
Abstract
The aim of this study is to provide a systematic review of the known epigenetic alterations caused by cigarette smoke; establish an evidence-based perspective of their clinical value for screening, diagnosis, and treatment of smoke-related disorders; and discuss the challenges and ethical concerns associated with epigenetic studies. A well-defined, reproducible search strategy was employed to identify relevant literature (clinical, cellular, and animal-based) between 2000 and 2019 based on AMSTAR guidelines. A total of 80 studies were identified that reported alterations in DNA methylation, histone modifications, and miRNA expression following exposure to cigarette smoke. Changes in DNA methylation were most extensively documented for genes including AHRR, F2RL3, DAPK, and p16 after exposure to cigarette smoke. Likewise, miR16, miR21, miR146, and miR222 were identified to be differentially expressed in smokers and exhibit potential as biomarkers for determining susceptibility to COPD. We also identified 22 studies highlighting the transgenerational effects of maternal and paternal smoking on offspring. This systematic review lists the epigenetic events/alterations known to occur in response to cigarette smoke exposure and identifies the major genes and miRNAs that are potential targets for translational research in associated pathologies. Importantly, the limitations and ethical concerns related to epigenetic studies are also highlighted, as are the effects on the ability to address specific questions associated with exposure to tobacco/cigarette smoke. In the future, improved interpretation of epigenetic signatures will lead to their increased use as biomarkers and/or in drug development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cigarette smoking is the most prevalent preventable cause of death and disease in the world. According to a recent report by the CDC (Centres for Disease Control and Prevention), approximately 480,000 adults in the US die due to active or passive smoking every year (Jamal et al. 2018). While the effects of active smoking are well documented and include cardiovascular diseases, respiratory illnesses (pneumonia, influenza, bronchitis, emphysema, and chronic airway obstruction), and cancer (lungs, mouth, pharynx, larynx, esophagus, stomach, pancreas, uterine cervix, kidney, ureter, and bladder) (MacKenzie et al. 1994); smoking-related health problems and neurocognitive deficits are also widely associated with second-hand smoke exposure (Heffernan 2016; Lubick 2011). In fact, as of late special emphasis is being given to the effect of third-hand smoke exposure, which refers to the residual tobacco smoke pollutants that accumulate on surfaces and in dust after tobacco use in closed environments. It is believed that third-hand smoke could stay on surfaces for months and may yield secondary pollutants that are considered to be potential health hazards, especially to infants and children (Burton 2011; Protano and Vitali 2011).
The challenge with studying smoke-related diseases is that not all smokers develop smoke-related disorders, nor are all non-smokers safe from its effects (Saha et al. 2007; Terzikhan et al. 2016). Considering this, it is not far-fetched to consider that smoke-related changes in gene expression are regulated epigenetically. Epigenetics is the study of the covalent modifications of DNA, protein, or RNA without alteration of their primary sequences. These alterations function to regulate gene expression during development or in response to environmental stimulus. As the link between genes (nature) and environment (nurture), epigenetics is responsible for normal growth and development in mammals. During the last decade, epigenetics has emerged as an interdisciplinary field with enormous scientific and therapeutic potential. Epigenetic changes have been implicated in cancer, cardiovascular diseases, behavioral disorders, inflammation, aging, neurodegenerative diseases, and diabetes (Portela and Esteller 2010). Understanding the complexity of the epigenetic signatures associated with cigarette smoke exposure may provide information about key therapeutic targets.
The aim of this systematic review of literature is to: (a) conduct evidence-based analysis of the role of epigenetics in regulating cellular signaling in response to cigarette smoke exposure, and (b) determine the clinical value of targeting epigenetic markers for the screening, diagnosis, and treatment of smoke-related disease risks among humans. To achieve this, we performed a scoping search using PubMed and ScienceDirect to identify the available literature between 2000 and 2019 highlighting epigenetic alterations following smoke exposure in cellular, clinical, and animal-based models. We identified a total of 80 studies that met our inclusion/exclusion criteria, out of which 35 investigated alterations in DNA methylation; 11 identified changes in histone modifications and chromatin structure, and 12 compared the miRNA profile among smokers and non-smokers. It should be noted that there are no previously published systematic reviews that draw together all aspects of epigenetic changes following smoke exposure, as is the focus of the current review. Moreover, this review will also include discussion of the technical challenges and ethical concerns related to epigenetic studies in smoking-related research. A careful review and solution to these concerns are necessary to design improved research strategies to determine the epigenetic interplay in smoke-related inflammation/pathologies and identify better diagnostic biomarkers and therapeutic targets.
Methods
For this review, we cross checked our study results against the ‘assessment tool of multiple systematic reviews’ (AMSTAR) guidelines (Shea et al. 2007). The AMSTAR tool includes the following 11 items: (1) an a priori statement of research questions and inclusion/exclusion criteria, (2) duplicate literature searches by two or more co-authors, (3) use of at least two electronic search engines followed by a supplemental search of reviews, textbooks and secondary references with keywords and MESH statements reported in the “Methods” section, (4) specification of status of publication (e.g., grey literature, which we define as reports that are not published in peer-reviewed journals or scientific monographs) as an inclusion/exclusion criterion, (5) a list of studies excluded from the review, (6) a summary of study characteristics that meet the inclusion criteria, (7) a formal assessment of strength or limitations of individual studies, (8) consideration of strength of evidence in drawing conclusions, (9) pooling of study results in a quantitative meta-analysis accompanied by a test for heterogeneity, if possible, (10) assessment of publication bias, and (11) a statement of sources of support (Shea et al. 2007). Except for items 9 and 10, which are contingent on the feasibility of a formal meta-analysis, the AMSTAR checklist is applicable to this systematic review. The aim of this review was to identify and summarize the studies that examine the alterations in epigenetic markers (DNA methylation, histone modifications, and miRNA expression) in response to cigarette smoking.
Study selection
Electronic data sources PubMed and ScienceDirect were used to conduct the initial literature search. For both the sources, the following keywords were used as a search criterion: “epigenetics + smoking”, “smoking + DNA methylation”, “smoking + histone modifications”, “smoking + chromatin remodelling”, “smoking + miRNA”, “epigenetic + maternal smoking”, “epigenetic + paternal smoking”, and “transgenerational epigenetics + smoking”. Secondary references of retrieved articles were reviewed to identify publications not captured by the electronic search. The search and selection of relevant studies were conducted independently by three study authors (GK, RB, and ST) with all disagreements resolved by consensus.
Inclusion/exclusion criteria
To be included in this review, a study must report an epigenetic change caused due to smoke exposure in the form of alterations in: (i) DNA methylation, (ii) histone modifying marks or, (iii) miRNA profile. The EWAS studies included in this review generally comprise of clinical data; however, wherever needed, in vivo and in vitro data have also been referred to explain the mechanistic implications of observed epigenetic alterations. The study data included in this review only relate to pulmonary health outcomes (like asthma, COPD, emphysema, allergy, etc.), while other health impacts are excluded. However, for the purpose of pointing out the transgenerational effects of paternal smoking, references showing male infertility have been included.
Data extracted
The data from each study were tabulated, and the resulting summary tables were again cross checked with disagreements resolved by consensus. Information extracted from each study for the purposes of this review included the following:
-
1.
Type of study: clinical, cell-based or animal-based.
-
2.
Description of the sample: type, size, composition, and source.
-
3.
Smoke exposure categorization: current, former or never smokers for human samples or smoke exposure duration for in vitro and in vivo studies.
-
4.
Endpoints of interest (see inclusion criteria).
-
5.
Results for each study were mentioned in terms of the number of genes/CpG sites/miRNAs altered on smoking and its implications. When the result was reported in a qualitative fashion, the corresponding text was extracted from the original publication and reproduced verbatim.
Measure of strengths and weaknesses
Epigenome-wide studies were assessed based on the sample size, the method used for analysis of epigenetic alterations and the statistical method applied to deduce results. However, since no epigenome-wide data were available for the studies investigating alterations in the post-translational modifications at histones, we relied on the cell-based and animal-based studies in this area.
Results
A total of 1280 studies were retrieved after searching for the keywords in the two databases. After excluding the duplicates and screening the studies as per the inclusion/exclusion criteria, a total of 80 studies were included in this review (Fig. 1). Out of these, 35 studies investigated the DNA methylation alterations associated with smoking among human subjects; 11 studies identified the smoking-related changes in histone modifications and chromatin structure while 12 compared the miRNA profile among smokers and non-smokers (Table 1). Interestingly, cigarette smoking was found to have transgenerational effects on the offspring as well. To study this effect, the epigenetic changes associated with paternal and maternal smoking are listed separately. For the purpose of this review, we have included 7 and 15 studies associated with paternal and maternal smoking, respectively (Table 2). In the subsequent sections, the findings from each of these studies have been summarized in brief.

AMSTAR flow diagram with reasons of exclusion. AMSTAR assessment tool of multiple systematic reviews
Smoking-mediated alterations in DNA methylation
DNA methylation is the addition of a methyl group at the fifth carbon atom of cytosine (5mC) in the DNA strand (Fig. 2a) and is the most extensively studied epigenetic mechanism. Further, this modification plays a crucial role in regulating gene expression. Importantly, DNA methylation marks special sites called CpG islands, which exert important effects on gene transcription (Lim and Maher 2011; Edwards et al. 2017). Generally, but not always, the hypermethylation of CpG islands on gene promoters leads to gene silencing (Yang and Schwartz 2011). This may be explained by the need for accessibility of the gene promoter to transcription factors and other regulatory units for induction of gene transcription, which can be restricted by the presence of DNA methylation (Lim and Maher 2011).

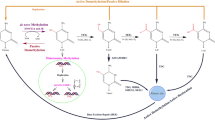
Mechanisms of epigenetic regulation. The addition of a methyl (–CH3) group to the cytosine residue on the DNA strand is termed as ‘DNA methylation’ which is the most widely studied epigenetic regulation. In general, it causes suppression of gene expression by hindering the binding of regulatory proteins onto the DNA strand (a). The second level of epigenetic regulation occurs at histone level by addition of chemical groups such as acetyl (ac), methyl (me), phosphoryl, ubiquitinyl, etc., to the lysine (K), arginine (R), serine or threonine residues. The figure depicts some of the major histone marks on histone 2A (H2A), 2B (H2B), 3 (H3), and 4 (H4) listing the position (numbers denoted at the bottom) and type of amino acid residue (K or R) and the nature of the modification [ac, me, me1 (mono-methylation), me2 (dimethylation), and me3 (trimethylation)]. The inducible histone modifications are depicted in green; while the repressive histone signatures are shown in red (b). MicroRNAs (miRNA) are the third level of epigenetic regulation within the cell. Like messenger RNAs (mRNA), miRNAs are also transcribed by RNA polymerase II. However, unlike mRNAs, the pri-miRNAs are translocated to the cytoplasm by the nuclear enzyme Drosha; where it is converted to miRNA by the action of Dicer enzyme as shown in the figure. The miRNA shows complementarity to the sequence of its target mRNA. On binding of the miRNA to the mRNA, a duplex is formed which prevents the binding of the translational machinery to the mature mRNA, and thus halts gene expression (c) (color figure online)
Functionally, DNA methylation is catalyzed by a protein family known as DNA methyltransferases (DNMTs) which transfer methyl groups from S-adenosyl-l-methionine (SAM) to the 5-carbon position of cytosine residues in DNA (Jin and Robertson 2013). On the contrary, DNA demethylation is mediated by the ten-eleven translocation (TET) enzymes, which add a hydroxyl group onto the methyl group of 5mC to form 5hmC (5-hydroxymethyl cytosine) (Moore et al. 2013). A brief description of the roles of methyltransferases and demethylases is provided in Table 3.
Several epigenome-wide association studies (EWAS) have shown an association between the modifications in DNA methylation in the blood of smokers and their smoking status (Breitling et al. 2011; de Vries et al. 2018a; Harlid et al. 2014; Li et al. 2018; Philibert et al. 2013; Prince et al. 2019; Su et al. 2016). These studies have identified several CpGs mapping to specific genes that are altered on smoke exposure. Some of the common hits identified from such studies include coagulation factor II (thrombin) receptor-like 3 (F2RL3) (Sun et al. 2013; Zaghlool et al. 2015), aryl hydrocarbon receptor repressor (AHRR) (Elliott et al. 2014; Fasanelli et al. 2015; Monick et al. 2012; Philibert et al. 2013; Zaghlool et al. 2015), cyclin-dependent kinase inhibitor (p16) (Belinsky et al. 2002; Soria et al. 2002), and death-associated protein kinase (DAPK) (Soria et al. 2002).
F2RL3 encodes a protein involved in platelet activation, intimal hyperplasia, and inflammation (Breitling et al. 2011; Fasanelli et al. 2015). It has been suggested to be a strong predictor of mortality during smoking-induced cardiovascular diseases and cancers (Breitling et al. 2011; Zhang et al. 2014). Likewise, hypomethylation at specific CpG loci of AHRR (the repressor of the aryl hydrocarbon receptor) has been associated with tobacco smoking and is linked to increased risk of lung cancer among smokers (Fasanelli et al. 2015; Prince et al. 2019; Shenker et al. 2013). Also, methylation at p16 and DAPK promoter has been reported in former patients with non-small cell lung carcinoma (NSCLC) (Soria et al. 2002). Such reports suggest that the smoking-associated DNA methylation changes could act as biomarkers to predict the susceptibilities toward major pulmonary diseases among smokers.
However, contradictory evidences exist with regard to DNA methylation. For instance, Qiu et al. (2012) studied DNA methylation changes in the WBCs from 1454 smokers with and without COPD. They identified 349 CpG sites that were associated with the presence and severity of COPD. Most of the associated CpG sites were hypomethylated and were found on genes associated with immune and inflammatory system pathways, responses to stress and external stimuli, wound healing, and coagulation cascades. These findings led the group to conclude that epigenetic changes might cause COPD (Qiu et al. 2012). On the contrary, de Vries et al. conducted similar investigations on the whole blood of 1561 individuals from a Dutch cohort comprising current and never smokers but found no association between the DNA methylation and the occurrence of COPD in the study groups. Few of the reasons described by the authors for such contradiction were the fact that the study group associated with the work of Qiu et al. had severe COPD with an FEV1/FVC ratio less than 0.7. Additionally, the platform used to study DNA methylation by both the groups varied and the former did not include confounding factors like age and sex in their analyses (de Vries et al. 2018b). Such contradictions are not uncommon when reviewing epigenetic studies and just highlight the highly dynamic nature of DNA methylation.
Evidence has suggested that various genetic variants mediate the smoking-associated DNA methylation changes (Dogan et al. 2017; Gupta et al. 2019; Leng et al. 2015; Siedlinski et al. 2012). This proves that gene and environment work at tandem to decide the disease fate in individuals. This is the reason behind variations observed in smoking-related methylation changes across populations (Elliott et al. 2014; Shenker et al. 2013; Dogan et al. 2014; Sun et al. 2013; Lee et al. 2016; Xu et al. 2010; Zaghlool et al. 2015; Zhu et al. 2016).
Interestingly smoking-related DNA methylation changes are strongly associated with the smoking habits and time since smoking cessation (Ambatipudi et al. 2016; Breitling et al. 2011; Li et al. 2018). Studies have found that there exists a marked reversibility of methylation changes after smoking cessation at certain gene loci (Ambatipudi et al. 2016; McCartney et al. 2018; Wan et al. 2012; Wilson et al. 2017), whereas differential DNA methylation for certain other genomic locations remain unaffected even years (up to 22 years) after smoking cessation (Ambatipudi et al. 2016). In one of the most extensive studies suggesting the reversion of DNA methylation marks on smoking cessation, a team of investigators led by Ambatipudi observed that CpGs that were hypermethylated in current smokers showed decreased methylation with increasing time since smoking cessation and vice versa. In contrast, four CpG sites [cg01940273 (ALPPL2), cg05951221 (ALPPL2), cg11554391 (AHRR), and cg21566642 (ALPPL2)], were recognized during this study that did not show any decrease in the methylation levels even 14.1–22 years after smoking cessation (Ambatipudi et al. 2016). Wan et al. classified the CpG methylations into two categories—‘rapidly reversible’ and ‘slowly reversible’—based on their differential methylation following smoking cessation in the peripheral blood from current, former, and never smokers. The genes myosin light chain kinase (MYLK) and leucine-rich repeat neuronal 3 (LRRN3) were identified as ‘rapidly-reversible’ while G-Protein-coupled receptor protein 15 (GPR15) and Fas ligand (FASLG) as ‘slowly-reversible’ CpG sites in this study (Wan et al. 2012). A slight contradiction to this view was provided in reports by Philibert et al. (2016) and Wilson et al. (2017), which showed that the magnitude of this reversion on smoking cessation is larger than normal individuals who have never smoked. This suggests overcorrection of DNA methylation marks on smoking cessation and adds another layer of complexity to the regulation of DNA methylation and gene expression in current and former smokers (Philibert et al. 2016; Wilson et al. 2017).
Despite our knowledge about the differential DNA methylation on smoke exposure, not many studies have been conducted to correlate the gene expression with CpG methylation changes. Most of the current investigations have been conducted using peripheral blood from smokers due to ease of sampling. However, DNA methylation changes on smoking may differ in multiple tissues (Hammons et al. 1999; Monick et al. 2012; Peters et al. 2007; Satta et al. 2008; Suzuki et al. 2007), which has not been examined in detail so far. Most of the current studies test genome-wide changes in DNA methylation signatures on smoking. The platforms and approaches used for each of these studies differ and so do the assumptions and statistics used to deduce outcomes. It, thus, becomes difficult to compare such genome-wide studies and find specific targets to serve as biomarkers or therapy. Finally, several of the EWAS investigations focus on the phenotypic outcome of cigarette smoking such as COPD, lung cancer, etc. (Carvalho et al. 2012; Fasanelli et al. 2015; Qiu et al. 2012), but they do not examine the effects of smoking per se. Such explorations are crucial to understand the molecular mechanisms affected on smoking and identifying potential biomarkers for early detection of various smoking-related disease outcomes (Lee and Pausova 2013).
Cigarette smoke-induced chromatin remodeling and changes in histone modifications
Histone modifications are chemical modifications in the form of methylation, acetylation, ubiquitination, and/or phosphorylation of specific amino acids [particularly lysine (Lys), serine (Ser), threonine (Thr), and tyrosine (Tyr)] on their N-terminal tail, which influence chromatin packaging and, in turn, transcriptional activity. Inducible histone modifications loosen the DNA association with histones, thus providing a permissive environment for transcription, whereas repressive histone modifications tighten the chromatin packaging, thus repressing gene expression. The inducible or repressive nature of a histone modification is determined based on the: (a) type of histone modification, (b) modified amino acid base, and (c) position of modification (Bannister and Kouzarides 2011; Moore et al. 2013), as depicted in Fig. 2b.
Of note, histone modifications not only regulate chromatin structure, but also recruit remodeling enzymes to reposition nucleosomes. Like DNA methylation, the addition/removal of chemical groups to histone tails is also catalyzed by a group of enzymes collectively termed as histone-modifying enzymes (HMEs) (Bannister and Kouzarides 2011). The names and functions of some of the common HMEs are listed in Table 3.
The in vitro and in vivo studies have shown downregulation of histone deacetylase 2 (HDAC2) expression and activity in smoking-induced lung inflammation (Ito et al. 2001; Marwick et al. 2004; Moodie et al. 2004; Sundar and Rahman 2016). In fact, reduction in HDAC2 has been reported in the lung tissues from COPD and severe/smoking asthma patients as well, thus stating its role in the disease pathophysiology (Barnes 2009; Ito et al. 2002; 2005). Furthermore, HDAC1 expression is often found to be reduced in smoke-challenged cells and bronchial biopsies from asthmatics; which is not surprising as it is associated with HDAC2 in the nucleus (Adenuga et al. 2009; Ito et al. 2002). Function of other HDACs in smoke-related inflammation is uncertain (Barnes 2009). Additionally, the expression of histone acetyltransferases (HATs)—CREB-binding protein (CBP) and p300-CBP-associated factor (PCAF)—has been found to be unaltered in response to cigarette smoke challenge in vitro (Ito et al. 2001).
Reports suggest that inactivation of HDAC2 leads to increased acetylation at histone 3 (H3) and 4 (H4) in the lungs of smokers, COPD patients, cigarette smoke-exposed animals (rat), and cellular (A549) models (Ito et al. 2005; Marwick et al. 2004; Moodie et al. 2004; Szulakowski et al. 2006). However, most of these studies investigate global changes in the histone marks in association with smoking and do not provide much information about the site-specific regulation of gene transcription (Marwick et al. 2004).
Nevertheless, cigarette smoke exposure does not just alter the histone acetylation marks, but also affects other histone modifications. This was reported by Sundar et al. in smoke-exposed mouse lungs and human bronchial epithelial (H292) cells. Using a bottom-up mass spectrometry approach, this group identified acetylation at H3K56, H4K8, H4K12, and H4K16 in smoke-exposed mouse lungs and H292 cells as compared to the controls. In the same study, distinct site-specific histone post-translational modifications at H3K27me1, H3K27me2, H4K31me2, and H4R35me2 were found to be associated with smoke exposure. These findings suggest a strong link between chromatin compaction, replication, and transcriptional control in response to cigarette smoke challenge (Sundar et al. 2014).
Contrary to the abovementioned studies, our group undertook a targeted approach to study the changes in the histone signatures at the promoter site of NLRP10 (family of NOD-like proteins) gene in response to smoke challenge (Kaur et al. 2018). We demonstrated NLRP10-mediated caspase-1 activation, cytokine/chemokine production (IL-1β, IL-18, MCP-1, and IL-17A), and NF-κB and MAPKs expression in the lungs of second-hand smoke-exposed C57Bl/6 mice and cigarette smoke extract-challenged human lung epithelial cells with type II characteristics. To identify upstream mediators of NLRP10 regulation, we investigated changes in the epigenetic signatures on the Nlrp10 promoter region following smoke exposure and observed significant changes in active (H3K4me3 and H3K9ac) as well as repressive (H3K9me3 and H4K20me3) gene markers on histone 3 and histone 4 both in vivo and in vitro. Furthermore, alterations in the respective histone acetyl- and methyltransferases (PCAF, SET1, ESET, SUV20H1) correlated well with the alterations observed in the histone markers (Kaur et al. 2018). Such targeted studies are important to understand the mechanism of regulation of inflammatory responses on smoke exposure at molecular level.
Despite all these evidences, establishing a direct link between altered expression of HATs/HDACs leading to chromatin remodeling and gene transcription at specific gene sites has many challenges (Marwick et al. 2004). The histone PTMs vary based on the duration of smoke exposure, type of cell/tissue exposed to smoke, and the experimental models used for the study; making it difficult to ascertain their exact role under physiological and disease state (Marwick et al. 2004; Sundar et al. 2014). Furthermore, there are contrasting evidences with regard to the expression and activity of various HDACs in smoke-related disease conditions (Ito et al. 2005; Sohal et al. 2013). The histone modifications are dynamic in nature and gene transcription is affected by a concerted effect of the histone modifications which adds to the complexity of studying such a phenomenon (Bannister and Kouzarides 2011; Sundar et al. 2014).
Smoke-associated alterations in miRNA expression
MicroRNA (miRNA) are a class of small endogenous RNAs approximately 22 nucleotides in length that play an important regulatory role by targeting specific mRNAs for degradation and/or translational repression in both animal and plant cells. These are single-stranded RNA molecules produced from hairpin-shaped precursors, also known as pri-miRNAs, by the action of two RNase III-type proteins: Drosha (in nucleus) and Dicer (in cytoplasm) (Wahid et al. 2010) (Fig. 2c).
There are currently a total of 1917 known mature human miRNAs in miR-Base, the central online repository for miRNA sequences and annotation, which accounts for 1–5% of all predicted human genes (Banerjee and Luettich 2012; Wang et al. 2015). Functionally, miRNAs regulate developmental processes, cellular homoeostasis, and responses to various stimuli by binding to a target mRNA and altering its protein expression. (Banerjee and Luettich 2012; Wahid et al. 2010).
Reports indicate distinct miRNA profiles among smokers and non-smokers (Advani et al. 2017; Andersson et al. 2018; Banerjee et al. 2015; Gross et al. 2014; Huang et al. 2014; Shen et al. 2017; Wang et al. 2015; Willinger et al. 2017). In general, these studies have found differences in the expression of several miRNAs following exposure to smoke. Most of the dysregulated miRNAs have been found to be associated with lung development, airway epithelium differentiation, inflammation, and cancer (Banerjee and Luettich 2012; Wang et al. 2015; Willinger et al. 2017).
However, just like DNA methylation, changes in the miRNA profile appear to be reversible in nature (Ambatipudi et al. 2016; Wang et al. 2015). Wang et al. observed that 3 months after quitting smoking, 22 of the 34 differentially expressed miRNAs returned to their normal levels, including the miRNAs related to cancer/inflammation (miR-181a), airway epithelium differentiation (miR-449b), and lung development (miR-214 and miR-127). Interestingly, the remaining 12 differentially expressed miRNAs associated with smoking did not revert to their normal levels even after smoking cessation. Further, the miRNAs with sustained dysregulated expression are associated with carcinogenesis and chronic airway diseases (miR-218, miR-133a, miR-133b, miR-487b, and miR-1246) (Wang et al. 2015). Importantly, the Wnt/β-catenin signaling pathway was found to be significantly enriched in the target genes linked with persistently dysregulated miRNAs, which is in accordance with the previous knowledge that Wnt pathway dysregulation is associated with diseases, including lung cancer and COPD (Heijink et al. 2013; Konigshoff and Eickelberg 2010; Wang et al. 2015).
Currently, miRNAs are being targeted as candidates for biomarkers and drug discovery for various disease conditions (Banerjee and Luettich 2012; Janssen et al. 2013). However, certain considerations must be made before making advances in this respect. Smoking-related dysregulation of miRNAs is dependent on multiple factors including the dose of exposure, duration of exposure, and cell types exposed. Confounding factors such as age, race, and other epigenetic changes could also affect miRNA expression (Banerjee and Luettich 2012; Izzotti et al. 2009, 2011; Wang et al. 2015). To date, miRNA-based research has been exploratory in nature. The main challenge ahead is the need for a targeted approach to narrow down and identify specific candidates for biomarker or drug development in smoke-related diseases. Of note, several of the miRNAs identified during such studies are regulated by DNA methylation, which signifies the cross talk between the epigenetic signatures and their regulatory mechanisms (Lyn-Cook et al. 2014; Wang et al. 2015). In the future, a detailed investigation of the cross talk between different epigenetic mechanisms will be necessary to fully appreciate their therapeutic potential.
Effects of maternal smoking
Altered DNA methylation and dysregulated miRNA expression have also been assessed to identify transgenerational effects of maternal smoking (Breton et al. 2009; Herberth et al. 2014; Jenkins et al. 2017; Knopik et al. 2012; Suter et al. 2011). In this regard, studies by Suter et al. 2010 and 2011 showed significant hypomethylation in the placentas of babies born to mothers who smoked during pregnancy compared to that of non-smoking mothers. This hypomethylation was found to correlate with increased placental CYP1A1 expression, which may have implications for xenobiotic metabolism in the offspring (Suter et al. 2010; 2011). Likewise, hypermethylation of the brain-derived neurotrophic factor (BDNF) might be responsible for its lowered expression with subsequent behavioral consequences in infants, children, and adolescents exposed in utero to maternal cigarette smoking (Knopik et al. 2012).
While assessing the gender-specific methylation differences in offspring in relation to maternal smoking, Murphy et al. (2012) tested the methylation profiles of two imprinted genes, H19 and IGF2, in cord blood. The results from this study suggest that there is a more pronounced epigenetic effect of maternal smoking on male offspring than on females (Murphy et al. 2012). This is not surprising as sex differences in DNA methylation patterns have been previously reported among Dutch famine victims in response to caloric restriction (Heijmans et al. 2007). Other population-wide studies of human disasters have further revealed heightened phenotypic responses and risk among males (Catalano et al. 2005; Khashan et al. 2011). One of the criticisms of this paper, however, was that it only tested the methylation status at two different regions of the imprinted genes. Nevertheless, methylation still holds significance as it plays an essential role in regulating growth and its deregulation may lead to disease and disorder in the growing fetus (Murphy et al. 2012). Another group of researchers studied the DNA methylation status at six CpG sites on the IL-13 gene in peripheral blood leukocytes of offspring to study the correlation between maternal smoking and asthma-related lung function. While a strong correlation between differential methylation at the cg13566430 site and maternal smoking during pregnancy was established, disease outcome in the offspring remained unclear due to lack of data relating to asthmatic traits (Patil et al. 2013). However, Herberth et al. were able to establish an association between maternal tobacco smoke exposure and cord blood miR223 expression, which was responsible for reduced Treg numbers, suggesting increased allergy risk in offsprings later in life (Herberth et al. 2014). Additionally, a recent study by Richmond et al. proved that maternal smoking during pregnancy is associated with persistent alterations in DNA methylation in the exposed offspring. These changes in the DNA methylation pattern were observed to exist even 18 years after prenatal exposure (Richmond et al. 2018). Overall, it can be concluded that changes in DNA methylation signatures and the miRNA profile caused by maternal smoking may not only increase disease susceptibilities in exposed offspring but are also transmitted to the next generation.
Effects of paternal smoking
Interestingly, the intergenerational effects of smoking are not limited to the smoking habits of mothers. Jenkins et al. identified 141 differentially methylated CpGs in the DNA of sperm from men who smoke compared with non-smokers. The differential methylation occurred more frequently at regions reported to display H3K4 and H3K27 methylation in mature spermatozoa (Jenkins et al. 2017). Functionally, H3K4 methylation is associated with gene activation while methylation at H3K27 is a gene repression signature. But despite opposite functions both these modifications are responsible for development, lineage commitment, and differentiation (Eissenberg and Shilatifard 2010; Nichol et al. 2016). These changes could not only account for reduced sperm count and motility in men who smoke, but also affect the fetal development of their offspring at later stages (Gunes et al. 2018). Another study tested the sperm quality of smoking and non-smoking males and reported differential miRNA expression in the spermatozoa from smokers vs. non-smokers. In fact, four (hsa-miR-146b-5p, hsa-miR-509-5p, hsa-miR-519d, and hsa-miR-652) of the differentially methylated miRNAs identified in this study are known to be altered in infertile men, thus suggesting that smoking might be associated with male sterility. Moreover, the major pathways affected by these differentially expressed miRNAs are known to be involved in cell differentiation, proliferation, and death. Thus, these pathways likely play a vital role during sperm and early embryo development (Marczylo et al. 2012). In a similar type of study, Hamad and his group used a whole-genome DNA methylation assay to study the differences in global DNA methylation among smokers and non-smokers. The results of this study revealed a significant increase in the levels of global DNA methylation in the sperm of smokers. Previous work showed that the production of low quality and apoptotic spermatozoa could be linked to altered spermatogenesis that resulted in global DNA hypermethylation (Hamad et al. 2018). These findings further suggest that global DNA methylation might affect normal spermatogenesis and thereby affect male fertility as well as the future progeny of men who smoke.
Challenges and ethical concerns related to epigenetic research
Despite the great potential of epigenetic research about the future of medicine, the research community is currently unable to tap its full potential. Hence, it is important to discuss the challenges and ethical concerns hindering advances in this area. While some of the challenges and ethical concerns may be directly encountered during research pertaining to smoke-related epigenetic alterations, most of these challenges are more generic in nature and are not restricted to any one disease condition.
Challenges
Study of the human epigenome is still in its infancy, as we are just beginning to understand the complexities of epigenetic signatures in disease and development (Zheleznyakova et al. 2017). Several challenges lie ahead on this venture of exploring the full potential of the epigenome. First, while there is only one genome in all individuals of a species, the epigenome exhibits tissue-specific variations (Costa et al. 2016; de Vries et al. 2018a; Leng et al. 2015). In this regard, it should be noted that most previous epigenetic studies were conducted using whole blood, which itself is comprised of multiple cell types. Therefore, it is likely that the epigenetic alterations detected in mixed tissues/whole blood arise from differences in cell composition between tissues/samples from diseased and control subjects (Weinhold 2006; Zheleznyakova et al. 2017). Thus, future experiments will be required to further assess tissue-specific differences in the epigenome.
Additionally, an individual’s epigenetic make-up could be affected by multiple confounding factors such as age, genetic background, environmental exposure, clinical conditions, etc. Such confounders cause discordance and discrepancies while comparing similar epigenetic cohort studies, thus affecting the end deductions (Knopik et al. 2012; Zheleznyakova et al. 2017). During our study, we found several instances where inclusion of confounding factors altered the study results completely. A good example to this is the contradictions in the findings by Qiu et al. (2012) and de Vries et al. (2018b) that have been explained earlier in this review.
Additional technical difficulties are known to arise when conducting epigenetic research. For example, bisulfite sequencing is the most extensively used technique to study DNA methylation. In fact, most of the studies listed in this review used this technique to study DNA methylation alterations with respect to smoke (Breitling et al. 2011; de Vries et al. 2018a; Prince et al. 2019; Sundar et al. 2017). However, bisulfite sequencing cannot differentiate between 5mC and 5hmC. This raises the possibility that the observed DNA methylation changes could be an overrepresentation due to the lack of assay specificity (Zheleznyakova et al. 2017).
In addition, few biological/chemical tools, such as antibodies and selective inhibitors or activators, exist to study epigenetic changes in vitro and in vivo. Thus, this limits the scope of understanding of this phenomenon. Currently scientists primarily use genetic association combined with molecular tools, including gene silencing, protein overexpression, and catalytic-inactive mutants, to determine relevant disease targets. However, each of these approaches has certain caveats which make them less than ideal (Campbell and Tummino 2014). We ourselves ran into this challenge when attempting to identify the HDACs/HATs responsible for histone modifications in vitro and in vivo (Kaur et al. 2018). We attempted to target the trimethylation on histone 3 and 4 during this study; however, finding an antibody that specifically binds to trimethyl and not mono- or di-methyl on histones remains a struggle.
Finally, improvements to overcome batch variability and to nullify the effects of confounding factors are needed in high-throughput technologies. These include computational capability, analytical techniques, mechanistic studies, and bioinformatic strategies (Campbell and Tummino 2014; Weinhold 2006).
Ethical concerns
Epigenetic research has largely been kept from the forefront of the drug development process due to ethical issues associated with the gathering of patient information. For example, it is well known that environmental factors including diet and exposure to chemical toxicants such as pesticides, diesel exhaust, and tobacco smoke increase disease risks; however, such exposures are frequently linked to poverty, standard of living, and working conditions of the exposed individuals, which places the onus on law and policymakers. Evidences related to epigenetic effects, including transgenerational effects, suggest that some individuals are predisposed to be more affected by adverse environmental conditions than other individuals. Thus, the focus shifts from populations with greater susceptibility to those receiving disproportionate exposure, thus resulting in calls for environmental justice advocates to address these injustices (Rothstein et al. 2009). Something that is important to mention here is the fact that during this literature review we found that most population-based studies were conducted on Caucasians (Ambatipudi et al. 2016; McCartney et al. 2018; Wan et al. 2012; Wilson et al. 2017). However, it is well known that some population is clearly predisposed to pulmonary health problems like COPD caused by smoking (El-Zein et al. 2012). Study of only specific population groups should thus be avoided both in research and during drug development, but it is something that is not practiced.
Another important aspect of extensive, ongoing epigenetic studies is the generation of a wealth of sensitive information regarding future health issues in patients and the possibility of transmitting those risks to offspring. Our investigations of maternal/paternal smoking clearly showed that offspring inherit not just genes, but also epigenes (Breton et al. 2009; Jenkins et al. 2017; Joubert et al. 2014; Marczylo et al. 2012). However, epigenes are not often considered, and thus, neither are the privacy and confidentiality issues surrounding such information. Another factor to consider is that unlike our genetic information, epigenetic effects are environmentally induced and might also be reversed, as seen in the case of smoke-induced DNA methylation and miRNA alterations (Ambatipudi et al. 2016; Wang et al. 2015). Though relevant, such questions have not yet been addressed (Rothstein et al. 2009; Shabani et al. 2018).
Related to the abovementioned challenges is the issue of equitable access to health care. Regarding genetics, both public and private providers are reluctant to approve various clinical genetic services on the grounds of these being experimental and not medically essential. Considering this, the success and popularity of epigenetic testing seems uncertain. Particularly, the issue of access to healthcare will be critical for individuals likely to work and live in hazardous environments (Rothstein et al. 2009). Moreover, the current medical system is treatment oriented and does not encourage means of disease prevention, which is where the true potential of epigenetic research lies.
Advancements in epigenetic studies have raised awareness of intergenerational equity and thereby broadened the scope of our duties to future generations. Ardent supporters of scientific advancements might believe that measures to prevent the transmission of epigenetically harmful signatures to future generations must be encouraged; however, critics may argue that this would interfere with the natural order of things (Rothstein et al. 2009). Thus, future policies must consider these concerns so that epigenetic information can be utilized as a diagnostic tool or treatment method for life-debilitating disease.
Discussion
Systematic review of the literature to identify epigenetic alterations on cigarette smoke exposure revealed heterogeneous results. Our search criteria identified 80 studies focusing mainly on 3 of the widely known epigenetic mechanisms, DNA methylation (35 studies), histone modifications/chromatin remodeling (11 studies), and miRNA expression (12 studies). Among these, most of the studies were conducted using human and/or clinical samples (69 studies); while 9 were employed in vitro and 8 used in vivo study models to identify epigenetic changes. Considerable evidence is available in the literature suggesting that cigarette smoking regulates DNA methylation signatures. In fact, several genes which have been commonly cited in multiple studies (e.g., AHRR, p16, F2RL3, and DAPK) could be developed as biomarkers to assess disease progression. These studies have several merits including large sample size, correlation with confounding factors such as smoking status; smoking history, smoking cessation, robust approach, and efficient summary statistics. Likewise, miRNA expression studies also have great potential to identify probable biomarkers and drug targets. However, unlike DNA methylation, other epigenetic markers have not been extensively investigated.
The major criticism of epigenetic studies related to smoke-associated diseases is the heterogeneity of sample size, sample type, and assessment techniques used to determine epigenetic alterations. Further, non-inclusion of comorbidities and factors such as demographics, genetic variations, BMI, and other health determinants for statistical corrections, and the lack of evidence regarding the consequences of the observed changes are the major drawbacks of these studies. In this context, three studies included in this review compared the DNA methylation alterations among smokers based on their demographics and provided contrasting outcomes. The study conducted by Zhu et al. measured methylation marks at > 485,000 CpGs in current, former, and never smokers from a Chinese cohort and compared their results to previous studies conducted on Europeans and African Americans. This group identified 161 CpGs annotated to 123 genes that were not associated with smoking in Europeans or African Americans and concluded that these sites were specific to the Chinese population (Zhu et al. 2016). Sun et al. conducted a methylome-wide study using 972 African Americans to identify DNA methylation sites associated with smoking in this population and compared their results with previous work done in Caucasians. They concluded that the two ethnic groups share common associations with cigarette smoking despite their distinct genetic backgrounds (Sun et al. 2013). While both of these studies compared their observation with data from a previously published study to draw conclusions, Elliot et al. performed a methylome-wide study in a population-based cohort including 1711 first-generation South Asian migrants and 1762 people of European origin aged 40–69 living in West London, UK. In this study, they found distinct smoking-associated methylation changes at both single CpG sites and overall smoking score (constructed based on methylation profile) based on ethnicity (Elliott et al. 2014). While most of the DNA methylation studies have been conducted in European populations, the studies conducted in other populations have not considered demographic differences in their results. Thus, there exists a wide knowledge gap with regard to the effect of genes and epigenes on smoking-associated disease susceptibilities among various populations. The abovementioned examples highlight the importance of inclusion of confounding factors like ethnicity in the DNA methylation studies. Future studies, thus, need to address these gaps to draw more meaningful conclusions.
Since a wide variety of cells/tissue samples have been used in epigenetic studies to deduce the effects of smoking, it is challenging to identify common epigenetic signatures and associated molecular mechanisms as these changes are predominantly tissue specific (Gutierrez-Arcelus et al. 2015). The information about various types of cellular and tissue models used in individual epigenetic studies is included in Tables 1 and 2. Likewise, it is difficult to compare the information about epigenetic changes from different studies due to the measurement of different variables (DNA methylation, histone modification or miRNA) in terms of individual signatures. While the studies included in our systematic review identify the top candidate genes, chromatin modifications, and miRNAs altered during smoke exposure, none of them explored the interconnections between various epigenetic markers. In future, such associations must be explored to tap the full potential of epigenetics in deducing disease mechanisms and developing therapies.
Conclusions
The evidence presented in this systematic review is suggestive of a vital role of epigenetic changes in regulating smoking-associated alterations and disease development. Better study design to interconnect different epigenetic marks, inclusion of confounding factors, and association with disease outcomes could improve the quality of future research.
References
Adenuga D, Yao H, March TH, Seagrave J, Rahman I (2009) Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol 40(4):464–473
Advani J, Subbannayya Y, Patel K et al (2017) Long-term cigarette smoke exposure and changes in MiRNA expression and proteome in non-small-cell lung cancer. OMICS 21(7):390–403
Alkhaled Y, Laqqan M, Tierling S, Lo Porto C, Amor H, Hammadeh ME (2018) Impact of cigarette-smoking on sperm DNA methylation and its effect on sperm parameters. Andrologia. https://doi.org/10.1111/and.12950
Ambatipudi S, Cuenin C, Hernandez-Vargas H et al (2016) Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics 8(5):599–618
Andersson BA, Sayardoust S, Lofgren S, Rutqvist LE, Laytragoon-Lewin N (2018) Cigarette smoking affects microRNAs and inflammatory biomarkers in healthy individuals and an association to single nucleotide polymorphisms is indicated. Biomarkers 24(2):180–185
Banerjee A, Luettich K (2012) MicroRNAs as potential biomarkers of smoking-related diseases. Biomark Med 6(5):671–684
Banerjee A, Waters D, Camacho OM, Minet E (2015) Quantification of plasma microRNAs in a group of healthy smokers, ex-smokers and non-smokers and correlation to biomarkers of tobacco exposure. Biomarkers 20(2):123–131
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395
Barnes PJ (2009) Histone deacetylase-2 and airway disease. Ther Adv Respir Dis 3(5):23543
Beach SRH, Lei MK, Ong ML, Brody GH, Dogan MV, Philibert RA (2017) MTHFR methylation moderates the impact of smoking on DNA methylation at AHRR for African American young adults. Am J Med Genet B Neuropsychiatr Genet 174(6):608–618
Belinsky SA, Palmisano WA, Gilliland FD et al (2002) Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res 62(8):2370–2377
Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H (2011) Tobacco-smoking-related differential DNA methylation: 27 K discovery and replication. Am J Hum Genet 88(4):450–457
Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD (2009) Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med 180(5):462–467
Burton A (2011) Does the smoke ever really clear? Thirdhand smoke exposure raises new concerns. Environ Health Perspect 119(2):A70–A74
Campbell RM, Tummino PJ (2014) Cancer epigenetics drug discovery and development: the challenge of hitting the mark. J Clin Invest 124(1):64–69
Carrozza MJ, Utley RT, Workman JL, Cote J (2003) The diverse functions of histone acetyltransferase complexes. Trends Genet 19(6):321–329
Carvalho RH, Haberle V, Hou J et al (2012) Genome-wide DNA methylation profiling of non-small cell lung carcinomas. Epigenet Chromatin 5(1):9
Catalano R, Bruckner T, Gould J, Eskenazi B, Anderson E (2005) Sex ratios in California following the terrorist attacks of September 11. Hum Reprod 20(5):1221–1227
Costa LA, da Silva ICB, Mariz B, da Silva MB, Freitas-Ribeiro GM, de Oliveira NFP (2016) Influence of smoking on methylation and hydroxymethylation levels in global DNA and specific sites of KRT14, KRT19, MIR-9-3 and MIR-137 genes of oral mucosa. Arch Oral Biol 72:56–65
de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370(Pt 3):737–749
de Vries M, van der Plaat DA, Nedeljkovic I et al (2018a) From blood to lung tissue: effect of cigarette smoke on DNA methylation and lung function. Respir Res 19(1):212
de Vries M, van der Plaat DA, Vonk JM, Boezen HM (2018b) No association between DNA methylation and COPD in never and current smokers. BMJ Open Respir Res 5(1):e000282
Dogan MV, Shields B, Cutrona C et al (2014) The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics 15:151
Dogan MV, Beach SRH, Philibert RA (2017) Genetically contextual effects of smoking on genome wide DNA methylation. Am J Med Genet B Neuropsychiatr Genet 174(6):595–607
Edwards JR, Yarychkivska O, Boulard M, Bestor TH (2017) DNA methylation and DNA methyltransferases. Epigenet Chromatin 10:23
Eissenberg JC, Shilatifard A (2010) Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol 339(2):240–249
Elliott HR, Tillin T, McArdle WL et al (2014) Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenet 6(1):4. https://doi.org/10.1186/1868-7083-6-4
El-Zein RA, Young RP, Hopkins RJ, Etzel CJ (2012) Genetic predisposition to chronic obstructive pulmonary disease and/or lung cancer: important considerations when evaluating risk. Cancer Prev Res (Phila) 5(4):522–527
Fa S, Larsen TV, Bilde K et al (2016) Assessment of global DNA methylation in the first trimester fetal tissues exposed to maternal cigarette smoking. Clin Epigenet 8:128
Fasanelli F, Baglietto L, Ponzi E et al (2015) Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat Commun 6:10192
Gross TJ, Powers LS, Boudreau RL et al (2014) A microRNA processing defect in smokers’ macrophages is linked to SUMOylation of the endonuclease DICER. J Biol Chem 289(18):12823–12834
Gu W, Yuan Y, Yang H et al (2018) Role of miR-195 in cigarette smoke-induced chronic obstructive pulmonary disease. Int Immunopharmacol 55:49–54
Gunes S, Metin Mahmutoglu A, Arslan MA, Henkel R (2018) Smoking-induced genetic and epigenetic alterations in infertile men. Andrologia 50(9):e13124
Gupta R, van Dongen J, Fu Y et al (2019) Epigenome-wide association study of serum cotinine in current smokers reveals novel genetically driven loci. Clin Epigenet 11(1):1
Gutierrez-Arcelus M, Ongen H, Lappalainen T et al (2015) Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet 11(1):e1004958
Hamad MF, Dayyih WAA, Laqqan M, AlKhaled Y, Montenarh M, Hammadeh ME (2018) The status of global DNA methylation in the spermatozoa of smokers and non-smokers. Reprod Biomed Online 37(5):581–589
Hammons GJ, Yan Y, Lopatina NG et al (1999) Increased expression of hepatic DNA methyltransferase in smokers. Cell Biol Toxicol 15(6):389–394
Harlid S, Xu Z, Panduri V, Sandler DP, Taylor JA (2014) CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the Sister Study. Environ Health Perspect 122(7):673–678
Heffernan T (2016) Editorial: the impact of active and passive smoking upon health and neurocognitive function. Front Psychiatry 7:148
Heijink IH, de Bruin HG, van den Berge M et al (2013) Role of aberrant WNT signalling in the airway epithelial response to cigarette smoke in chronic obstructive pulmonary disease. Thorax 68(8):709–716
Heijmans BT, Kremer D, Tobi EW, Boomsma DI, Slagboom PE (2007) Heritable rather than age-related environmental and stochastic factors dominate variation in DNA methylation of the human IGF2/H19 locus. Hum Mol Genet 16(5):547–554
Herberth G, Bauer M, Gasch M et al (2014) Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J Allergy Clin Immunol 133(2):543–550
Hillemacher T, Frieling H, Moskau S et al (2008) Global DNA methylation is influenced by smoking behaviour. Eur Neuropsychopharmacol 18(4):295–298
Huang J, Wu J, Li Y et al (2014) Deregulation of serum microRNA expression is associated with cigarette smoking and lung cancer. Biomed Res Int 2014:364316
Hyun K, Jeon J, Park K, Kim J (2017) Writing, erasing and reading histone lysine methylations. Exp Mol Med 49(4):e324
Ito K, Lim S, Caramori G, Chung KF, Barnes PJ, Adcock IM (2001) Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J 15(6):1110–1112
Ito K, Caramori G, Lim S et al (2002) Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med 166(3):392–396
Ito K, Ito M, Elliott WM et al (2005) Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 352(19):1967–1976
Izzotti A, Calin GA, Steele VE, Croce CM, De Flora S (2009) Relationships of microRNA expression in mouse lung with age and exposure to cigarette smoke and light. FASEB J 23(9):3243–3250
Izzotti A, Larghero P, Longobardi M et al (2011) Dose-responsiveness and persistence of microRNA expression alterations induced by cigarette smoke in mouse lung. Mutat Res 717(1–2):9–16
Jamal A, Phillips E, Gentzke AS et al (2018) Current cigarette smoking among adults—United States. MMWR Morb Mortal Wkly Rep 67(2):53–59
Janssen HL, Reesink HW, Lawitz EJ et al (2013) Treatment of HCV infection by targeting microRNA. N Engl J Med 368(18):1685–1694
Jenkins TG, James ER, Alonso DF et al (2017) Cigarette smoking significantly alters sperm DNA methylation patterns. Andrology 5(6):1089–1099
Jin B, Robertson KD (2013) DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol 754:3–29
Joubert BR, Haberg SE, Nilsen RM et al (2012) 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 120(10):1425–1431
Joubert BR, Haberg SE, Bell DA et al (2014) Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer Epidemiol Biomark Prev 23(6):1007–1017
Kaneda M, Okano M, Hata K et al (2004) Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429(6994):900–903
Kaur G, Bagam P, Pinkston R, Singh DP, Batra S (2018) Cigarette smoke-induced inflammation: NLRP10-mediated mechanisms. Toxicology 398–399:52–67
Khashan AS, McNamee R, Henriksen TB et al (2011) Risk of affective disorders following prenatal exposure to severe life events: a Danish population-based cohort study. J Psychiatr Res 45(7):879–885
Knopik VS, Maccani MA, Francazio S, McGeary JE (2012) The epigenetics of maternal cigarette smoking during pregnancy and effects on child development. Dev Psychopathol 24(4):1377–1390
Konigshoff M, Eickelberg O (2010) WNT signaling in lung disease: a failure or a regeneration signal? Am J Respir Cell Mol Biol 42(1):21–31
Laqqan M, Tierling S, Alkhaled Y, Porto CL, Solomayer EF, Hammadeh ME (2017) Aberrant DNA methylation patterns of human spermatozoa in current smoker males. Reprod Toxicol 71:126–133
Lee KW, Pausova Z (2013) Cigarette smoking and DNA methylation. Front Genet 4:132
Lee MK, Hong Y, Kim SY, London SJ, Kim WJ (2016) DNA methylation and smoking in Korean adults: epigenome-wide association study. Clin Epigenet. 8:103
Leng S, Wu G, Collins LB et al (2015) Implication of a Chromosome 15q15.2 Locus in Regulating UBR1 and Predisposing Smokers to MGMT Methylation in Lung. Cancer Res 75(15):3108–3117
Li S, Wong EM, Bui M et al (2018) Causal effect of smoking on DNA methylation in peripheral blood: a twin and family study. Clin Epigenet 10:18
Lim D, Maher E (2011) DNA methylation: a form of epigenetic control of gene expression. Obstet Gynaecol 12(1):37–42
Lubick N (2011) Global estimate of SHS burden. Environ Health Perspect 119(2):A66–A67
Lyn-Cook L, Word B, George N, Lyn-Cook B, Hammons G (2014) Effect of cigarette smoke condensate on gene promoter methylation in human lung cells. Tob Induc Dis 12(1):15
Maccani MA, Avissar-Whiting M, Banister CE, McGonnigal B, Padbury JF, Marsit CJ (2010) Maternal cigarette smoking during pregnancy is associated with downregulation of miR-16, miR-21, and miR-146a in the placenta. Epigenetics 5(7):583–589
MacKenzie TD, Bartecchi CE, Schrier RW (1994) The human costs of tobacco use (2). N Engl J Med 330(14):975–980
Marczylo EL, Amoako AA, Konje JC, Gant TW, Marczylo TH (2012) Smoking induces differential miRNA expression in human spermatozoa: a potential transgenerational epigenetic concern? Epigenetics 7(5):432–439
Markunas CA, Xu Z, Harlid S et al (2014) Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ Health Perspect 122(10):1147–1153
Marwick JA, Kirkham PA, Stevenson CS et al (2004) Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 31(6):633–642
McCartney DL, Stevenson AJ, Hillary RF et al (2018) Epigenetic signatures of starting and stopping smoking. EBioMedicine 37:214–220
Milutinovic S, Brown SE, Zhuang Q, Szyf M (2004) DNA methyltransferase 1 knock down induces gene expression by a mechanism independent of DNA methylation and histone deacetylation. J Biol Chem 279(27):27915–27927
Monick MM, Beach SR, Plume J et al (2012) Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am J Med Genet B Neuropsychiatr Genet 159B(2):141–151
Moodie FM, Marwick JA, Anderson CS et al (2004) Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J 18(15):1897–1899
Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacology 38(1):23–38
Morales E, Vilahur N, Salas LA et al (2016) Genome-wide DNA methylation study in human placenta identifies novel loci associated with maternal smoking during pregnancy. Int J Epidemiol 45(5):1644–1655
Murphy SK, Adigun A, Huang Z et al (2012) Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene 494(1):36–43
Nichol JN, Dupere-Richer D, Ezponda T, Licht JD, Miller WH Jr (2016) H3K27 methylation: a focal point of epigenetic deregulation in cancer. Adv Cancer Res 131:59–95
Ostrow KL, Michailidi C, Guerrero-Preston R et al (2013) Cigarette smoke induces methylation of the tumor suppressor gene NISCH. Epigenetics 8(4):383–388
Patil VK, Holloway JW, Zhang H et al (2013) Interaction of prenatal maternal smoking, interleukin 13 genetic variants and DNA methylation influencing airflow and airway reactivity. Clin Epigenet 5(1):22
Peluso ME, Munnia A, Bollati V et al (2014) Aberrant methylation of hypermethylated-in-cancer-1 and exocyclic DNA adducts in tobacco smokers. Toxicol Sci 137(1):47–54
Peters I, Vaske B, Albrecht K, Kuczyk MA, Jonas U, Serth J (2007) Adiposity and age are statistically related to enhanced RASSF1A tumor suppressor gene promoter methylation in normal autopsy kidney tissue. Cancer Epidemiol Biomark Prev 16(12):2526–2532
Philibert RA, Beach SR, Lei MK, Brody GH (2013) Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenet 5(1):19
Philibert R, Hollenbeck N, Andersen E et al (2016) Reversion of AHRR demethylation is a quantitative biomarker of smoking cessation. Front Psychiatry 7:55
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28(10):1057–1068
Prince C, Hammerton G, Taylor AE et al (2019) Investigating the impact of cigarette smoking behaviours on DNA methylation patterns in adolescence. Hum Mol Genet 28(1):155–165
Protano C, Vitali M (2011) The new danger of thirdhand smoke: why passive smoking does not stop at secondhand smoke. Environ Health Perspect 119(10):A422
Qiu W, Baccarelli A, Carey VJ et al (2012) Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med 185(4):373–381
Reynolds LM, Lohman K, Pittman GS et al (2017) Tobacco exposure-related alterations in DNA methylation and gene expression in human monocytes: the Multi-Ethnic Study of Atherosclerosis (MESA). Epigenetics 12(12):1092–1100
Richmond RC, Simpkin AJ, Woodward G et al (2015) Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet 24(8):2201–2217
Richmond RC, Suderman M, Langdon R, Relton CL, Davey Smith G (2018) DNA methylation as a marker for prenatal smoke exposure in adults. Int J Epidemiol 47(4):1120–1130
Rothstein MA, Cai Y, Marchant GE (2009) The ghost in our genes: legal and ethical implications of epigenetics. Health Matrix Clevel 19(1):1–62
Saha SP, Bhalla DK, Whayne TF Jr, Gairola C (2007) Cigarette smoke and adverse health effects: an overview of research trends and future needs. Int J Angiol 16(3):77–83
Satta R, Maloku E, Zhubi A et al (2008) Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc Natl Acad Sci USA 105(42):16356–16361
Shabani M, Borry P, Smeers I, Bekaert B (2018) Forensic epigenetic age estimation and beyond: ethical and legal considerations. Trends Genet 34(7):489–491
Shea BJ, Grimshaw JM, Wells GA et al (2007) Development of AMSTAR: a measurement tool to assess the methodological quality of systematic reviews. BMC Med Res Methodol 7:10
Shen W, Liu J, Zhao G et al (2017) Repression of Toll-like receptor-4 by microRNA-149-3p is associated with smoking-related COPD. Int J Chron Obstruct Pulmon Dis 12:705–715
Shenker NS, Polidoro S, van Veldhoven K et al (2013) Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet 22(5):843–851
Shi B, Gao H, Zhang T, Cui Q (2016) Analysis of plasma microRNA expression profiles revealed different cancer susceptibility in healthy young adult smokers and middle-aged smokers. Oncotarget 7(16):21676–21685
Siedlinski M, Klanderman B, Sandhaus RA et al (2012) Association of cigarette smoking and CRP levels with DNA methylation in alpha-1 antitrypsin deficiency. Epigenetics 7(7):720–728
Sohal SS, Reid D, Soltani A et al (2013) Changes in airway histone deacetylase2 in smokers and COPD with inhaled corticosteroids: a randomized controlled trial. PLoS One 8(5):e64833
Soria JC, Rodriguez M, Liu DD, Lee JJ, Hong WK, Mao L (2002) Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res 62(2):351–355
Su D, Wang X, Campbell MR et al (2016) Distinct epigenetic effects of tobacco smoking in whole blood and among leukocyte subtypes. PLoS One 11(12):e0166486
Sun YV, Smith AK, Conneely KN et al (2013) Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet 132(9):1027–1037
Sundar IK, Rahman I (2016) Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: implications for COPD and lung cancer. Am J Physiol Lung Cell Mol Physiol 311(6):L1245–L1258
Sundar IK, Chung S, Hwang JW et al (2012) Mitogen- and stress-activated kinase 1 (MSK1) regulates cigarette smoke-induced histone modifications on NF-kappaB-dependent genes. PLoS One 7(2):e31378
Sundar IK, Nevid MZ, Friedman AE, Rahman I (2014) Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer. J Proteome Res 13(2):982–996
Sundar IK, Yin Q, Baier BS et al (2017) DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin Epigenet 9:38
Suter M, Abramovici A, Showalter L et al (2010) In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism 59(10):1481–1490
Suter M, Ma J, Harris A et al (2011) Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics 6(11):1284–1294
Suzuki M, Shigematsu H, Shames DS et al (2007) Methylation and gene silencing of the Ras-related GTPase gene in lung and breast cancers. Ann Surg Oncol 14(4):1397–1404
Szulakowski P, Crowther AJ, Jimenez LA et al (2006) The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 174(1):41–50
Tehranifar P, Wu HC, McDonald JA et al (2018) Maternal cigarette smoking during pregnancy and offspring DNA methylation in midlife. Epigenetics 13(2):129–134
Terzikhan N, Verhamme KM, Hofman A, Stricker BH, Brusselle GG, Lahousse L (2016) Prevalence and incidence of COPD in smokers and non-smokers: the Rotterdam Study. Eur J Epidemiol 31(8):785–792
UniProt (2019a) UniProtKB - Q8NFU7 (TET1_HUMAN). In. https://www.uniprot.org/uniprot/Q8NFU7. Accessed Jan 4 2019
UniProt (2019b) UniProtKB - Q9UBC3 (DNM3B_HUMAN). In. https://www.uniprot.org/uniprot/Q9UBC3. Accessed Jan 4 2019
Van Pottelberge GR, Mestdagh P, Bracke KR et al (2011) MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 183(7):898–906
Wahid F, Shehzad A, Khan T, Kim YY (2010) MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta 11:1231–1243
Wan ES, Qiu W, Baccarelli A et al (2012) Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet 21(13):3073–3082
Wang G, Wang R, Strulovici-Barel Y et al (2015) Persistence of smoking-induced dysregulation of miRNA expression in the small airway epithelium despite smoking cessation. PLoS One 10(4):e0120824
Weinhold B (2006) Epigenetics: the science of change. Environ Health Perspect 114(3):A160–A167
Willinger CM, Rong J, Tanriverdi K et al (2017) MicroRNA signature of cigarette smoking and evidence for a putative causal role of microRNAs in smoking-related inflammation and target organ damage. Circ Cardiovasc Genet 10(5):e001678
Wilson R, Wahl S, Pfeiffer L et al (2017) The dynamics of smoking-related disturbed methylation: a two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genom 18(1):805
Xu Q, Ma JZ, Payne TJ, Li MD (2010) Determination of methylated CpG sites in the promoter region of catechol-O-methyltransferase (COMT) and their involvement in the etiology of tobacco smoking. Front Psychiatry 1:16
Xu W, Fang P, Zhu Z et al (2013) Cigarette smoking exposure alters pebp1 DNA methylation and protein profile involved in MAPK signaling pathway in mice testis. Biol Reprod 89(6):142
Yang IV, Schwartz DA (2011) Epigenetic control of gene expression in the lung. Am J Respir Crit Care Med 183(10):1295–1301
Yang SR, Chida AS, Bauter MR et al (2006) Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol 291(1):L46–L57
Yao H, Hwang JW, Moscat J et al (2010) Protein kinase C zeta mediates cigarette smoke/aldehyde- and lipopolysaccharide-induced lung inflammation and histone modifications. J Biol Chem 285(8):5405–5416
Zaghlool SB, Al-Shafai M, Al Muftah WA, Kumar P, Falchi M, Suhre K (2015) Association of DNA methylation with age, gender, and smoking in an Arab population. Clin Epigenet 7:6
Zhang Y, Yang R, Burwinkel B et al (2014) F2RL3 methylation in blood DNA is a strong predictor of mortality. Int J Epidemiol 43(4):1215–1225
Zheleznyakova GY, Piket E, Marabita F et al (2017) Epigenetic research in multiple sclerosis: progress, challenges, and opportunities. Physiol Genom 49(9):447–461
Zhu X, Li J, Deng S et al (2016) Genome-wide analysis of DNA methylation and cigarette smoking in a Chinese population. Environ Health Perspect 124(7):966–973
Acknowledgements
The funding has been received from FAMRI with Grant No. 123253_YCSA_Faculty; NIH with Grant No. 7 R15 ES023151 02 and SUS Foundation with Grant No. FY 2018-020.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The employment affiliation of the authors is shown on the cover page of the manuscript. The authors declare no conflict of interest. All the authors participated in the study design and interpretation of the findings. We declare that none of the authors have participated in any regulatory or legal proceedings related to the contents of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaur, G., Begum, R., Thota, S. et al. A systematic review of smoking-related epigenetic alterations. Arch Toxicol 93, 2715–2740 (2019). https://doi.org/10.1007/s00204-019-02562-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02562-y