
Abstract
Cancer cells display an altered metabolic circuitry that is directly regulated by oncogenic mutations and loss of tumor suppressors. Mounting evidence indicates that altered glutamine metabolism in cancer cells has critical roles in supporting macromolecule biosynthesis, regulating signaling pathways, and maintaining redox homeostasis, all of which contribute to cancer cell proliferation and survival. Thus, intervention in these metabolic processes could provide novel approaches to improve cancer treatment. This review summarizes current findings on the role of glutaminolytic enzymes in human cancers and provides an update on the development of small molecule inhibitors to target glutaminolysis for cancer therapy.
Similar content being viewed by others


Introduction
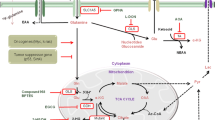
Cancer cells display enhanced and unusual metabolic activities compared with normal differentiated cells, as they reprogram their metabolic machinery in order to satisfy their bioenergetic and biosynthetic requirements.1 One of these metabolic abnormalities is that cancer cells take up glucose at higher rates than normal tissue, yet use less glucose for oxidative phosphorylation (OXPHOS) and favor the incomplete oxidation of glucose through the glycolytic pathway even in the presence of oxygen (aerobic glycolysis or Warburg effect).2, 3 Pyruvate generated from the glycolytic pathway is converted to lactate, rather than being used in the tricarboxylic acid (TCA) cycle. Although the requirement for mitochondrial ATP production is decreased in tumor cells, the demand for biosynthetic precursors and NADPH is increased.4 To compensate for these changes and to maintain a functional TCA cycle, cancer cells often rely on elevated glutaminolysis.5, 6, 7, 8 Glutamine, transported into the cells through transporters such as SLC1A5 and SLC7A5, is converted to glutamate and further to alpha-ketoglutarate (α-KG) by glutaminase (GLS), GDH and other enzymes to enable ATP production through the TCA cycle and to provide nitrogen, sulfur and carbon skeletons for producing necessary biosynthetic precursors for growing and proliferating cancer cells (Figure 1).5, 6, 7, 8

Glutaminolysis in cancer cells. Glutamine is transported through transporters (i.e., SLC1A5 and SLC7A5) to enter the glutaminolysis pathway. Enzymes in the glutaminolysis pathway suggested to be potential anticancer targets are shown in blue and inhibitors of these enzymes are listed in red. Dotted arrows show transcription factors involved in the regulation of relevant enzymes. α-KG, α-ketoglutarate; ASCT2, ASC amino acid transporter 2; AOA, aminooxyacetate; BCH, 2- aminobicyclo(2,2,1)-heptane-2-carboxylic acid; BPTES, bis-2-[5-phenylacetamido-1, 2, 4-thiadiazol-2-yl] ethyl sulfide; DON, 6-diazo-5-oxo-l-norleucine; EGCG, epigallocatechin gallate; GDH1, glutamate dehydrogenase 1; GLS, glutaminase; GOT2, glutamate oxaloacetate transaminase 2; GPNA, γ-l-glutamyl-p-nitroanilide; GPT2, glutamate pyruvate transaminase 2; LAT1, L-type amino acid transporter 1.
Glutaminolysis, which catabolizes glutamine to generate ATP and lactate, is a metabolic pathway that involves the initial deamination of glutamine by GLS, yielding glutamate and ammonia. Glutamate is then converted to α-KG, a TCA cycle intermediate, to produce both ATP and anabolic carbons for the synthesis of amino acids, nucleotides and lipids (Figure 1).5 The conversion of glutamate to α-KG is catalyzed by either glutamate dehydrogenase (GDH) or transaminases, such as glutamate pyruvate transaminases (GPTs, that is, alanine aminotransferases) and glutamate oxaloacetate transaminases (GOTs, that is, aspartate aminotransferase), which convert α-keto acids into their corresponding amino acids (Figure 1). Glutamine not only provides a major substrate for respiration but also for the synthesis of other macromolecules, such as nucleotides, proteins and hexosamines.9, 10 In addition to its critical role in providing carbon and nitrogen for macromolecule biosynthesis, glutaminolysis also has an important role in regulating redox balance, mTOR signaling, apoptosis and autophagy.11, 12, 13, 14, 15
For decades, researchers have shown that glutamine metabolism is also critical for cancer cell growth and survival by supporting macromolecular biosynthesis and maintaining bioenergetics and redox balance.16, 17, 18, 19, 20, 21 High extracellular glutamine concentrations stimulate tumor growth and are essential for cell transformation.22, 23, 24 Glutamine utilization is higher in tumor cells as well as rapidly diving cells including lymphocytes.25 Moreover, the flux of mitochondrial enzymes involved in glutamine/glutamate oxidation is elevated in tumor cells compared with normal cells.21, 26 Glutaminolysis contributes to tumor growth in two distinct but connected ways, by promoting cell proliferation and inhibiting cell death. The major function of glutaminolysis is to supply intermediary metabolites in the TCA cycle for cell growth. For example, glutaminolysis produces α-KG and replenishes the TCA cycle, which not only provides intermediates for other biosynthetic pathways but also supports energy production.7 Glutamine is required for nucleotide biosynthesis through donating its nitrogen to purines and pyrimidines. In addition, glutamine contributes to the biosynthesis of hexosamine and certain nonessential amino acids.20 Thus, glutamine is indispensable for cell proliferation by providing nitrogen and carbon skeletons for macromolecule biosynthesis.23 In addition to these well-recognized roles, glutamine contributes to sustaining cell proliferation. Recent studies have uncovered several novel functions that account for its roles in regulating cell proliferative events. For instance, cancer cells under hypoxia or with defective mitochondria can use glutamine-derived α-KG to produce citrate through reductive carboxylation, which is critical for lipid synthesis in these cells.27, 28, 29 Glutamine flux is reported to regulate mTOR activation to coordinate cell growth and proliferation.30 Furthermore, glutaminolysis mediates lysosomal translocation and subsequent activation of mTORC1.11 Collectively, these findings expand the role of glutaminolysis in metabolic rewiring to support cancer cell proliferation and tumor growth.
Equally important, glutaminolysis is also involved in many metabolic processes and signaling pathways that inhibit cell death. Cancer cells constantly encounter a variety of stress signals in vivo such as reactive oxygen species (ROS) and nutrient limitations. Consequently, cancer cells must rewire their metabolic pathways under these environmental conditions. For example, mounting evidence suggests that cancer cells show elevated levels of ROS compared with normal cells. A moderate increase in ROS promotes cell proliferation and differentiation, whereas excessive amounts of ROS inevitably cause oxidative damage to proteins, lipid and nucleotides.31, 32 Glutaminolysis is directly involved in maintaining ROS homeostasis, which is crucial for cell growth and survival. Glutamine supports the production of glutathione (GSH) and nicotinamide adenine dinucleotide phosphate (NADPH), which are two major reducing powers in the cell.33, 34 In addition, glutamine-derived fumarate has been shown to be important for the control of oxidative stress through several distinct mechanisms including upregulating the activity of ROS scavenging enzyme glutathione peroxidase 1 (GPx1) and activating Nrf2 antioxidant signaling.12, 35, 36 The diverse contributions of glutaminolysis to redox homeostasis highlight the complexity of metabolic rewiring in response to environmental cues.
In this review article, we highlight recent advances in our understanding of glutaminolytic enzymes in regulating cancer cell metabolism, cell signaling, and redox homeostasis. Furthermore, we will discuss their potential therapeutic application for treating human cancers.
Glutamine transporter
Mammalian cells are able to synthesize glutamine through glutamine synthetase (GLUL), which renders exogenous glutamine dispensable. However, GLUL is not highly expressed in all tissues.23 Moreover, glutamine becomes conditionally essential when the demand for glutamine surpasses the supply, especially for rapidly proliferating cells such as cancer cells.37 Glutamine uptake is mediated through membrane-anchored amino acid transporters.38 Mammalian cells express more than two-dozen amino acid transporters, the expression of which is tightly controlled in a tissue-specific manner.38 Many of the transporters are overexpressed in cancer cells and it has been reported that certain oncogenes or tumor suppressors regulate their expression. For example, SLC1A5 (ASCT2), a Na+-coupled transporter for glutamine, alanine, serine and cysteine, is upregulated by Myc33 and downregulated by retinoblastoma protein (Rb).39 Also, SLC7A5 (LAT1), a bidirectional transporter that regulates simultaneous efflux of glutamine out of cells and influx of leucine into cells, is upregulated by HIF-2α and Myc, and is highly expressed in renal cell carcinoma and prostate cancer.33, 40 These observations suggest the existence of a functional link between oncogenes and glutamine uptake that promotes glutaminolysis and tumor growth. Indeed, Nicklin et al.30 reported that SLC1A5 is coupled with SLC7A5 to activate mTOR signaling. Glutamine is transported into the cells through SLC1A5 and subsequently the efflux of glutamine out of the cells via SLC7A5 is coupled to the entry of leucine, which activates mTOR signaling and coordinates cell proliferation and growth.30 Several studies further demonstrate that glutamine transporters, such as SLC1A5 and SLC7A5, are required for the growth of a variety of tumors including acute myeloid leukemia (AML),41 lung cancer,42, 43 kidney cancer40 and melanoma.44 In addition, targeting SLC1A5 by either RNAi or small molecule inhibitors, benzylserine and l-γ-glutamyl-p-nitroanilide, suppresses cancer cell proliferation in vitro and tumor growth in vivo.42, 43 More importantly, the expression of glutamine transporters inversely correlates with the prognosis of cancer patients, indicating the potential of glutamine transporters as a prognostic biomarker for cancer treatment.
Glutaminase
Glutaminolysis starts with the conversion of glutamine to glutamate, which is catalyzed by the GLSs in mitochondria.45 There are two forms of GLS in humans: kidney-type glutaminase (GLS, KGA or GAC) encoded by GLS and liver-type glutaminase (GLS2, LGA or GAB) encoded by GLS2. GLS is expressed ubiquitously, whereas GLS2 is expressed primarily in the liver.46 Previous studies have shown that GLS is expressed in a wide variety of tumors and its upregulation correlates with tumor growth.46 However, the regulation of GLS in cancer cells is not well understood. A few studies indicate a functional link between MYC, an oncogene, and glutamine metabolism. Yuneva et al.47 reported that depletion of glutamine induces apoptosis that depends on activity of ectopic MYC. Wise et al.48 and Gao et al.33 reported that Myc induces a transcriptional program that promotes glutaminolysis in cancer cells. Further metabolomic studies confirm that glutaminolysis is profoundly affected by induction of Myc.49, 50 Moreover, it has been shown that the mTOR pathway upregulates GLS expression through enhancing translation of c-Myc protein.51 mTOR signaling stimulates glutaminolysis by not only upregulating GLS but also enhancing the activity of another glutaminolytic enzyme, glutamate dehydrogenase 1 (GDH1),52 which indicates the complexity of the crosstalk between glutaminolysis and mTOR signaling.
In addition to its transcriptional regulation by c-Myc, GLS can be regulated through the modulation of its enzymatic activity or protein stability.53, 54, 55 NF-κB53 or Raf-MEK-ERK signaling54 regulates GLS activity without affecting its protein expression. Colombo et al.55 reported that GLS is targeted for destruction by APC/C-Cdh1 in a cell cycle-dependent fashion. Moncada et al. reported that APC/C (anaphase-promoting complex/cyclosome)-Cdh1, the ubiquitin ligase that controls the G1 to S phase transition, regulates GLS1 and the glycolytic enzyme PFKFB3. Decreased APC/C-Cdh1 in G1 phase stabilizes both GLS1 and PFKFB3, providing a proliferative advantage to cancer cells.56 Collectively, these findings suggest that the activity of GLS is regulated by diverse oncogenes and survival signals in cancer cells.
GLS can be inhibited using RNAi or small molecule inhibitors such as bis-2-[5-phenylacetamido-1, 2, 4-thiadiazol-2-yl] ethyl sulfide (BPTES),57 CB-83958 and compound 968.53 Inhibition of GLS has been shown to significantly suppress tumor growth in several experimental models including breast cancer and lymphoma.33, 53 Reduction of GLS expression by RNAi decreases ATP level and increases ROS level. Both the TCA cycle metabolite oxaloacetate (OAA) and ROS scavenger N-acetylcysteine (NAC) partially rescue the decreased proliferation of cancer cells induced by GLS silencing.33 This finding emphasizes the significance of glutaminolysis in replenishing TCA cycle intermediates and maintaining redox balance in cancer cells, further suggesting that targeting GLS could be a novel therapeutic strategy for the treatment of human cancers. In certain glioblastoma cells, however, silencing GLS inhibits cell proliferation but fails to eliminate the cancer cells. A detailed study revealed that a compensatory anaplerotic mechanism catalyzed by pyruvate carboxylase is induced to enable the cells to bypass the need for glutamine.59 Glucose-derived pyruvate provided by pyruvate carboxylase is necessary when GLS is silenced, indicating that targeting both GLS and pyruvate carboxylase could produce synthetic lethality. In addition, two recent studies have identified GLS as a critical player in mediating resistance to therapies in glioblastoma and T-cell acute lymphoblastic leukemia. Tanaka et al.60 reported that GLS expression and glutamate levels are elevated following mTOR kinase inhibitor treatment in glioblastoma cells and targeting both mTOR kinase and GLS results in synergistic tumor cell death. In T-cell acute lymphoblastic leukemia, Herranz et al. identified glutaminolysis as a crucial pathway for cell growth downstream of NOTCH1. Knockout of GLS led to significant anti-leukemic effects, which is further enhanced by anti-NOTCH1 therapy in NOTCH1-induced T-ALL.61
Unlike GLS, the exact role of GLS2 in cancer is still controversial and not completely understood. Early reports have shown that GLS2 is a p53 target gene that is important for the control of cellular ROS level through regulating GSH production.62, 63 Overexpression of GLS2 in cancer cells inhibits tumor growth in vivo, which is consistent with a tumor-suppressing function. In contrast, later studies demonstrated that GLS2 is overexpressed in certain type of cancers and that silencing GLS2 inhibits tumor growth.64, 65 Nevertheless, it is well accepted that like GLS, GLS2 also has an important role in maintaining cellular redox homeostasis. Cancer cells must maintain redox homeostasis to ensure survival in vivo by regulating endogenous and exogenous oxidative stress. Inhibiting GLS2 can disrupt the redox balance in cancer cells and sensitize them to oxidative stress.66 Moreover, a recent study revealed that glutaminolysis has a crucial role in ferroptosis, an iron-dependent, non-apoptotic cell death.67 Genetic and pharmacological inhibition studies demonstrated that GLS2 but not GLS is required for ferroptosis. The function and regulation of GLS in cancer is likely context dependent and warrants further investigation.
Glutamate dehydrogenase
The initial deamination of glutamine by GLS yields glutamate and ammonia. Glutamate is then converted to α-KG, a TCA cycle intermediate, to produce ATP and anabolic carbons for the synthesis of amino acids, nucleotides, and lipids. Several enzymes are involved in the conversion of glutamate to α-KG in the mitochondria. These include GDH1, GOT2 and GPT2. Jin et al.12 recently reported that RNAi-mediated knockdown of GDH1, but not GPT2 or GOT2, results in significantly decreased α-KG production accompanied by reduced glutaminolysis, decreased anabolic glutamine-dependent RNA biosynthesis and elevated ROS levels in breast cancer and lung cancer cells. Previous studies support that the activation of GDH tightly correlates with increased glutaminolysis, further supporting GDH as a critical regulator of glutamine metabolism.68 Conversion of glutamate to α-KG by GDH is carried out via oxidative deamination. There are two isozymes of GDH: GDH1 is expressed ubiquitously whereas GDH2 has only been found in the retina, testis and brain.69 The activity of mammalian GDH, one of the most allosterically regulated enzymes, is subject to complex allosteric regulation that depends on the metabolic status of the cell.70 It has been shown that palmitoyl CoA, GTP and ATP can inhibit GDH activity, whereas ADP and leucine serve as allosteric activators.71 In addition to allosteric regulation, GDH is also regulated through a post-translational modification. Previous reports demonstrate that SIRT4, a mitochondrial enzyme, utilizes NAD to ADP-ribosylate and downregulate GDH activity in pancreatic β-cells.72 Loss of SIRT4 activates GDH and therefore promotes amino acid-stimulated insulin secretion.72 The complexity of regulation of GDH in cells places this enzyme in a unique metabolic regulatory step.
The importance of GDH regulation is underscored by the finding that dysregulation of GDH caused by a dominant mutation leads to hyperinsulinism/hyperammonemia (HI/HA).73 The function of GDH in cancer has been elusive until recently. Yang et al. reported that GDH1 is required for glioblastoma cells to maintain survival under limited glucose supply. Glucose deprivation activates GDH activity and inhibition of GDH1 sensitizes glioblastoma cells to glucose withdrawal.68 Similarly, Choo et al. also reported that inhibition of GDH1 leads to cell death in TSC−/− cells under glucose deprivation, while suppression of GOT and GPT shows no effect. Treatment with the mTOR inhibitor rapamycin prevents glucose deprivation-induced cell death but fails to do so when GDH1 activity was blocked.74 These studies suggest a pro-survival role of GDH1 in cancer cells under nutrient stress such as low glucose.
Furthermore, recent reports have shown that GDH is critically involved in the regulation of redox homeostasis in many types of cancer.12, 75 Jin et al. showed that GDH1 expression is upregulated in breast and lung cancers. Silencing GDH1 in human cancer cells led to elevated ROS level and decreased cell proliferation both in vitro and in vivo, indicating that cellular redox homeostasis is disrupted by inhibition of GDH1.12 Fumarate, the subsequent metabolite of α-KG, directly binds and activates GPx1, a ROS scavenging enzyme, in cancer cells. Inhibition of GDH1 by shRNA or R162, a GDH1-specific inhibitor, decreases fumarate levels, which leads to decreased GPx activity and reduced ROS scavenging capabilities in cancer cells.12 These findings position GDH1 at a critical node of cellular ROS regulation and further highlight the functional link between glutaminolysis and redox homeostasis.
The involvement of GDH2 in cancer metabolism and growth has also been suggested. A recent report showed that GDH2 is important for supporting the growth of tumors driven by isocitrate dehydrogenase 1 (IDH1) mutations.76 The authors found that somatic mutation of IDH1, the most common initiating event for secondary glioblastoma, exerts growth inhibitory effects that can be abrogated by GDH2 but not GDH1 overexpression.76 These findings suggest that targeting GDH2 may selectively benefit cancer patients with IDH1 mutations.
Aspartate transaminase
In addition to GDH, the conversion of glutamate to α-KG can also be carried out through a reversible transamination process, producing α-KG as well as nonessential amino acids such as alanine and aspartate.19 In a recent report, aspartate transaminases (GOTs) were identified as critical metabolic enzymes in human pancreatic ductal adenocarcinoma (PDAC) for maintaining redox homeostasis. Genetic inhibition of GOT1 and GOT2 led to elevated ROS levels and suppression of tumor growth in vivo.34 In another study, Yang et al. found that GOT2 activity is regulated through acetylation. Acetylation of GOT2 supports ATP production and stimulates production of NADPH to suppress ROS generation.77 Collectively, these studies have identified GOTs as an important player in redox regulation in human pancreatic cancer.
Therapeutic opportunities
The interest in finding small molecules that target glutamine metabolism started as early as the 1950s, when several glutamine analogs including acivicin, 6-diazo-5-oxo-l-norleucine (L-DON) and azaserine were tested for their anti-tumor activity in both preclinical and clinical studies.78 All three analogs showed certain degrees of anti-proliferation effects in vitro as well as in xenograft animal models. These analogs non-specifically target several glutaminolytic enzymes such as GLS and therefore inhibit glutamine-dependent nucleotide biosynthesis. Although glutamine analogs introduced the possibility of therapeutically exploiting glutamine addiction for cancer treatment, dose-limiting toxicities including neurotoxicity, gastrointestinal toxicity and myelosuppression prevent the further development of these glutamine analogs in the clinic.78
As a greater appreciation of the role of glutamine in cancer metabolism has emerged in recent years, new efforts have been made to characterize compounds that inhibit specific steps of glutaminolysis, especially those with oncogene-associated glutamine dependency (Table 1).79, 80, 81 For example, SLC1A5 has been identified as the primary glutamine transporter in certain types of cancers and is indispensable for cancer cell proliferation. Therefore, specific inhibition of the transporter to block glutamine uptake could be a promising therapeutic approach for cancer treatment. Indeed, benzylserine and l-γ-glutamyl-p-nitroanilide have been shown to effectively inhibit SLC1A5 and suppress tumor growth in lung cancer and melanoma.43, 44 In addition, it has been shown that 2-aminobicyclo(2,2,1)-heptane-2-carboxylic acid (BCH), an inhibitor of SLC7A5, inhibits mTOR signaling.30
GLS is perhaps so far the most extensively studied drug target in the glutaminolysis pathway because of its critical role in glutamine metabolism. Therefore, characterization of new specific inhibitors for GLS has recently become a field of intense research and a variety of small molecule inhibitors have been reported.57, 82 The best characterized GLS inhibitor is bis-2-[5-phenylacetamido-1, 2, 4-thiadiazol-2-yl] ethyl sulfide (BPTES). BPTES allosterically inhibits the dimer-to-tetramer transition of GLS,54, 57 which is critical for activation of the enzyme. Crystal structure analysis of GLS-BPTES showed that BPTES binds to the dimer interface of GLS and consequently stabilizes the inactive tetrameric form of the enzyme.54 In addition, a number of derivatives of BPTES have been developed,83 such as CB-839 which is currently in Phase I clinical trials.58, 81 BPTES effectively inhibits the growth of several types of tumors, including Myc-dependent hepatocellular carcinoma,46 renal cell carcinomas84 and lymphoma.49 Consistent with the critical role of glutaminolysis in cellular energetic and redox homeostasis, inhibition of GLS by BPTES decreases ATP levels and profoundly increases ROS levels.49 In addition, the combination of BPTES and heat shock protein 90 inhibitors has been shown to selectively induce cell death in mTORC1-driven tumor cells.85 Interestingly, the expression of pyruvate carboxylase negatively correlates with BPTES sensitivity in lung cancer cell lines, indicating that the sensitivity to GLS inhibition could be affected by the mode of carbon entry into the TCA.86 Compound 968 (benzophenanthridinone 968) represents another class of GLS inhibitor that has drawn great attention recently.53, 87 Unlike BPTES, 968 is less effective at inhibiting GLS enzyme activity when added to GLS activated by pretreatment with inorganic phosphate, an allosteric activator.88 Indeed, further study showed that unlike BPTES, 968 does not stabilize an inactive tetrameric state of GLS but rather preferentially binds to an inactive, monomeric state of GLS and inhibits the active conformational changes of the enzyme.88 968 treatment suppressed oncogenic transformation induced by Rho GTPases in fibroblast and breast cancer and showed synergistic anti-tumor activity in combination with mTOR inhibition in glioblastoma.53, 60 Since BPTES and 968 regulate GLS activity through distinct allosteric mechanisms, these compounds could be further examined for combination treatment for enhanced GLS inhibition and tumor suppression.
The possibility of targeting the step that converts glutamate to α-KG has also been explored and several inhibitors have been described. It has been shown that epigallocatechin gallate, a flavonoid from green tea, allosterically inhibits GDH and effectively suppresses tumor growth in many types of cancers that rely on glutamine.71 However, given the fact that epigallocatechin gallate shows numerous pharmacological activities and potentially has multiple targets in the cell, it is unclear how much the inhibition of GDH by epigallocatechin gallate accounts for its anti-tumor activity in vivo. Jin et al.12 recently identified purpurin and its cell permeable derivative R162 as novel GDH inhibitors. Purpurin directly binds and specifically inhibits GDH1 activity in vitro. More importantly, purpurin shows no effect on the activity of other NADPH-dependent enzymes such as 6-phosphogluconate dehydrogenase and fumarate hydratase, whereas epigallocatechin gallate dramatically affects the activity of both enzymes. Inhibition of GDH by R162 in cancer cells induces elevated ROS level and inhibits tumor growth in vivo, further supporting a critical role of glutaminolysis in maintaining redox balance in cancer cells and indicating GDH1 as a promising anticancer target.12
In addition to GDH, glutamate-dependent transaminases have also been considered as drug targets for modulating glutaminolysis. Aminooxyacetate, which non-specifically inhibits transaminases, has been shown to be effective in inhibiting cell proliferation and tumor growth in several preclinical studies.89, 90
Concluding remarks
Glutamine metabolism in cancer has drawn a substantial amount of interest over the past decade. It has become increasingly clear that glutaminolysis is involved in diverse aspects of cellular functions in cancer cells, many of which are beyond the metabolic role of glutamine. Given the complex interplay between oncogenic signaling and metabolic rewiring, additional metabolic and non-metabolic functions of glutaminolysis in cancer cells will be uncovered. Elucidating how the cancer cells coordinate these functions and reprogram their metabolic pathways in response to environmental stress is of great importance, as these new findings could provide opportunities for therapeutic intervention. Challenges, however, still remain. Because other fast-proliferating cells, such as immune cells, often share similar metabolic pathways with cancer cells,91 more specific pharmacological interventions that target the particular vulnerabilities of cancer cells will be the future focus of the field.
References
Hsu PP, Sabatini DM . Cancer cell metabolism: Warburg and beyond. Cell 2008; 134: 703–707.
Warburg O . On the origin of cancer cells. Science 1956; 123: 309–314.
Kim JW, Dang CV . Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006; 66: 8927–8930.
Frezza C, Gottlieb E . Mitochondria in cancer: not just innocent bystanders. Semin Cancer Biol 2009; 19: 4–11.
Wise DR, Thompson CB . Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010; 35: 427–433.
Medina MA . Glutamine and cancer. J Nutr 2001; 131: 2539S–2542S.
Reitzer LJ, Wice BM, Kennell D . Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 1979; 254: 2669–2676.
Lu W, Pelicano H, Huang P . Cancer metabolism: is glutamine sweeter than glucose? Cancer Cell 2010; 18: 199–200.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 2007; 104: 19345–19350.
Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 2010; 24: 2784–2799.
Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 2012; 47: 349–358.
Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015; 27: 257–270.
Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell 2014; 56: 205–218.
Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012; 22: 631–644.
Eng CH, Yu K, Lucas J, White E, Abraham RT . Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal 2010. 3ra31.
Medina MA, Sanchez-Jimenez F, Marquez J, Rodriguez Quesada A, Nunez de Castro I . Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem 1992; 113: 1–15.
Kovacevic Z, McGivan JD . Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev 1983; 63: 547–605.
Souba WW . Glutamine and cancer. Ann Surg 1993; 218: 715–728.
DeBerardinis RJ, Cheng T . Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010; 29: 313–324.
Hensley CT, Wasti AT, DeBerardinis RJ . Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 2013; 123: 3678–3684.
Moreadith RW, Lehninger AL . The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J Biol Chem 1984; 259: 6215–6221.
Medina MA, Nunez de Castro I . Glutaminolysis and glycolysis interactions in proliferant cells. Int J Biochem 1990; 22: 681–683.
Dang CV . Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle 2010; 9: 3884–3886.
McKeehan WL . Glycolysis, glutaminolysis and cell proliferation. Cell Biol Int Rep 1982; 6: 635–650.
Newsholme EA, Crabtree B, Ardawi MS . Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q J Exp Physiol 1985; 70: 473–489.
Friday E, Oliver R 3rd, Welbourne T, Turturro F . Glutaminolysis and glycolysis regulation by troglitazone in breast cancer cells: relationship to mitochondrial membrane potential. J Cell Physiol 2011; 226: 511–519.
Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep 2014; 7: 1679–1690.
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012; 481: 380–384.
Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012; 481: 385–388.
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009; 136: 521–534.
Gorrini C, Harris IS, Mak TW . Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013; 12: 931–947.
Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015; 27: 211–222.
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458: 762–765.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013; 496: 101–105.
Adam J, Hatipoglu E, O'Flaherty L, Ternette N, Sahgal N, Lockstone H et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011; 20: 524–537.
Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011; 20: 511–523.
Lacey JM, Wilmore DW . Is glutamine a conditionally essential amino acid? Nutr Rev 1990; 48: 297–309.
Bhutia YD, Babu E, Ramachandran S, Ganapathy V . Amino acid transporters in cancer and their relevance to ‘glutamine addiction’: novel targets for the design of a new class of anticancer drugs. Cancer Res 2015; 75: 1782–1788.
Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014; 33: 556–566.
Elorza A, Soro-Arnaiz I, Melendez-Rodriguez F, Rodriguez-Vaello V, Marsboom G, de Carcer G et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell 2012; 48: 681–691.
Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013; 122: 3521–3532.
Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res 2013; 19: 560–570.
Hassanein M, Qian J, Hoeksema MD, Wang J, Jacobovitz M, Ji X et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int J Cancer 2015; 137: 1587–1597.
Wang Q, Beaumont KA, Otte NJ, Font J, Bailey CG, van Geldermalsen M et al. Targeting glutamine transport to suppress melanoma cell growth. Int J Cancer 2014; 135: 1060–1071.
Mates JM, Segura JA, Martin-Rufian M, Campos-Sandoval JA, Alonso FJ, Marquez J . Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med 2013; 13: 514–534.
Xiang Y, Stine ZE, Xia J, Lu Y, O'Connor RS, Altman BJ et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest 2015; 125: 2293–2306.
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y . Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 2007; 178: 93–105.
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 2008; 105: 18782–18787.
Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012; 15: 110–121.
Murphy TA, Dang CV, Young JD . Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab Eng 2013; 15: 206–217.
Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol 2014; 24: 2274–2280.
Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013; 153: 840–854.
Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010; 18: 207–219.
Thangavelu K, Pan CQ, Karlberg T, Balaji G, Uttamchandani M, Suresh V et al. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci USA 2012; 109: 7705–7710.
Colombo SL, Palacios-Callender M, Frakich N, Carcamo S, Kovacs I, Tudzarova S et al. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc Natl Acad Sci USA 2011; 108: 21069–21074.
Moncada S, Higgs EA, Colombo SL . Fulfilling the metabolic requirements for cell proliferation. Biochem J 2012; 446: 1–7.
Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J 2007; 406: 407–414.
Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 2014; 13: 890–901.
Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA 2011; 108: 8674–8679.
Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest 2015; 125: 1591–1602.
Herranz D, Ambesi-Impiombato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med 2015; 21: 1182–1189.
Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z . Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA 2010; 107: 7455–7460.
Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA 2010; 107: 7461–7466.
Martin-Rufian M, Nascimento-Gomes R, Higuero A, Crisma AR, Campos-Sandoval JA, Gomez-Garcia MC et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J Mol Med (Berl) 2014; 92: 277–290.
Xiang L, Xie G, Liu C, Zhou J, Chen J, Yu S et al. Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim Biophys Acta 2013; 1833: 2996–3005.
Giacobbe A, Bongiorno-Borbone L, Bernassola F, Terrinoni A, Markert EK, Levine AJ et al. p63 regulates glutaminase 2 expression. Cell Cycle 2013; 12: 1395–1405.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X . Glutaminolysis and Transferrin regulate Ferroptosis. Mol Cell 2015; 59: 298–308.
Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ . Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 2009; 69: 7986–7993.
Plaitakis A, Latsoudis H, Spanaki C . The human GLUD2 glutamate dehydrogenase and its regulation in health and disease. Neurochem Int 2011; 59: 495–509.
Smith TJ, Stanley CA . Untangling the glutamate dehydrogenase allosteric nightmare. Trends Biochem Sci 2008; 33: 557–564.
Li C, Allen A, Kwagh J, Doliba NM, Qin W, Najafi H et al. Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. J Biol Chem 2006; 281: 10214–10221.
Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006; 126: 941–954.
Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 1998; 338: 1352–1357.
Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO et al. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell 2010; 38: 487–499.
Lorin S, Tol MJ, Bauvy C, Strijland A, Pous C, Verhoeven AJ et al. Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy 2013; 9: 850–860.
Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc Natl Acad Sci USA 2014; 111: 14217–14222.
Yang H, Zhou L, Shi Q, Zhao Y, Lin H, Zhang M et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J 2015; 34: 1110–1125.
Ahluwalia GS, Grem JL, Hao Z, Cooney DA . Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther 1990; 46: 243–271.
Elhammali A, Ippolito JE, Collins L, Crowley J, Marasa J, Piwnica-Worms D . A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies Zaprinast as a glutaminase inhibitor. Cancer Discov 2014; 4: 828–839.
Schulze A, Harris AL . How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012; 491: 364–373.
DeLaBarre B, Hurov J, Cianchetta G, Murray S, Dang L . Action at a distance: allostery and the development of drugs to target cancer cell metabolism. Chem Biol 2014; 21: 1143–1161.
Katt WP, Cerione RA . Glutaminase regulation in cancer cells: a druggable chain of events. Drug Discov Today 2014; 19: 450–457.
Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem 2012; 55: 10551–10563.
Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci USA 2015; 112: 6539–6544.
Li J, Csibi A, Yang S, Hoffman GR, Li C, Zhang E et al. Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1-driven tumor cells. Proc Natl Acad Sci USA 2015; 112: E21–E29.
Ulanet DB, Couto K, Jha A, Choe S, Wang A, Woo HK et al. Mesenchymal phenotype predisposes lung cancer cells to impaired proliferation and redox stress in response to glutaminase inhibition. PLoS One 2014; 9: e115144.
Katt WP, Ramachandran S, Erickson JW, Cerione RA . Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol Cancer Ther 2012; 11: 1269–1278.
Stalnecker CA, Ulrich SM, Li Y, Ramachandran S, McBrayer MK, DeBerardinis RJ et al. Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc Natl Acad Sci USA 2015; 112: 394–399.
Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res 2008; 10: R84.
Korangath P, Teo WW, Sadik H, Han L, Mori N, Huijts CM et al. Targeting glutamine metabolism in breast cancer with aminooxyacetate. Clin Cancer Res 2015; 21: 3263–3273.
Ghesquiere B, Wong BW, Kuchnio A, Carmeliet P . Metabolism of stromal and immune cells in health and disease. Nature 2014; 511: 167–176.
Acknowledgements
We acknowledge Dr Anthea Hammond for editorial assistance. We apologize to authors whose contributions were not directly cited in this review due to space limitations. This study is supported in part by ACS grant RSG-11-081-01 and NIH grants R01 CA175316 (S.K.) and F31 CA183365 (G.A.). S.K. is a Georgia Cancer Coalition Scholar, Robbins Scholar, and an American Cancer Society Basic Research Scholar.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Jin, L., Alesi, G. & Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 35, 3619–3625 (2016). https://doi.org/10.1038/onc.2015.447
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.447
- Springer Nature Limited
This article is cited by
-
The new insights into autophagy in thyroid cancer progression
Journal of Translational Medicine (2023)
-
TRPM2-mediated Ca2+ signaling as a potential therapeutic target in cancer treatment: an updated review of its role in survival and proliferation of cancer cells
Cell Communication and Signaling (2023)
-
Regulation of tumor metabolism by post translational modifications on metabolic enzymes
Cancer Gene Therapy (2023)
-
RBM4 dictates ESCC cell fate switch from cellular senescence to glutamine-addiction survival through inhibiting LKB1-AMPK-axis
Signal Transduction and Targeted Therapy (2023)
-
Early onset of urea synthesis and ammonia detoxification pathways in three terrestrially developing frogs
Journal of Comparative Physiology B (2023)