase and de novo glutathione synthesis linked to a high-mesenchymal cell state. Cell Metab 28(3):383–399
Dang CV, Le A, Gao P (2009) MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res 15(21):6479–6483
DeBerardinis RJ (2020) Tumor microenvironment, metabolism, and immunotherapy. N Engl J Med 382(9):869–871
DeBerardinis RJ, Cheng T (2010) Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29(3):313–324
de Guzzi Cassago CA, Dias MM, Pinheiro MP et al (2018) glutaminase affects the transcriptional activity of peroxisome proliferator-activated receptor γ (PPARγ) via direct interaction. Biochemistry 57:6293–6307
Dias MM, Adamoski D, Dos Reis LM et al (2020) GLS2 is protumorigenic in breast cancers. Oncogene 39(3):690–702
Dilip A, Cheng G, Joseph J et al (2013) Mitochondria-targeted antioxidant and glycolysis inhibition: synergistic therapy in hepatocellular carcinoma. Anticancer Drugs 24(9):881–888
Dorai T, Dorai B, Pinto JT, Grasso M, Cooper AJL (2020) High levels of glutaminase II pathway enzymes in normal and cancerous prostate suggest a role in ‘glutamine addiction’. Biomolecules 10(1):2
Dorai T, Shah A, Summers F et al (2018) NRH: quinone oxidoreductase 2 (NQO2) and glutaminase (GLS) both play a role in large extracellular vesicles (LEV) formation in preclinical LNCaP-C4-2B prostate cancer model of progressive metastasis. Prostate 78(15):1181–1195
Draoui N, de Zeeuw P, Carmeliet P (2017) Angiogenesis revisited from a metabolic perspective: role and therapeutic implications of endothelial cell metabolism. Open Biol 7(12):170219
Du X, Zhang P, Fu H, Ahsan HM, Gao J, Chen Q (2019) Smart mitochondrial-targeted cancer therapy: subcellular distribution, selective TrxR2 inhibition accompany with declined antioxidant capacity. Int J Pharm 555:346–355
El-Hout M, Cosialls E, Mehrpour M, Hamaï A (2020) Crosstalk between autophagy and metabolic regulation of cancer stem cells. Mol Cancer 19:27
Engblom C, Pfirschke C, Pittet MJ (2016) The role of myeloid cells in cancer therapies. Nat Rev Cancer 16:447–462
Eriksson SE, Ceder S, Bykov VJN, Wiman KG (2019) p53 as a hub in cellular redox regulation and therapeutic target in cancer. J Mol Cell Biol 11(4):330–341
Fatima I, El-Ayachi I, Playa HC et al (2019) Simultaneous multi-organ metastases from chemo-resistant triple-negative breast cancer are prevented by interfering with WNT-signaling. Cancers (Basel) 11(12):2039
Faubert B, Solmonson A, DeBerardinis RJ (2020) Metabolic reprogramming and cancer progression. Science 368(6487):eaaw5473
Ferreira AP, Cassago A, de Gonçalves K et al (2013) Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J Biol Chem 288(39):28009–28020
Gao P, Tchernyshyov I, Chang T-C et al (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765
Galan-Cobo A, Sitthideatphaiboon P, Qu X et al (2019) LKB1 and KEAP1/NRF2 Pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res 79(13):3251–3267
Gentric G, Mieulet V, Mechta-Grigoriou F (2017) Heterogeneity in cancer metabolism: new concepts in an old field. Antioxid Redox Signal 26(9):462–485
Giacobbe A, Bongiorno-Borbone L, Bernassola F et al (2013) p63 regulates glutaminase 2 expression. Cell Cycle 12:1395–1405
Gregory MA, Nemkov T, Park HJ et al (2019) Targeting glutamine metabolism and redox state for leukemia therapy. Clin Cancer Res 25(13):4079–4090
Gregory MA, Nemkov T, Reisz JA et al (2018) Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp Hematol 58:52–58
Halama A, Kulinski M, Dib SS et al (2018) Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett 430:133–147
Hammouda MB, Ford AE, Liu Y, Zhang JY (2020) The JNK signaling pathway in inflammatory skin disorders and cancer. Cells 9(4):857
Han T, Guo M, Zhang T, Gan M, Xie C, Wang JB (2017) A novel glutaminase inhibitor-968 inhibits the migration and proliferation of non-small cell lung cancer cells by targeting EGFR/ERK signaling pathway. Oncotarget 8(17):28063–28073
Hayano M, Yang WS, Corn CK et al (2016) Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ 23:270–278
Hensley CT, DeBerardinis RJ (2015) In vivo analysis of lung cancer metabolism: nothing like the real thing. J Clin Investig 25:495–497
Hensley CT, Wasti AT, DeBerardinis RJ (2013) Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 123(9):3678–3684
Herranz D, Ambesi-Impiombato A, Sudderth J et al (2015) Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med 21(10):1182–1189
Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z (2010) Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA 107(16):7455–7460
Ippolito L, Giannoni E, Chiarugi P, Parri M (2020) Mitochondrial redox hubs as promising targets for anticancer therapy. Front Oncol 10:256
Jiang J, Srivastava S, Zhang J (2019) Starve cancer cells of glutamine: break the spell or make a hungry monster? Cancers (Basel) 11(6):E804
Jiang H, Zhang N, Tang T, Feng F, Sun H, Qu W (2020) Target the human alanine/serine/cysteine transporter 2(ASCT2): achievement and future for novel cancer therapy. Pharmacol Res 158:104844
Kamarajan P, Rajendiran TM, Kinchen J, Bermúdez M, Danciu T, Kapila YL (2017) Head and neck squamous cell carcinoma metabolism draws on glutaminolysis, and stemness is specifically regulated by glutaminolysis via aldehyde dehydrogenase. J Proteome Res 16(3):1315–1326
Katt WP, Ramachandran S, Erickson JW, Cerione RA (2012) Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol Cancer Ther 11(6):1269–1278
Kitano Y, Baba Y, Nakagawa S et al (2018) Nrf2 promotes oesophageal cancer cell proliferation via metabolic reprogramming and detoxification of reactive oxygen species. J Pathol 244(3):346–357
Koch K, Hartmann R, Tsiampali J et al (2020) A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov 6:20
Kodama M, Oshikawa K, Shimizu H (2020) A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat Commun 11(1):1320
Kreitz J, Schönfeld C, Seibert M et al (2019) Metabolic plasticity of acute myeloid leukemia. Cells 8(8):80
Kuo TC, Chen CK, Hua KT et al (2016) Glutaminase 2 stabilizes Dicer to repress Snail and metastasis in hepatocellular carcinoma cells. Cancer Lett 383:282–294
Lamonte G, Tang X, Chen JL et al (2013) Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab 1(1):23
LeBoeuf SE, Wu WL, Karakousi TR et al (2020) Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non-essential amino acids. Cell Metab 31(2):339–350
Lee JS, Kang JH, Lee SH et al (2016a) Dual targeting of glutaminase 1 and thymidylate synthase elicits death synergistically in NSCLC. Cell Death Dis 7(12):e2511
Lee YM, Lee G, Oh TI et al (2016b) Inhibition of glutamine utilization sensitizes lung cancer cells to apigenin-induced apoptosis resulting from metabolic and oxidative stress. Int J Oncol 48(1):399–408
Lee P, Malik D, Perkons N et al (2020) Targeting glutamine metabolism slows soft tissue sarcoma growth. Nat Commun 11:498
Lee YZ, Yang CW, Chang HY et al (2014) Discovery of selective inhibitors of glutaminase-2, which inhibit mTORC1, activate autophagy and inhibit proliferation in cancer cells. Oncotarget 5(15):6087–6101
Leone RD, Zhao L, Englert JM et al (2019) Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366:1013–1021
Li B, Cao Y, Meng G et al (2019a) Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 39:239–254
Li J, Csibi A, Yang S et al (2015) Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1-driven tumor cells. Proc Natl Acad Sci USA 112(1):E21–29
Li J, Li X, Wu L, Pei M, Li H, Jiang Y (2019b) miR-145 inhibits glutamine metabolism through c-myc/GLS1 pathways in ovarian cancer cells. Cell Biol Int 43(8):921–930
Li C, Liu H, Duan D, Zhou Z, Liu Z (2018) Preclinical study of an (18)F-labeled glutamine derivative for cancer imaging. Nucl Med Biol 64–65:34–40
Liu Z, Wang J, Li Y, Fan J, Chen L, Xu R (2017) MicroRNA-153 regulates glutamine metabolism in glioblastoma through targeting glutaminase. Tumour Biol 39:1010428317691429
López de la Oliva AR, Campos-Sandoval JA, Gómez-García MC et al (2020) Nuclear translocation of glutaminase GLS2 in human cancer cells associates with proliferation arrest and differentiation. Sci Rep 10(1):2259
Lora J, Alonso FJ, Segura JA, Lobo C, Márquez J, Matés JM (2004) Antisense glutaminase inhibition decreases glutathione antioxidant capacity and increases apoptosis in Ehrlich ascitic tumour cells. Eur J Biochem 271(21):4298–4306
Luan W, Zhou Z, Zhu Y, Xia Y, Wang J, Xu B (2018) miR-137 inhibits glutamine catabolism and growth of malignant melanoma by targeting glutaminase. Biochem Biophys Res Commun 495:46–52
Lukey MJ, Greene KS, Erickson JW, Wilson KF, Cerione RA (2016) The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat Commun 7:11321
Lukey MJ, Cluntun AA, Katt WP et al (2019) Liver-type glutaminase GLS2 is a druggable metabolic node in luminal-subtype breast cancer. Cell Rep 29(1):76–88
Luo Y, Li W, Ling Z et al (2020) ASCT2 overexpression is associated with poor survival of OSCC patients and ASCT2 knockdown inhibited growth of glutamine-addicted OSCC cells. Cancer Med 9(10):3489–3499
Majewska E, Márquez J, Albrecht J, Szeliga M (2019) Transfection with GLS2 glutaminase (GAB) sensitizes human glioblastoma cell lines to oxidative stress by a common mechanism involving suppression of the PI3K/AKT pathway. Cancers (Basel) 11(1):115
Majewska E, Rola R, Barczewska M, Márquez J, Albrecht J, Szeliga M (2017) Transcription factor GATA3 expression is induced by GLS2 overexpression in a glioblastoma cell line but is GLS2-independent in patient-derived glioblastoma. J Physiol Pharmacol 68(2):209–214
Marin-Valencia I, Yang C, Mashimo T et al (2012) Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab 15(6):827–837 (Erratum in: 16(5):686)
Márquez J, Alonso FJ, Matés JM, Segura JA, Martín-Rufián M, Campos-Sandoval JA (2017) Glutamine addiction in gliomas. Neurochem Res 42:1735–1746
Márquez J, Matés JM, Campos-Sandoval JA (2016) Glutaminases. Adv Neurobiol 13:133–171
Martín-Rufián M, Nascimento-Gomes R, Higuero A et al (2014) Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J Mol Med (Berl) 92(3):277–290
Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP (2017) Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14(1):11–31
Martínez-Reyes I, Chandel NS (2020) Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 11(1):102
Masisi BK, El Ansari R, Alfarsi L, Rakha EA, Green AR, Craze ML (2020) The role of glutaminase in cancer. Histopathology 76(4):498–508
Matés JM, Campos-Sandoval JA, de los Santos-Jiménez J, Segura JA, Alonso FJ, Márquez J (2020a) Metabolic reprogramming of cancer by chemicals that target glutaminase isoenzymes. Curr Med Chem 27(32):5317–5339
Matés JM, Campos-Sandoval JA (1870) Márquez J (2018) Glutaminase isoenzymes in the metabolic therapy of cancer. Biochim Biophys Acta Rev Cancer 1870(2):158–164
Matés JM, Campos-Sandoval JA, de los Santos-Jiménez J, Márquez J (2019) Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett 467:29–39
Matés JM, Di Paola FJ, Campos-Sandoval JA, Mazurek S, Márquez J (2020b) Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin Cell Dev Biol 98:34–43
Matés JM, Segura JA, Alonso FJ, Márquez J (2008) Intracellular redox status and oxidative stress: implications for cell proliferation, apoptosis, and carcinogenesis. Arch Toxicol 82(5):273–299
Matés JM, Segura JA, Alonso FJ, Márquez J (2012a) Oxidative stress in apoptosis and cancer: an update. Arch Toxicol 86(11):1649–1665
Matés JM, Segura JA, Alonso FJ, Márquez J (2012b) Sulphur-containing non enzymatic antioxidants: therapeutic tools against cancer. Front Biosci (Schol Ed) 4:722–748
Matés JM, Segura JA, Campos-Sandoval JA et al (2009) Glutamine homeostasis and mitochondrial dynamics. Int J Biochem Cell Biol 41(10):2051–2061
Matés JM, Segura JA, Martín-Rufián M, Campos-Sandoval JA, Alonso FJ, Márquez J (2013) Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med 13(4):514–534
McBrayer SK, Mayers JR, DiNatale GJ et al (2018) Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175(1):101–116
Morotti M, Bridges E, Valli A (2019) Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc Natl Acad Sci USA 116(25):12452–12461
Muir A, Danai LV, Vander Heiden MG (2018) Microenvironmental regulation of cancer cell metabolism: implications for experimental design and translational studies. Dis Model Mech 11(8):dmm035758
Mukhopadhyay S, Goswami D, Adiseshaiah PP et al (2020) Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res 80(8):1630–1643
Niu Y, Zhang J, Tong Y, Li J, Liu B (2019) Physcion 8-O-β-glucopyranoside induced ferroptosis via regulating miR-103a-3p/GLS2 axis in gastric cancer. Life Sci 237:116893
Obara-Michlewska M, Szeliga M (2020) Targeting glutamine addiction in gliomas. Cancers (Basel) 12(2):310
Okazaki A, Gameiro PA, Christodoulou D (2017) Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J Clin Invest 127(5):1631–1645
Olalla L, Gutiérrez A, Campos JA et al (2002) Nuclear localization of L-type glutaminase in mammalian brain. J Biol Chem 277(41):38939–38944
Olson KA, Schell JC, Rutter J (2016) Pyruvate and metabolic flexibility: illuminating a path toward selective cancer therapies. Trends Biochem Sci 41:219–230
Ortiz-Pedraza Y, Muñoz-Bello JO, Olmedo-Nieva L et al (2020) Non-coding RNAs as key regulators of glutaminolysis in cancer. Int J Mol Sci 21(8):2872
Pérez-Gómez C, Matés JM, Gómez-Fabre PM, del Castillo-Olivares A, Alonso FJ, Márquez J (2003) Genomic organization and transcriptional analysis of the human l-glutaminase gene. Biochem J 370:771–784
Peterse EFP, Niessen B, Addie RD et al (2018) Targeting glutaminolysis in chondrosarcoma in context of the IDH1/2 mutation. Br J Cancer 118(8):1074–1083
Pillai SR, Damaghi M, Marunaka Y, Spugnini EP, Fais S, Gillies RJ (2019) Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev 38(1–2):205–222
Rajeshkumar NV, Yabuuchi S, Pai SG et al (2017) Treatment of pancreatic cancer patient-derived xenograft panel with metabolic inhibitors reveals efficacy of phenformin. Clin Cancer Res 23(18):5639–5647
Ramirez-Peña E, Arnold J, Shivakumar V et al (2019) The epithelial to mesenchymal transition promotes glutamine independence by suppressing GLS2 expression. Cancers (Basel) 11(10):E1610
Rashida Gnanaprakasam JN, Wu R, Wang R (2018) Metabolic reprogramming in modulating T cell reactive oxygen species generation and antioxidant capacity. Front Immunol 9:1075
Ren L, Ruiz-Rodado V, Dowdy T (2020) Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab 8:4
Romero R, Sayin VI, Davidson SM et al (2017) Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23(11):1362–1368
Saha SK, Islam SMR, Abdullah-Al-Wadud M, Islam S, Ali F, Park KS (2019) Multiomics analysis reveals that GLS and GLS2 differentially modulate the clinical outcomes of cancer. J Clin Med 8(3):355
Schoonjans CA, Joudiou N, Brusa D, Corbet C, Feron O, Gallez B (2020) Acidosis-induced metabolic reprogramming in tumor cells enhances the anti-proliferative activity of the PDK inhibitor dichloroacetate. Cancer Lett 470:18–28
Schulze A, Harris AL (2012) How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491:364–373
Schulte ML, Fu A, Zhao P et al (2018) Pharmacological blockade of ASCT2-dependent glutamine transport leads to anti-tumor efficacy in preclinical models. Nat Med 24(2):194–202
Scialò F, Fernández-Ayala DJ, Sanz A (2017) Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol 8:428
Sengupta D, Cassel T, Teng KY et al (2020) Regulation of hepatic glutamine metabolism by miR-122. Mol Metab 34:174–186
Son J, Lyssiotis CA, Ying H et al (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443):101–105
Sourbier C, Ricketts CJ, Liao PJ et al (2019) Proteasome inhibition disrupts the metabolism of fumarate hydratase- deficient tumors by downregulating p62 and c-Myc. Sci Rep 9(1):18409
Sullivan LB, Martinez-Garcia E, Nguyen H et al (2013) The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell 51(2):236–248
Suzuki S, Tanaka T, Poyurovsky MV et al (2010) Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA 107(16):7461–7466
Szeliga M, Bogacińska-Karaś M, Kuźmicz K, Rola R, Albrecht J (2016) Downregulation of GLS2 in glioblastoma cells is related to DNA hypermethylation but not to the p53 status. Mol Carcinog 55(9):1309–1316
Teng R, Liu Z, Tang H et al (2019) HSP60 silencing promotes Warburg-like phenotypes and switches the mitochondrial function from ATP production to biosynthesis in ccRCC cells. Redox Biol 24:101218
Torresano L, Nuevo-Tapioles C, Santacatterina F, Cuezva JM (2020) Metabolic reprogramming and disease progression in cancer patients. Biochim Biophys Acta Mol Basis Dis 1866(5):165721
Udupa S, Nguyen S, Hoang G et al (2019) Upregulation of the glutaminase II pathway contributes to glutamate production upon glutaminase 1 inhibition in pancreatic cancer. Proteomics 19(21–22):1800451
Vaziri-Gohar A, Zarei M, Brody JR, Winter JM (2018) Metabolic dependencies in pancreatic cancer. Front Oncol 8:617
Venneti S, Dunphy MP, Zhang H et al (2015) Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med 7(274):274ra17
Wang JB, Erickson JW, Fuji R et al (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18:207–219
Wang D, Meng G, Zheng M et al (2016) The glutaminase-1 inhibitor 968 enhances dihydroartemisinin-mediated antitumor efficacy in hepatocellular carcinoma cells. PLoS ONE 11(11):e0166423
Wang Q, Hardie RA, Hoy AJ et al (2015) Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol 236(3):278–289
Weinberg F, Hamanaka R, Wheaton WW et al (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci 107:8788–8793
Wu C, Chen L, Jin S, Li H (2018) Glutaminase inhibitors: a patent review. Expert Opin Ther Pat 28(11):823–835
Wu WC, Sun HW, Chen J et al (2019) Immunosuppressive immature myeloid cell generation is controlled by glutamine metabolism in human cancer. Cancer Immunol Res 7(10):1605–1618
Xia HL, Lv Y, Xu CW et al (2017) MiR-513c suppresses neuroblastoma cell migration, invasion, and proliferation through direct targeting glutaminase (GLS). Cancer Biomark 20:589–596
Xiang L, Xie G, Liu C et al (2013) Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim Biophys Acta 1833(12):2996–3005
Xiao D, Ren P, Su H et al (2015) Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 6(38):40655–40666
Ye J, Huang Q, Xu J et al (2018) Targeting of glutamine transporter ASCT2 and glutamine synthetase suppresses gastric cancer cell growth. J Cancer Res Clin Oncol 144(5):821–833
Yuan M, Kremer DM, Huang H et al (2019) Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC-MS/MS. Nat Protoc 14(2):313–330
Yuneva M (2008) Finding an “Achilles’ heel” of cancer: the role of glucose and glutamine metabolism in the survival of transformed cells. Cell Cycle 7(14):2083–2089
Yuneva MO, Fan TW, Allen TD (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15(2):157–170
Zhang Z, Gao Z, Rajthala S et al (2020) Metabolic reprogramming of normal oral fibroblasts correlated with increased glycolytic metabolism of oral squamous cell carcinoma and precedes their activation into carcinoma associated fibroblasts. Cell Mol Life Sci 77(6):1115–1133
Zhang ZZ, Lee EE, Sudderth J et al (2016a) Glutathione depletion, pentose phosphate pathway activation, and hemolysis in erythrocytes protecting cancer cells from vitamin C-induced oxidative stress. J Biol Chem 291(44):22861–22867
Zhang C, Liu J, Zhao Y et al (2016b) Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife 5:e10727
Zhang J, Wang C, Chen M et al (2013) Epigenetic silencing of glutaminase 2 in human liver and colon cancers. BMC Cancer 13:601
Zhdanov AV, Waters AH, Golubeva AV, Dmitriev RI, Papkovsky DB (2014) Availability of the key metabolic substrates dictates the respiratory response of cancer cells to the mitochondrial uncoupling. Biochim Biophys Acta 1837(1):51–62
Zhou R, Pantel AR, Li S et al (2017) [(18)F](2S,4R)4-Fluoroglutamine PET detects glutamine pool size changes in triple-negative breast cancer in response to glutaminase inhibition. Cancer Res 77:1476–1484
Zhu H, Liu F, Zhang Y et al (2019a) (2S,4R)-4-[(18)F]Fluoroglutamine as a PET indicator for bone marrow metabolism dysfunctional: from animal experiments to clinical application. Mol Imaging Biol 21(5):945–953
Zhu L, Tang F, Lei Z et al (2019b) Antiapoptotic properties of MALT1 protease are associated with redox homeostasis in ABC-DLBCL cells. Mol Carcinog 58(12):2340–2352
Acknowledgements
This work was financially supported by Ministerio de Ciencia y Tecnología of Spain, RTI2018-096866-B-I00. JDS-J is granted by FPU17/04084, Ministerio de Ciencia, Innovación y Universidades.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors, except for those published previously and approved by respective ethic committees.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Matés, J.M., Campos-Sandoval, J.A., de los Santos-Jiménez, J. et al. Glutaminases regulate glutathione and oxidative stress in cancer. Arch Toxicol 94, 2603–2623 (2020). https://doi.org/10.1007/s00204-020-02838-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-020-02838-8