
Abstract
In the past decade, an increasing urge to develop new and novel methods for the treatment of degenerative diseases where there is currently no effective therapy has lead to the emerging of the cell therapy or cellular therapeutics approach for the management of those conditions where organ functions are restored through transplantation of healthy and functional cells. Stem cells, because of their nature, are currently considered among the most suitable cell types for cell therapy. There are an increasing number of studies that have tested the stromal stem cell functionality both in vitro and in vivo. Consequently, stromal (mesenchymal) stem cells (MSCs) are being introduced into many clinical trials due to their ease of isolation and efficacy in treating a number of disease conditions in animal preclinical disease models. The aim of this review is to revise MSC biology, their potential translation in therapy, and the challenges facing their adaptation in clinical practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSCs) (also known as skeletal stem cells or bone marrow stromal stem cells) are plastic adherent, non-hematopoietic cells that reside in a perivascular niche in the bone marrow stroma, that possess self-renewal and multi-lineage differentiation capacity (Bianco et al. 2001, 2006, 2013). Friedenstein et al. was the first to demonstrate that within the stromal fraction of bone marrow, there exist stem cells with the ability to create heterotopic bone and bone marrow microenvironment upon in vivo transplantation in mice (Friedenstein et al. 1966). In a subsequent publication, they described “bone marrow osteogenic stem cells” as fibroblast colony-forming cells that serve as common precursors for bone and cartilage formation (Friedenstein et al. 1987). The widely used name of mesenchymal stem cell was coined by Caplan et al. to describe cells responsible for bone and cartilage formation, repair and turnover during embryonic development and adulthood (Caplan 1991). However, the accuracy of the term “mesenchymal” has been debated (Bianco et al. 2013), and alternative names for the same cell population have been proposed, e.g., skeletal stem cells (SSC) or stromal stem cells.
Isolation and definition of MSC
Traditionally, MSCs have been isolated using plastic adherence (Kassem et al. 1993). However, this method leads to growth of a heterogenous cell population with a mixture of true stem cells as well as their committed progenitors (Kuznetsov et al. 1997). Some recent studies have attempted to isolate an MSC population based on specific criteria (Houlihan et al. 2012; Mabuchi et al. 2013) including surface markers (Tormin et al. 2011). We have employed DNA microarrays to define a set of non-canonical MSC markers predictive for their in vivo bone-forming capacity and stemness (Larsen et al. 2010).
Mesenchymal stem cells are defined as plastic adherent cells, expressing a variety of surface markers, e.g., CD44, CD63, CD105, CD146, with the capacity for in vitro differentiation into osteoblast, adipocyte and chondrocyte. Based on these criteria, isolation of MSC-like cells has been reported from different tissues including adipose tissue, umbilical cord, dental pulp, skeletal muscle, synovium, periodontal ligament and even brain (Bianco et al. 2001; Harkness et al. 2010; Kermani et al. 2008; Lian et al. 2010; Mahmood et al. 2010, 2012; Orbay et al. 2012; Paul et al. 2012). However, as shown by our group, these MSC-like populations are not identical and exhibit differences in their molecular phenotype and differentiation responses (Al-Nbaheen et al. 2013). Currently, only bone marrow-derived MSCs have documented evidence of stemness including the ability to form bone and bone marrow organ upon serial transplantation in vivo (Sacchetti et al. 2007), although direct demonstration of these “stemness characteristics” of MSC-like populations, isolated from other tissues, is still needed.
Regulation of MSC differentiation
MSC has a great potential for use in cellular therapeutics targeting skeletal tissue regeneration. A prerequisite for their efficient use in therapy is to identify the molecular mechanisms controlling lineage-specific differentiation. MSC lineage specification is based on activation of lineage-specific transcription factors, e.g., Runx2, PPARγ and Sox9 for osteoblastic, adipocytic and chondrocytic lineages, respectively. The expression and activity of these transcription factors are regulated by micro-environmental conditions that include hormonal (e.g., PTH, vitamin D3, and estrogen), growth factors (e.g., BMPs, TGFβs, IGF), and mechanical forces (Cook and Genever 2013). These micro-environmental factors induce a number of intracellular signaling pathways that involve protein kinases that on activation mediate the effects of different stimuli on transcription factors. In addition, there is an increasing interest in the role of non-coding RNAs (e.g., miRNAs) and epigenetic mechanisms in regulating the expression and function of transcription factors that determine the differentiation fate of MSC. Here, we will update on the biology of the main transcription factors that regulate differentiation of MSCs into osteoblasts, adipocytes, and chondrocytes.
Osteoblast differentiation
Runt-related transcription factor-2 (Runx2, also known as Cbfa1) is the master regulator of osteogenesis which also has a role in hypertrophic cartilage formation (Ducy et al. 1997; Hinoi et al. 2006). During skeletal development, expression of Runx2 starts at sites of mesenchymal condensation and its expression is detectable throughout different stages of bone formation (Franceschi et al. 2007). In addition to transcriptional regulation, the role of Runx2 is controlled by post-translational modification (such as phosphorylation, acetylation) and through interactions with other nuclear co-activators and co-repressors (Franceschi et al. 2003; Huang et al. 2007; Wang et al. 2013; Xiao et al. 2000). Runx2 has been shown to be necessary and sufficient to commit mesoderm-type cells into the osteogenic lineage (Franceschi et al. 2007; Marie 2008). In vivo overexpression of Runx2 in chondrocytes leads to skeletal malformation, due to ossification of permanent cartilage (Ueta et al. 2001). Mouse fetuses with loss of Runx2 function lack calcified bones and die at birth, due to respiratory failure (Franceschi et al. 2007). Haploinsufficiency of Runx2 in humans leads to the human disease of cleidocranial dysplasia characterized by hypoplastic clavicles, open cranial fontanels, and decreased bone mass (Huang et al. 2007). The Runx2 consensus sequence (PuACCPuCA) is present in gene promoters of the majority of osteoblastic genes such as osteopontin (OPN), bone sialoprotein (BSP), type 1 collagen alpha 1 chain (Col1a1), and osteocalcin (OC) and thus acts as an activator of the osteoblast differentiation program (Marie 2008). In addition, Runx2 plays a vital role in regulating osteoblast proliferation and survival, through regulation of cell cycle and PI3 K-Akt signaling (Fujita et al. 2004; Pratap et al. 2003).
Expression of Runx2 is regulated by several transcription factors, such as beta-catenin (β-catenin), msh homeobox 2 (Msx2), and distal-less homeobox 5 (Dlx5). (Huang et al. 2007). Recently, small heterodimer partner-interacting leucine zipper protein (SMILE), an orphan nuclear receptor, has been reported to physically interact with and to negatively regulate Runx2 transcriptional activity (Jang et al. 2014). The physical interaction between Runx2 and the glucocorticoid receptor leads to inhibition of Runx2 function and impaired osteogenesis which is one of the possible mechanisms through which prolonged glucocorticoid treatment induces decreased bone formation (Koromila et al. 2014). ESET, a histone methyltransferase, has been shown to interact with Runx2 and negatively regulate its transcriptional activity (Lawson et al. 2013); jumonji domain-containing 3 (Jmjd3), a histone demethylase which specifically catalyzes the removal of trimethylation of histone H3 at lysine 27 (H3K27me3), is necessary for promoter activities of Runx2 and Osterix (Yang et al. 2013); and p300/CBP-associated factor (PCAF) directly binds to and acetylates Runx2, leading to an increased in its transcriptional activity and enhanced osteogenesis (Wang et al. 2013). Interestingly, Runx2 is target for several miRNAs (miRs). miRNA-15b promotes osteoblast differentiation by targeting Smurf1 and protecting Runx2 from Smurf1-mediated proteasomal degradation (Vimalraj et al. 2014). Loss of function of miRNA-17-92 cluster is associated with impaired bone formation and reduced expression of Runx2 in bones in miR-17-92 +/Δ mice (Zhou et al. 2013). miR-3077-5p has been shown to be responsible for the reduced level of Runx2 protein and therefore reduced osteoblast differentiation of MSC isolated from osteoporotic patients (Liao et al. 2013).
Osterix (Osx) is another major transcription factor that regulates osteogenesis. Osx is also known as specificity protein 7 (Sp7). It is a zinc-finger osteoblast-specific transcription factor. Osterix induces the promoter activity of osteoblast differentiation genes such as OC, Col1a1, OPN, and ALP (Huang et al. 2007; Koga et al. 2005). The presence of the Runx2 responsive element in the Osx promoter (Nishio et al. 2006), and normal expression of Runx2 in Osx-deficient mice that exhibit severely defective bone formation (Nakashima et al. 2002), indicates that Osx functions downstream of Runx2. Expression of chondrocyte markers (e.g., Sox9 and Col2a1) by osteoprogenitor cells of Osx-deficient mice suggests that Runx2-expressing osteoprogenitor cells have the potential to differentiate into either osteoblasts or chondrocytes and that Osx functions downstream of Runx2 to induce the bipotential osteo-chondro-progenitors to differentiate toward the osteoblastic lineage (Nakashima et al. 2002).
The function of Osterix is modulated by post-translational modifications such as phosphorylation and ubiquitination (Li et al. 2013a; Ortuno et al. 2010; Peng et al. 2013). Phosphorylation of Osx by Erk1/2 and p38 MAP kinases, and Akt increase its transcriptional activity (Choi et al. 2011b, c; Ortuno et al. 2010). In addition, glycogen synthase kinase 3 alpha (GSK3α) and calmodulin-dependent kinase II (CaMKII) enhance transcriptional activity, protein levels, and protein stability of Osx (Choi et al. 2013; Li et al. 2013a). In addition, interaction of Osx with other transcription factors such as NFTAc, TFII, p300, and Brg1 promotes Osx activity (Sinha and Zhou 2013). Similar to Runx2, miRNAs regulate Osx expression. Expression of Osx has been shown to be negatively regulated by miR-93, 125, 135, 138, 143, 145, 214, 322, and 637 (Eskildsen et al. 2011; Gamez et al. 2013; Goettsch et al. 2011; Jia et al. 2013; Li et al. 2014; Schaap-Oziemlak et al. 2010; Shi et al. 2013; Yang et al. 2012a; Zhang et al. 2011a). Moreover, epigenetic regulation of Osx transcription by histone demethylases Jmjd3 and NO66 has recently been reported (Sinha et al. 2013; Yang et al. 2013). Finally, it has been shown that Osx binding sites are present in the promoter region of Osx and auto-regulation is a major mechanism by which expression of Osx is controlled (Barbuto and Mitchell 2013).
Other osteoblast-associated transcriptional factors
Activator protein-1 (AP-1), β-catenin, activating transcription factor 4 (ATF4), and members of Msx/Dlx family are transcription factors that have role in regulation of osteoblast differentiation and bone formation, but their expression in not limited to skeletal tissue (Cook and Genever 2013; Marie 2008).
Adipocyte differentiation
Peroxisome proliferator-activated receptor-γ (PPARγ) is known as master regulator of adipogenesis. It is a nuclear hormone receptor transcriptional factor, which is sufficient and indispensable for adipogenic differentiation of MSC (Nuttall et al. 2014; Tontonoz et al. 1994). In vitro treatment of MSC with thiazolidinediones (TZD), which are ligand agonists of PPARγ, leads to enhanced adipogenesis and inhibition of osteoblastogenesis of MSC (Gimble et al. 1996). In vivo studies involving chronic exposure of rodents to TZDs demonstrated an increase in bone marrow fat content and decreased bone mass upon treatment with several but not all thiazolidinediones (Lazarenko et al. 2007; Tornvig et al. 2001).
Among the regulators of PPARγ that are relevant to MSC biology is the canonical Wnt-β-catenin pathway which inhibits the mRNA expression of PPARγ. Non-canonical Wnt signaling activates histone methyl-transferase SETDB1 that represses PPARγ transactivation through methylation of histone H3K9 of the target genes (Takada et al. 2009b). TNF-α- or IL-1-induced TAK1/TAB 1/NIK signaling cascade decreases PPARγ-mediated adipogenesis by inhibiting the binding of PPARγ to the DNA response element (Takada et al. 2009a). Nocturnin (NOC), which is a nutrient-responsive gene, binds to PPARγ and increases its nuclear translocation and transcriptional activity, thereby enhancing adipogenesis (Kawai et al. 2010). Snail, a transcription factor from the zinc-finger family, inhibits the transcriptional activity of the PPARγ gene by directly binding to the E-box motifs in the PPARγ promoter (Lee et al. 2013). Sterol regulatory binding element protein-1 (SREBP1) is a transcription factor that regulates adipocyte differentiation and cholesterol homeostasis. SREBP1 positively regulates the expression of PPARγ through interaction with E-box domains in the PPARγ promoter (Fajas et al. 1999). Lipin 1, a co-regulator of transcription factors that also has phosphatidate phosphatase activity, functions as a key regulator of PPARγ activity through its ability to release co-repressors and recruit co-activators (Kim et al. 2013). Both GATA2 and GATA3 negatively regulate adipogenesis through direct binding to PPARγ (Tong et al. 2000). In addition, a number of miRNAs such as miRNA-130b and miR-20a have been shown to negatively regulate adipogenesis by targeting PPARγ (Pan et al. 2013; Zhang et al. 2011b).
CAAT/enhancer binding protein-α (C/EBPα) is another key transcription factor that is involved in regulation of adipogenesis (Samuelsson et al. 1991). Overexpression of C/EBPα induces adipogenesis in fibroblasts, and loss of C/EBPα function inhibits adipogenesis (Freytag et al. 1994; Lin and Lane 1992). C/EBPs expression is regulated by a positive feedback loop that includes PPARγ expression (Park et al. 2012). There are C/EBP binding sites within the promoter of PPARγ, and expression of PPARγ is thought to activate C/EBPα (Park et al. 2012). C/EBP homologous proteins (CHOPs) negatively regulate adipogenesis through interactions with C/EBPs (Tang and Lane 2000). In addition, the negative regulatory role of GATA2 and 3 on adipogenesis is partly mediated through formation of protein complexes with C/EBPα or β (Tong et al. 2005). It has recently been demonstrated that post-translational modification and epigenetic mechanisms have a role in the regulation of C/EBPα expression and function (Borengasser et al. 2013; Li et al. 2013b; Pal et al. 2013). E6AP, an E3 ubiquitin ligase, inhibits adipogenesis through ubiquitination of C/EBPα and targets it to ubiquitin–proteasome pathways for degradation (Pal et al. 2013). In addition, increased propensity for adipogenesis in the male offspring of the overfeeding-induced obese rats is associated with increased in vivo expression of adipogenic regulators such as C/EBPα and alterations in DNA methylation of CpG sites and CGI shores of developmentally important genes, including key pro-adipogenic factors (Borengasser et al. 2013). Moreover, dexamethasone-induced osteoporosis characterized by decreased bone formation and increased marrow fat is associated with inhibition of C/EBPα promoter methylation leading to enhanced expression of C/EBPα and adipogenic differentiation of MSC (Li et al. 2013b).
Chondrocyte differentiation
SRY-box containing gene 9 (Sox9), a high-mobility-group (HMG) box containing transcription factor, is known as the master regulator of chondrogenesis. Sox9 activates the expression of chondrocyte-specific genes such as Col2a1 and Agc1 and direct concomitant positive and negative transcriptional control by SOX9 ensures differentiation phase-specific gene expression in chondrocytes (Cook and Genever 2013; Leung et al. 2011; Yamashita et al. 2012). Moreover, Sox5 and Sox6 act in redundancy with each other to robustly enhance the functions of Sox9 (Lefebvre et al. 1998). Regulation of chondrogenesis, chondrocyte proliferation, and transition to a non-mitotic hypertrophic state by Sox9 is required for development of cartilage and endochondral bone (Leung et al. 2011). Heterozygous mutations in SOX9 cause campomelic dysplasia, a severe skeletal dysmorphology syndrome in humans characterized by a generalized hypoplasia of endochondral bones (Oh et al. 2010).
Expression and function of Sox9 are regulated through recruitment of diverse transcriptional co-activators, histone-modifying enzymes, subunits of the mediator complex, and components of the general transcriptional machinery (e.g., Med12, Med25, and CBP/p300) to the transactivation domain of Sox9 (Akiyama and Lefebvre 2011). AT-rich interactive domain 5b (Arid5b), a transcriptional co-regulator of Sox9, physically interacts with Sox9 and synergistically induces chondrogenesis by facilitating the Phf2-mediated histone demethylation of Sox9-regulated chondrogenic gene promoters (Hata et al. 2013). Suppression of Sox9 transcriptional activity by Twist1 is the mechanism by which canonical Wnt signaling inhibits chondrogenesis (Gu et al. 2012). Notch signaling negatively regulates chondrogenesis by repressing Sox9 transcription through recruitment of the Rbpj/NICD transcription complex to the Rbpj-binding sites upstream of the Sox9 promoter (Chen et al. 2013). miR-145 has been shown to be a direct regulator of SOX9 in normal healthy human articular chondrocytes (Martinez-Sanchez et al. 2012). miR-101 has role in IL-1β-induced chondrocyte ECM degradation by targeting 3′UTR of Sox9 (Dai et al. 2012).
New source for MSC: generation of MSC-like cells from human pluripotent cells
The use of bone marrow-derived MSCs in therapeutic applications has been hampered by the limited ability to obtain a sufficient number of cells as the cells undergo replicative senescence during ex vivo culture expansion (Kassem and Marie 2011; Stenderup et al. 2003). Thus, alternative sources for generating MSC-like cells with increased proliferation potential have been studied. One of the most promising cell types are pluripotent stem cells (PSCs) either from embryonic (ESC) or induced (iPSCs) sources. These cells have an unlimited proliferation capability and ability to differentiate into all cells of the body including MSC-like cells (Harkness et al. 2011; Tremoleda et al. 2008). Differentiation of human PSC toward MSC-like cells has been performed through a number of different methods including recapitulation of gastrulation-like stages via embryoid body formation (EB) (Sottile et al. 2003; Tremoleda et al. 2008); direct addition of morphogens to PSC culture media (Boyd et al. 2009; Evseenko et al. 2010); co-culture of PSC with osteoprogenitors such as OP9 cells (Barberi et al. 2005; de Peppo et al. 2010; Inanc et al. 2007); or isolation of cells spontaneously differentiated at the edges of feeder-free colonies where an epithelial-to-mesenchymal transition (EMT) takes place (Harkness et al. 2011; Olivier and Bouhassira 2011; Trivedi and Hematti 2008). The most commonly employed methods for differentiation are via EB formation combined with addition of growth factors during culture (Mahmood et al. 2010; Schuldiner et al. 2000) or addition of growth factors and morphogens directly to PSC monolayer cultures. The later method may lack the 3-D structure and microenvironment provided by EB formation (Matsumoto et al. 2011). When PSCs are induced into MSC-like cells through co-culture with differentiated osteoblastic cells, the differentiated cells provide selective micro-environmental cues conducive for lineage specification (Fengming Yue et al. 2013).
Each of the methods mentioned has been reported in a number of publications (see (Abdallah Basem et al. 2011) for review). However, during the initial differentiation period, most methods (excluding cells undergoing EMT) demonstrate a degree of cellular heterogeneity. Repeated passaging (de Peppo et al. 2010; Karp et al. 2006), cell sorting (Brown et al. 2009; Lian et al. 2007), or selective isolation methods based on adhesion to specific extracellular matrix components (Harkness et al. 2011; Liu et al. 2012) have all been used to achieve a more homogeneous populations with MSC characteristics (Harkness et al. 2011). Nevertheless, the functional ability for these cells to regenerate bone and cartilage (in preclinical animal models) needs to be fully determined.
From basic biology to clinical applications
Stromal stem cells therapy
Cellular therapy is an emerging field in clinical medicine aimed at using cells (and in particular stem cells) for treatment of chronic and degenerative diseases. As can be seen in Table 1, several cell types have been suggested in clinical applications based on their phenotype and functionality. Bone marrow-derived MSCs are among the most suitable candidates for cellular therapeutics because of their ease of isolation, differentiation potential into skeletal tissues, and their excellent safety record (Lepperdinger et al. 2008) as well as their immunomodulatory and regeneration promoting properties (Nauta and Fibbe 2007; Zhao et al. 2010). MSCs have been employed in an increasing number of clinical studies for enhancing tissue regeneration following injury of both skeletal damage, e.g., bone (Gangji and Hauzeur 2005; Le Blanc et al. 2005), cartilage (Wakitani et al. 2007), and non-skeletal diseases, e.g., type I diabetes mellitus (Bhansali et al. 2009; Estrada et al. 2008), Crohn’s diseases (Duijvestein et al. 2010; Liang et al. 2012), and following myocardial infarction (Chen et al. 2004; Hare et al. 2009).
The clinical use of MSC in therapy has employed both local and systemic injections. Systemic infusion of MSC for tissue repair is a clinically attractive approach and is similar to route used for hematopoietic stem cell transplantation. However, the mechanisms that govern migration of MSCs to injured tissues are still poorly understood (Karp and Leng Teo 2009). A limited degree of MSC homing to damaged tissues has been described in many preclinical studies using animal models of brain injury (Ji et al. 2004), skeletal disorders (Devine et al. 2001; Shi et al. 2007), and acute radiation syndrome (Lange et al. 2011; Yang et al. 2012b). Although human MSCs do express several chemokine receptors and adhesion molecules (Sordi et al. 2005; Wu and Zhao 2012) known to mediate homing of leukocytes to inflamed tissues (Mohle et al. 1998; Quesenberry and Becker 1998), their precise role in MSCs homing is still under investigation.
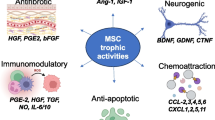
Due to the limited homing capacity of MSC to injured tissues, the positive initial clinical effects of MSC therapy are thought to be due to “humoral” factors secreted by MSC that enhance tissue regeneration. MSCs are known to secrete a plethora of autocrine and paracrine chemokines and growth factors (TGFB, TSG6, PGE2) that stimulate endogenous/resident cells, exhibit anti-apoptotic and immuno-modulatory effects as well as enhance vasculogenesis (Gnecchi et al. 2008; Mirotsou et al. 2011). While it was reported that systemically injected MSC may get entrapped in the lungs (Bentzon et al. 2005; Schrepfer et al. 2007), their paracrine effects ensure that MSCs still exercise a positive influence through secretion of factors that exert favorable actions on distant, damaged tissues (Choi et al. 2011a). Currently, more than 3,800 stem cell-based clinical trials are registered worldwide with the NIH (USA) clinical trials database (USA 62.4 %, Europe 20.15 %, China 7.5 %, Canada 4.3 %) (Database UNIoHNctrar In. 2014), and the initial results of many Phase I or Phase I-II trials are encouraging.
Examples of the use of MSC in clinical therapy
Skeletal tissue regeneration
Regeneration of bone tissue is needed in a growing number of skeletal diseases, e.g., local non-union bone defects following tumor removal or complicated fractures. Transplantation of stem cells that are capable of bone generation in vivo is therefore an attractive and alternative approach to bone autograft or allograft techniques.
The efficacy of use of BM MSC for repair of bone defects or complicated fractures has been tested in animal models and in some phase I/II clinical trials. Bone marrow MSCs over-expressing VEGF and BMP2 were systemically administered in mice with surgically induced tibial bone defects. In mice injected with overexpressing cells, enhanced bone formation was observed and was associated with enhanced tissue vascularity at fracture site when compared with controls (Kumar et al. 2010). Similarly, murine bone marrow-derived MSCs overexpressing Osx were implanted in mice calvarial critical size bone defects and resulted in efficient healing (Tu et al. 2007).
In the past decade, clinical studies have employed a variety of cell types, most commonly bone marrow-derived mononuclear cells (MNC) that contain MSC in addition to other hematopoietic cells. Hernigou et al. (2005) have demonstrated that injection of an autologous bone marrow aspirate-derived MNC into the site of bone non-union fractures in 60 patients did result in bone union in the 53 (88.3 %) of treated individuals. In the seven patients that exhibited failure of bone union, a low CFU-F (fibroblastic colony-forming unit) count was observed (which a surrogate measure of the number of MSC in the injected cells), suggesting a role of MSC and progenitor cell numbers in determining the outcome of cell therapy (Hernigou et al. 2005). In a small case series, autologous bone marrow MSCs were extracted and cultured in platelet rich plasma (PRP) followed by transplantation to sites of bone defects in individuals with achondroplasia or limb hypoplasia undergoing distraction osteogenesis for limb lengthening. Healing was observed in the treated patients with new bone formation during femoral lengthening as a consequence of the cell transplant in these patients (Kitoh et al. 2004). Also, promising preliminary results for treatment of femoral head osteonecrosis have been reported (Gangji and Hauzeur 2005; Kawate et al. 2006).
Osteoarthritis is common degenerative joint disease and among the most frequent causes of joint pain and disability. In a recent pilot study, twelve osteoarthritic patients with chronic knee pain were treated with autologous bone marrow MSCs. Culture expanded MSCs (40 × 106 cells) were locally administrated by intra-articular injection after which patients exhibited rapid and progressive improvement of functional recovery of the joint function with improvement of cartilage quality in most of the patients (Orozco et al. 2013). In another study, the effects of local injection with either autologous BM MSC or cultured chondrocytes on disease progression were evaluated in 72 patients suffering from osteoarthritis (OA). Patients in both groups showed significant improvement in “quality of life,” but no differences could be observed between both groups (Nejadnik et al. 2010).
Myocardial regeneration
The myocardium has a limited capacity for regeneration, thus, following myocardial infarction; myocardial repair is carried out by formation of scar tissue that has negative effects on the myocardial contractility and function. Cardiovascular diseases caused by such impairment of myocardium functions leading to hear failure are among the major causes of mortality worldwide (Fuster et al. 2011). MSCs have been tested for their ability to enhance myocardial regeneration following acute myocardial infarction (AMI) or chronic ischemic heart failure (CHF). Results from studies where undifferentiated MSCs were injected with aim of regenerating the myocardium have demonstrated that engraftment and/or differentiation of the injected cells into newly generated cardiomyocytes is very limited or nonexistent despite observed beneficial effects (Noiseux et al. 2006; Perez-Ilzarbe et al. 2008). Thus, it is considered that the beneficial effects of MSC in cardiac regeneration are mediated by “humoral” factors secreted by MSC that enhance tissue regeneration (Mirotsou et al. 2011) or stimulate and activate resident cardiac stem cells (CSCs) (Hatzistergos et al. 2010).
Modification of MSC to enhance production of cytokines or growth factors, known to enhance myocardial regeneration, has also been tested. For example, rat MSCs overexpressing IGF 1 were locally injected in a rat model of acute myocardial infarction. The injected cells led to increased local production of stromal derived factor-1 (SDF-1), reduction in infarct size, and increased ejection fraction (Haider et al. 2008). Chemokine receptor type 1 (CCR1), a member of the chemokine family, was overexpressed in mouse bone marrow MSC and injected locally in a mouse model of acute myocardial infarction and led to reduced apoptosis, increased vascularity, restoration of cardiac function, and reduction in the infarct size (Huang et al. 2010). In order to enhance survival of transplanted stem cells in the myocardium, overexpression of survival-enhancing factors, e.g., VEGF (Tao et al. 2011) and Akt (Shiojima and Walsh 2006), was tried and resulted in improved survival of injected rat MSC within the tissue. When transplanted in a rat model of AMI, a significant reduction in infarct size and improved left ventricular function were observed (Mangi et al. 2003; Shujia et al. 2008). In another study, rat MSCs overexpressing survival protein B cell lymphoma 2 (Bcl-2) were injected locally (intra-cardiac) into a rat model of AMI. The genetically modified cells exhibited long-term survival at the infarction site and resulted in 17 % reduction in infarct size (Li et al. 2007).
In the past decade, many clinical trials utilizing a number of bone marrow-derived cell preparations (including bone marrow-derived MNC) have been conducted (reviewed in (Jeevanantham et al. 2012; Zimmet et al. 2012). A non-randomized study evaluating the effects of repeated intracoronary BMSC infusions in 32 patients with CHF (LV ejection fraction less than 40 %) demonstrated encouraging results. These patients received BMSC infusion at baseline and after four months. Follow-up consisted of serial echocardiograms (four, eight, and twelve months) after the first intervention, measurements of the ratio of transmitral flow (E) velocity to early mitral annulus (e’) velocity (E/e’), left atrial (LA) volume, and plasma levels of N-terminal pro-brain natriuretic peptide (NT-pro-BNP). During the initial treatment phase, there were no changes in main outcome but after treatment with intracoronary BMSC, a significant decrease was observed in E/e’ ratio, LA volume, and plasma NT-pro-BNP. The effect was greatest in patients who received the largest amount of CD34 (+) cells (Diederichsen et al. 2010). A placebo-controlled clinical study of intra-coronary injection of autologous MSC within twelve hours after the onset of acute myocardial infarction was conducted in 69 patients. No side effects or toxicity were reported during the six month follow-up. Positive effects of increased left ventricular ejection fraction and left ventricular end diastolic volume that improve contractility and enhance infarct viability were reported (Chen et al. 2004). Hera et al. (2009) performed a double-blind, placebo-controlled, dose-ranging (0.5, 1.6 or 5 × 106 cells\kg) safety trial of intravenous allogeneic MSC’s in 53 patients with anterior myocardial infarction. Global symptom score and ejection fraction (an estimated of left ventricular function) were significantly improved in MSC-treated group compared with controls. In another study, 33 patients with dilated cardiomyopathy underwent intracoronary infusion of BMC using balloon catheter. After 3 months of cell administration, regional wall motion of the affected myocardium and global left ventricular ejection fraction were improved. The authors reported that the increase in regional contractile function was directly related to the functionality of the infused cells as measured by their colony-forming capacity (Fischer-Rasokat et al. 2009).
Graft versus host disease
Graft versus host disease (GvhD) is a potentially fatal disease that develops as a consequence of allogenic hematopoietic stem cell transplantation. Human bone marrow derived and adipose tissue-derived MSCs (Fang et al. 2007) were tried out for treatment of GvhD with success based on their immunoregulatory characteristics as mentioned above. In a recent study, nineteen patients suffering from chronic GVHD were treated with MSCs (0.6 × 106 cells/kg). Fourteen (74 %) of these patients demonstrated partial or complete responses and five patients (25 %) discontinued immunosuppressive agents (Weng et al. 2010). The 2-year survival rate was 77.7 % in this study. Clinical improvement was accompanied by the increasing ratio of CD5+CD19+/CD5−CD19+ B cells and CD8+CD28−/CD8+CD28+ T cells. No patients reported side effects from the MSC therapy (Weng et al. 2010). The beneficial effects of MSC were also observed in a phase II clinical trial of 55 children and adult patients with acute severe and steroid resistant GvhD. Intravenous infusion of autologous MSC was safe and resulted in higher survival rates in patients with complete response and significantly lower transplantation-related mortality (Le et al. 2008). In another study, MSC was employed to treat nine patients (eight patients with steroid refractory acute GVHD and one patient with chronic GVHD). MSCs obtained from either identical siblings, haploidentical donors, and HLA-mismatched donors were systematically injected and caused clinical recovery in six out of the eight patients (Ringden et al. 2006). In a recent randomized clinical trial of 32 patients with grade II-IV GvhD that either received intravenous autologous MSC (2 or 8 × 106 cells/kg) or standard therapy, 77 % of patients that received MSC transplantation exhibited complete response and no MSC infusion-related toxicities were observed (Kebriaei et al. 2009).
MSC as therapeutic vehicle
The ability and ease of genetic modification of MSCs have encouraged their use as a vehicle for gene transfer and/or secretion of ectopic proteins. Potential transplantation of modified MSC as vehicles for secretion of therapeutic factors has been suggested in a number of studies (Porada and Almeida-Porada 2010; Sarkar et al. 2010). In a recent study, mRNA transfection was utilized to generate MSCs that simultaneously expressed P-selectin glycoprotein ligand-1 (PSGL-1), Sialyl-Lewisx (SLeX)) and secreted interleukin-10 (IL-10). Using membrane dyes, these cells were tracked in vivo following systemic injection, and a rapid homing of MSCs to the site of inflammation was observed with a higher anti-inflammatory effect which significantly decreased local inflammation (Levy et al. 2013).
Heile et al. (2009) tested the efficacy of human bone marrow-derived MSC transduced with the human telomerase reverse transcriptase gene (hTERT) (hMSC-TERT) (Simonsen et al. 2002) overexpressing GLP-1 (glucaogon-like peptide 1), a protein known to enhance neuronal tissue regeneration, in a rat brain injury model. By assessment of MAP-2 and GFAP expression, implanted hMSC-TERT-GLP1 cells resulted in reduction in hippocampal cell loss as well as reduction in cortical and glial defects (Heile et al. 2009; Klinge et al. 2011). Based on these promising results, a phase I trial in patients with cerebral hemorrhage that required surgery was initiated where encapsulated GLP-1-overexpressing hMSC-TERT cells were transplanted within a retrievable mesh device (described as “tea-bag” approach) into the brain following removal of bleeding. Following a treatment period of 14 days, the “tea-bag” was removed (http://www.biocompatibles.com/media/press-releases/first-ever-treatment-of-stroke-patient-with-stem-cell-therapy-product). The feasibility and safety of this approach have been documented in this trial, and thus, the efficacy of treatment needs to be determined.
Concerns regarding use of stem cells in therapy
The safety record of human MSC is excellent, and during ex vivo culture expansion, the cells exhibit a stable phenotype with no risk of spontaneous transformation (Stenderup et al. 2003; Wang et al. 2012). While PSCs are an attractive source for generating a large number of phenotypically stable cells suitable for therapy, there are a number of safety aspects relating to the use of human PSC and their derivatives in clinical applications that also apply to MSC. One of these is immune rejection. While autologous transplantation from MSC remains the safest method, allogeneic MSC transplantation is also possible since MSC exhibit immune suppressive properties (De Miguel et al. 2012; Le Blanc and Ringden 2007). The recent development of patient specific iPSC allows the generation of cells compatible with the donor; and thus, generation and expansion prior to transplantation become less of an issue.
Another important concern is the risk for malignant transformation of the transplanted stem cells. As mentioned above, MSCs do not transform during in vitro culture and no history of development of cancer has been reported following administration of MSC in patients. Conversely, PSCs have a propensity for forming tumors when implanted as undifferentiated cells in vivo and thus the contamination of differentiated PSC with undifferentiated cells may pose a risk (Miura et al. 2009). However, it is currently unknown how many (or few) undifferentiated PSC would be acceptable within cells for clinical transplantation. Hentze et al. (2009) reported a detection limit of 1:4,000 for tumor formation when injecting single cells into immune compromised mice. However, the current ability to form tumors in vivo depends more on the strain of mice used (degree of immunocompromisation) (Quintana et al. 2008) than other factors such as site of injection (Cunningham et al. 2012), and thus, this method may not be predictive for the behavior of the transplanted cells in vivo.
Development of assays that screen stem cells for their safety prior to their transplantation is being developed. Demonstration of genetic alterations in cultured cell suggests a malignant transformation potential (Bentivegna et al. 2013; Nouspikel 2013). Two routine procedures have been utilized to assess chromosomal and genetic abnormalities: karyotyping and identification of gross morphological changes (such as acquisition, deletion, or inversions) using G-banded karyotyping or the creation of a virtual karyotype using single nucleotide polymorphism (SNP) or comparative genomic hybridization (CGH), (Hagenkord et al. 2008). We have recently demonstrated the possible use of noninvasive Raman spectroscopy (Harkness et al. 2012) as a method for detecting transformed cells among cultured cells.
Several techniques have been proposed to eliminate undifferentiated PSC prior to transplantation. These include selective apoptosis of PSC (Bieberich et al. 2004), removal of PSC through flow cytometry (Schriebl et al. 2012) or through mechanical removal (Tang et al. 2012). It would be more efficient if the differentiation protocols utilized were robust enough to induce a homogeneous cell type without pluripotent cells remaining.
References
Abdallah Basem M, Harkness L, Mahmood A, Kassem M (2011) Direct differentiation of human embryonic stem cells toward osteoblasts and chondrocytes through an intermediate mesenchyme progenitor lineage. In: Atwood C (ed) Embryonic stem cells: the hormonal regulation of pluripotency and embryogenesis, InTech Europe, Rijeka, Croatia, p 607–618. http://www.intechopen.com
Akiyama H, Lefebvre V (2011) Unraveling the transcriptional regulatory machinery in chondrogenesis. J Bone Miner Metab 29(4):390–395. doi:10.1007/s00774-011-0273-9
Al-Nbaheen M, Vishnubalaji R, Ali D et al (2013) Human stromal (mesenchymal) stem cells from bone marrow, adipose tissue and skin exhibit differences in molecular phenotype and differentiation potential. Stem Cell Rev 9(1):32–43. doi:10.1007/s12015-012-9365-8
Barberi T, Willis LM, Socci ND, Studer L (2005) Derivation of multipotent mesenchymal precursors from human embryonic stem cells. PLoS Med 2(6):e161
Barbuto R, Mitchell J (2013) Regulation of the osterix (Osx, Sp7) promoter by osterix and its inhibition by parathyroid hormone. J Mol Endocrinol 51(1):99–108. doi:10.1530/JME-12-0251
Bentivegna AMM, Riva G, Foudah D, Butta V, Dalprà L, Tredici G (2013) DNA methylation changes during in vitro propagation of human mesenchymal stem cells: implications for their genomic stability? Stem Cells Int 2013:192425. doi:10.1155/2013/192425
Bentzon JF, Stenderup K, Hansen FD et al (2005) Tissue distribution and engraftment of human mesenchymal stem cells immortalized by human telomerase reverse transcriptase gene. Biochem Biophys Res Commun 330(3):633–640. doi:10.1016/j.bbrc.2005.03.072
Bhansali A, Upreti V, Khandelwal N et al (2009) Efficacy of autologous bone marrow-derived stem cell transplantation in patients with type 2 diabetes mellitus. Stem Cells Dev 18(10):1407–1416. doi:10.1089/scd.2009.0164
Bianco P, Riminucci M, Gronthos S, Robey PG (2001) Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells 19(3):180–192
Bianco P, Kuznetsov SA, Riminucci M, Gehron RP (2006) Postnatal skeletal stem cells. Methods Enzymol 419:117–148
Bianco P, Cao X, Frenette PS et al (2013) The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med 19(1):35–42. doi:10.1038/nm.3028
Bieberich E, Silva J, Wang G, Krishnamurthy K, Condie BG (2004) Selective apoptosis of pluripotent mouse and human stem cells by novel ceramide analogues prevents teratoma formation and enriches for neural precursors in ES cell-derived neural transplants. J Cell Biol 167(4):723–734
Borengasser SJ, Zhong Y, Kang P et al (2013) Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology 154(11):4113–4125. doi:10.1210/en.2012-2255
Boyd NL, Robbins KR, Dhara SK, West FD, Stice SL (2009) Human embryonic stem cell-derived mesoderm-like epithelium transitions to mesenchymal progenitor cells. Tissue Eng Part A 15(8):1897–1907
Brown SE, Tong W, Krebsbach PH (2009) The derivation of mesenchymal stem cells from human embryonic stem cells. Cells Tissues Organs 189(1–4):256–260
Caplan AI (1991) Mesenchymal stem cells. J Orthop Res Off Publ Orthop Res Soc 9(5):641–650. doi:10.1002/jor.1100090504
Chen SL, Fang WW, Ye F et al (2004) Effect on left ventricular function of intracoronary transplantation of autologous bone marrow mesenchymal stem cell in patients with acute myocardial infarction. Am J Cardiol 94(1):92–95
Chen S, Tao J, Bae Y et al (2013) Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. J Bone Miner Res Off J Am Soc Bone Miner Res 28(3):649–659. doi:10.1002/jbmr.1770
Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ (2011a) Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-kappaB signaling in resident macrophages. Blood 118(2):330–338. doi:10.1182/blood-2010-12-327353
Choi YH, Gu YM, Oh JW, Lee KY (2011b) Osterix is regulated by Erk1/2 during osteoblast differentiation. Biochem Biophy Res Commun 415(3):472–478. doi:10.1016/j.bbrc.2011.10.097
Choi YH, Jeong HM, Jin YH, Li H, Yeo CY, Lee KY (2011c) Akt phosphorylates and regulates the osteogenic activity of Osterix. Biochem Biophys Res Commun 411(3):637–641. doi:10.1016/j.bbrc.2011.07.009
Choi YH, Choi JH, Oh JW, Lee KY (2013) Calmodulin-dependent kinase II regulates osteoblast differentiation through regulation of Osterix. Biochem Biophys Res Commun 432(2):248–255. doi:10.1016/j.bbrc.2013.02.005
Cook D, Genever P (2013) Regulation of mesenchymal stem cell differentiation. Adv Exp Med Biol 786:213–229. doi:10.1007/978-94-007-6621-1_12
Cunningham JJ, Ulbright TM, Pera MF, Looijenga LHJ (2012) Lessons from human teratomas to guide development of safe stem cell therapies. Nat Biotech 30(9):849–857
Dai L, Zhang X, Hu X, Zhou C, Ao Y (2012) Silencing of microRNA-101 prevents IL-1beta-induced extracellular matrix degradation in chondrocytes. Arthritis Res Ther 14(6):R268. doi:10.1186/ar4114
Database UNIoHNctrar In. (2014) http://clinicaltrials.gov/ Accessed 6 February 2014
De Miguel MP, F-JS Blázquez-Martínez A, Pascual CY, Aller MA, Arias J, Arnalich-Montiel F (2012) Immunosuppressive properties of mesenchymal stem cells: advances and applications. Curr Mol Med 12(5):17. doi:10.2174/156652412800619950
de Peppo GM, Sjovall P, Lennerås M, et al (2010) osteogenic potential of human mesenchymal stem cells and human embryonic stem cell-derived mesodermal progenitors: a tissue engineering perspective. Tissue Eng Part A 16(11). doi: 10.1089/ten.tea.2010.0052
Devine SM, Bartholomew AM, Mahmud N et al (2001) Mesenchymal stem cells are capable of homing to the bone marrow of non-human primates following systemic infusion. Exp Hematol 29(2):244–255
Diederichsen AC, Moller JE, Thayssen P et al (2010) Changes in left ventricular filling patterns after repeated injection of autologous bone marrow cells in heart failure patients. Scand Cardiovasc J 44(3):139–145. doi:10.3109/14017430903556294
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89(5):747–754
Duijvestein M, Vos AC, Roelofs H et al (2010) Autologous bone marrow-derived mesenchymal stromal cell treatment for refractory luminal Crohn’s disease: results of a phase I study. Gut 59(12):1662–1669. doi:10.1136/gut.2010.215152
Eskildsen T, Taipaleenmaki H, Stenvang J et al (2011) MicroRNA-138 regulates osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. Proc Natl Acad Sci USA 108(15):6139–6144. doi:10.1073/pnas.1016758108
Estrada EJ, Valacchi F, Nicora E et al (2008) Combined treatment of intrapancreatic autologous bone marrow stem cells and hyperbaric oxygen in type 2 diabetes mellitus. Cell Transpl 17(12):1295–1304
Evseenko D, Zhu Y, Schenke-Layland K et al (2010) Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc Natl Acad Sci 107(31):13742–13747
Fajas L, Schoonjans K, Gelman L et al (1999) Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism. Mol Cell Biol 19(8):5495–5503
Fang B, Song Y, Lin Q et al (2007) Human adipose tissue-derived mesenchymal stromal cells as salvage therapy for treatment of severe refractory acute graft-vs.-host disease in two children. Pediatr Transpl 11(7):814–817
Fengming Yue SS, Ichikawa H, Yoshie S, Akimi Mogi SM, Nagai M, Yokohama T, Sasaki TDaK (2013) Induce differentiation of embryonic stem cells by co-culture system. In: Andrades PJA (ed) Regenerative medicine and tissue engineering. InTech Europe, Rijeka, Croatia, p 117–139. http://www.intechopen.com
Fischer-Rasokat U, Assmus B, Seeger FH et al (2009) A pilot trial to assess potential effects of selective intracoronary bone marrow-derived progenitor cell infusion in patients with nonischemic dilated cardiomyopathy: final 1-year results of the transplantation of progenitor cells and functional regeneration enhancement pilot trial in patients with nonischemic dilated cardiomyopathy. Circ Heart Fail 2(5):417–423. doi:10.1161/CIRCHEARTFAILURE.109.855023
Franceschi RT, Xiao G, Jiang D, Gopalakrishnan R, Yang S, Reith E (2003) Multiple signaling pathways converge on the Cbfa1/Runx2 transcription factor to regulate osteoblast differentiation. Connect Tissue Res 44(Suppl 1):109–116
Franceschi RT, Ge C, Xiao G, Roca H, Jiang D (2007) Transcriptional regulation of osteoblasts. Ann N Y Acad Sci 1116:196–207. doi:10.1196/annals.1402.081
Freytag SO, Paielli DL, Gilbert JD (1994) Ectopic expression of the CCAAT/enhancer-binding protein alpha promotes the adipogenic program in a variety of mouse fibroblastic cells. Genes Dev 8(14):1654–1663
Friedenstein AJ, Piatetzky S II, Petrakova KV (1966) Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol 16(3):381–390
Friedenstein AJ, Chailakhyan RK, Gerasimov UV (1987) Bone marrow osteogenic stem cells: in vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet 20(3):263–272
Fujita T, Azuma Y, Fukuyama R et al (2004) Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3 K-Akt signaling. J Cell Biol 166(1):85–95. doi:10.1083/jcb.200401138
Fuster V, Kelly BB, Vedanthan R (2011) Global cardiovascular health: urgent need for an intersectoral approach. J Am Coll Cardiol 58(12):1208–1210
Gamez B, Rodriguez-Carballo E, Bartrons R, Rosa JL, Ventura F (2013) MicroRNA-322 (miR-322) and its target protein Tob2 modulate Osterix (Osx) mRNA stability. J Biol Chem 288(20):14264–14275. doi:10.1074/jbc.M112.432104
Gangji V, Hauzeur JP (2005) Treatment of osteonecrosis of the femoral head with implantation of autologous bone-marrow cells. Surgical technique. J Bone Joint Surg Am 87(Suppl 1 Pt 1):106–112. doi:10.2106/JBJS.D.02662
Gimble JM, Robinson CE, Wu X et al (1996) Peroxisome proliferator-activated receptor-gamma activation by thiazolidinediones induces adipogenesis in bone marrow stromal cells. Mol Pharmacol 50(5):1087–1094
Gnecchi M, Zhang Z, Ni A, Dzau VJ (2008) Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res 103(11):1204–1219. doi:10.1161/CIRCRESAHA.108.176826
Goettsch C, Rauner M, Pacyna N, Hempel U, Bornstein SR, Hofbauer LC (2011) miR-125b regulates calcification of vascular smooth muscle cells. Am J Pathol 179(4):1594–1600. doi:10.1016/j.ajpath.2011.06.016
Gu S, Boyer TG, Naski MC (2012) Basic helix-loop-helix transcription factor Twist1 inhibits transactivator function of master chondrogenic regulator Sox9. J Biol Chem 287(25):21082–21092. doi:10.1074/jbc.M111.328567
Hagenkord J, Parwani A, Lyons-Weiler M et al (2008) Virtual karyotyping with SNP microarrays reduces uncertainty in the diagnosis of renal epithelial tumors. Diagn Pathol 3(1):44
Haider HK, Jiang S, Idris NM, Ashraf M (2008) IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1alpha/CXCR4 signaling to promote myocardial repair. Circ Res 103(11):1300–1308
Hare JM, Traverse JH, Henry TD et al (2009) A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol 54(24):2277–2286
Harkness L, Mahmood A, Ditzel N, Abdallah BM, Nygaard JV, Kassem M (2010) Selective isolation and differentiation of a stromal population of human embryonic stem cells with osteogenic potential. Bone 48(2):231–241. doi:10.1016/j.bone.2010.09.023
Harkness L, Mahmood A, Ditzel N, Abdallah BM, Nygaard JV, Kassem M (2011) Selective isolation and differentiation of a stromal population of human embryonic stem cells with osteogenic potential. Bone 48(2):231–241
Harkness LNS, Beermann J, Bozhevolnyi SI, Kassem M (2012) Identification of abnormal stem cells using Raman spectroscopy. Stem Cells Dev 21(12):8. doi:10.1089/scd.2011.0600
Hata K, Takashima R, Amano K et al (2013) Arid5b facilitates chondrogenesis by recruiting the histone demethylase Phf2 to Sox9-regulated genes. Nat Commun 4:2850. doi:10.1038/ncomms3850
Hatzistergos KE, Quevedo H, Oskouei BN et al (2010) Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ Res 107(7):913–922. doi:10.1161/CIRCRESAHA.110.222703
Heile AM, Wallrapp C, Klinge PM et al (2009) Cerebral transplantation of encapsulated mesenchymal stem cells improves cellular pathology after experimental traumatic brain injury. Neurosci Lett 463(3):176–181
Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR (2009) Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res 2(3):198–210. doi:10.1016/j.scr.2009.02.002
Hernigou P, Poignard A, Beaujean F, Rouard H (2005) Percutaneous autologous bone-marrow grafting for nonunions. Influence of the number and concentration of progenitor cells. J Bone Joint Surg Am 87(7):1430–1437
Hinoi E, Bialek P, Chen YT et al (2006) Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. Genes Dev 20(21):2937–2942. doi:10.1101/gad.1482906
Houlihan DD, Mabuchi Y, Morikawa S et al (2012) Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-1 and PDGFR-α. Nat Protoc 7(12):2103–2111
Huang W, Yang S, Shao J, Li YP (2007) Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front Biosci 12:3068–3092
Huang J, Zhang Z, Guo J et al (2010) Genetic modification of mesenchymal stem cells overexpressing CCR1 increases cell viability, migration, engraftment, and capillary density in the injured myocardium. Circ Res 106(11):1753–1762
Inanc B, Elcin AE, Elcin YM (2007) Effect of osteogenic induction on the in vitro differentiation of human embryonic stem cells cocultured with periodontal ligament fibroblasts. Artif Organs 31(11):792–800
Jang H, Kim EJ, Park JK et al (2014) SMILE inhibits BMP-2-induced expression of osteocalcin by suppressing the activity of the RUNX2 transcription factor in MC3T3E1 cells. Bone. doi:10.1016/j.bone.2013.12.028
Jeevanantham V, Butler M, Saad A, Abdel-Latif A, Zuba-Surma EK, Dawn B (2012) Adult bone marrow cell therapy improves survival and induces long-term improvement in cardiac parameters: a systematic review and meta-analysis. Circulation 126(5):551–568. doi:10.1161/CIRCULATIONAHA.111.086074
Ji JF, He BP, Dheen ST, Tay SS (2004) Interactions of chemokines and chemokine receptors mediate the migration of mesenchymal stem cells to the impaired site in the brain after hypoglossal nerve injury. Stem Cells 22(3):415–427
Jia J, Tian Q, Ling S, Liu Y, Yang S, Shao Z (2013) miR-145 suppresses osteogenic differentiation by targeting Sp7. FEBS Lett 587(18):3027–3031. doi:10.1016/j.febslet.2013.07.030
Karp JM, Leng Teo GS (2009) Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell 4(3):206–216
Karp JM, Ferreira LS, Khademhosseini A, Kwon AH, Yeh J, Langer RS (2006) Cultivation of human embryonic stem cells without the embryoid body step enhances osteogenesis in vitro. Stem Cells 24(4):835–843
Kassem M, Marie PJ (2011) Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell 10(2):191–197
Kassem M, Mosekilde L, Eriksen EF (1993) 1,25-dihydroxyvitamin D3 potentiates fluoride-stimulated collagen type I production in cultures of human bone marrow stromal osteoblast-like cells. J Bone Miner Res 8(12):1453–1458
Kawai M, Green CB, Lecka-Czernik B et al (2010) A circadian-regulated gene, Nocturnin, promotes adipogenesis by stimulating PPAR-gamma nuclear translocation. Proc Natl Acad Sci USA 107(23):10508–10513. doi:10.1073/pnas.1000788107
Kawate K, Yajima H, Ohgushi H et al (2006) Tissue-engineered approach for the treatment of steroid-induced osteonecrosis of the femoral head: transplantation of autologous mesenchymal stem cells cultured with beta-tricalcium phosphate ceramics and free vascularized fibula. Artif Organs 30(12):960–962
Kebriaei P, Isola L, Bahceci E et al (2009) Adult human mesenchymal stem cells added to corticosteroid therapy for the treatment of acute graft-versus-host disease. Biol Blood Marrow Transpl 15(7):804–811
Kermani AJ, Fathi F, Mowla SJ (2008) Characterization and genetic manipulation of human umbilical cord vein mesenchymal stem cells: potential application in cell-based gene therapy. Rejuvenation Res 11(2):379–386. doi:10.1089/rej.2008.0674
Kim HE, Bae E, Jeong DY et al (2013) Lipin1 regulates PPARgamma transcriptional activity. Biochem J 453(1):49–60. doi:10.1042/BJ20121598
Kitoh H, Kitakoji T, Tsuchiya H et al (2004) Transplantation of marrow-derived mesenchymal stem cells and platelet-rich plasma during distraction osteogenesis–a preliminary result of three cases. Bone 35(4):892–898. doi:10.1016/j.bone.2004.06.013
Klinge PM, Harmening K, Miller MC et al (2011) Encapsulated native and glucagon-like peptide-1 transfected human mesenchymal stem cells in a transgenic mouse model of Alzheimer’s disease. Neurosci Lett 497(1):6–10
Koga T, Matsui Y, Asagiri M et al (2005) NFAT and Osterix cooperatively regulate bone formation. Nat Med 11(8):880–885. doi:10.1038/nm1270
Koromila T, Baniwal SK, Song YS, Martin A, Xiong J, Frenkel B (2014) Glucocorticoids antagonize RUNX2 during osteoblast differentiation in cultures of ST2 pluripotent mesenchymal cells. J Cell Biochem 115(1):27–33. doi:10.1002/jcb.24646
Kumar S, Wan C, Ramaswamy G, Clemens TL, Ponnazhagan S (2010) Mesenchymal stem cells expressing osteogenic and angiogenic factors synergistically enhance bone formation in a mouse model of segmental bone defect. Mol Ther 18(5):1026–1034
Kuznetsov SA, Krebsbach PH, Satomura K et al (1997) Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res Off J Am Soc Bone Miner Res 12(9):1335–1347
Lange C, Brunswig-Spickenheier B, Cappallo-Obermann H et al (2011) Radiation rescue: mesenchymal stromal cells protect from lethal irradiation. PLoS One 6(1):e14486
Larsen KH, Frederiksen CM, Burns JS, Abdallah BM, Kassem M (2010) Identifying a molecular phenotype for bone marrow stromal cells with in vivo bone-forming capacity. J Bone Miner Res 25(4):796–808. doi:10.1359/jbmr.091018
Lawson KA, Teteak CJ, Gao J et al (2013) ESET histone methyltransferase regulates osteoblastic differentiation of mesenchymal stem cells during postnatal bone development. FEBS Lett 587(24):3961–3967. doi:10.1016/j.febslet.2013.10.028
Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B (2007) Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology 148(6):2669–2680. doi:10.1210/en.2006-1587
Le Blanc K, Ringden O (2007) Immunomodulation by mesenchymal stem cells and clinical experience. J Intern Med 262(5):509–525
Le Blanc K, Gotherstrom C, Ringden O et al (2005) Fetal mesenchymal stem-cell engraftment in bone after in utero transplantation in a patient with severe osteogenesis imperfecta. Transplantation 79(11):1607–1614
Le BK, Frassoni F, Ball L et al (2008) Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet 371(9624):1579–1586
Lee YH, Kim SH, Lee YJ et al (2013) Transcription factor Snail is a novel regulator of adipocyte differentiation via inhibiting the expression of peroxisome proliferator-activated receptor gamma. Cell Mol Life Sci 70(20):3959–3971. doi:10.1007/s00018-013-1363-8
Lefebvre V, Li P, de Crombrugghe B (1998) A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J 17(19):5718–5733. doi:10.1093/emboj/17.19.5718
Lepperdinger G, Brunauer R, Jamnig A, Laschober G, Kassem M (2008) Controversial issue: is it safe to employ mesenchymal stem cells in cell-based therapies? Exp Gerontol 43(11):1018–1023
Leung VY, Gao B, Leung KK et al (2011) SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet 7(11):e1002356. doi:10.1371/journal.pgen.1002356
Levy O, Zhao W, Mortensen LJ et al (2013) mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood. doi:10.1182/blood-2013-04-495119
Li W, Ma N, Ong LL et al (2007) Bcl-2 engineered MSCs inhibited apoptosis and improved heart function. Stem Cells 25(8):2118–2127
Li H, Jeong HM, Choi YH et al (2013a) Glycogen synthase kinase 3 alpha phosphorylates and regulates the osteogenic activity of Osterix. Biochem Biophys Res Commun 434(3):653–658. doi:10.1016/j.bbrc.2013.03.137
Li J, Zhang N, Huang X et al (2013b) Dexamethasone shifts bone marrow stromal cells from osteoblasts to adipocytes by C/EBPalpha promoter methylation. Cell Death Dis 4:e832. doi:10.1038/cddis.2013.348
Li E, Zhang J, Yuan T, Ma B (2014) miR-143 suppresses osteogenic differentiation by targeting Osterix. Mol Cell Biochem. doi:10.1007/s11010-013-1957-3
Lian Q, Lye E, Suan Yeo K et al (2007) Derivation of clinically compliant MSCs from CD105 + , CD24 − differentiated human ESCs. Stem Cells 25(2):425–436
Lian Q, Zhang Y, Zhang J et al (2010) Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation 121(9):1113–1123. doi:10.1161/CIRCULATIONAHA.109.898312
Liang J, Zhang H, Wang D et al (2012) Allogeneic mesenchymal stem cell transplantation in seven patients with refractory inflammatory bowel disease. Gut 61(3):468–469. doi:10.1136/gutjnl-2011-300083
Liao L, Yang X, Su X et al (2013) Redundant miR-3077-5p and miR-705 mediate the shift of mesenchymal stem cell lineage commitment to adipocyte in osteoporosis bone marrow. Cell Death Dis 4:e600. doi:10.1038/cddis.2013.130
Lin FT, Lane MD (1992) Antisense CCAAT/enhancer-binding protein RNA suppresses coordinate gene expression and triglyceride accumulation during differentiation of 3T3-L1 preadipocytes. Genes Dev 6(4):533–544
Liu Y, Goldberg AJ, Dennis JE, Gronowicz GA, Kuhn LT (2012) One-Step derivation of mesenchymal stem cell (MSC)-like cells from human pluripotent stem cells on a fibrillar collagen coating. PLoS One 7(3):e33225. doi:10.1371/journal.pone.0033225
Mabuchi Y, Houlihan DD, Akazawa C, Okano H, Matsuzaki Y (2013) Prospective isolation of murine and human bone marrow mesenchymal stem cells based on surface markers. Stem Cells Int 2013:507301. doi:10.1155/2013/507301
Mahmood A, Harkness L, Schroder HD, Abdallah BM, Kassem M (2010) Enhanced differentiation of human embryonic stem cells to mesenchymal progenitors by inhibition of TGF-beta/activin/nodal signaling using SB-431542. J Bone Miner Res 25(6):1216–1233. doi:10.1002/jbmr.34
Mahmood A, Harkness L, Abdallah BM et al (2012) Derivation of stromal (skeletal and mesenchymal) stem-like cells from human embryonic stem cells. Stem Cells Dev 21(17):3114–3124. doi:10.1089/scd.2012.0035
Mangi AA, Noiseux N, Kong D et al (2003) Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med 9(9):1195–1201
Marie PJ (2008) Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys 473(2):98–105. doi:10.1016/j.abb.2008.02.030
Martinez-Sanchez A, Dudek KA, Murphy CL (2012) Regulation of human chondrocyte function through direct inhibition of cartilage master regulator SOX9 by microRNA-145 (miRNA-145). J Biol Chem 287(2):916–924. doi:10.1074/jbc.M111.302430
Matsumoto Y, Ivasaki H, Suda T (2011) Maintenance of adult stem cells: role of the stem cell niche. In: Phinney DG (ed) Adult stem cells: biology and methods of analysis. Stem cell biology and regenerative medicine. Springer, Berlin, pp 35–55
Mirotsou M, Jayawardena TM, Schmeckpeper J, Gnecchi M, Dzau VJ (2011) Paracrine mechanisms of stem cell reparative and regenerative actions in the heart. J Mol Cell Cardiol 50(2):280–289. doi:10.1016/j.yjmcc.2010.08.005
Miura K, Okada Y, Aoi T, et al (2009) Variation in the safety of induced pluripotent stem cell lines. Nat Biotech 27(8):743–745 http://www.nature.com/nbt/journal/v27/n8/suppinfo/nbt.1554_S1.html
Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L (1998) The chemokine receptor CXCR-4 is expressed on CD34 + hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood 91(12):4523–4530
Nakashima K, Zhou X, Kunkel G et al (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108(1):17–29
Nauta AJ, Fibbe WE (2007) Immunomodulatory properties of mesenchymal stromal cells. Blood 110(10):3499–3506. doi:10.1182/blood-2007-02-069716
Nejadnik H, Hui JH, Feng Choong EP, Tai BC, Lee EH (2010) Autologous bone marrow-derived mesenchymal stem cells versus autologous chondrocyte implantation: an observational cohort study. Am J Sports Med 38(6):1110–1116. doi:10.1177/0363546509359067
Nishio Y, Dong Y, Paris M, O’Keefe RJ, Schwarz EM, Drissi H (2006) Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene 372:62–70. doi:10.1016/j.gene.2005.12.022
Noiseux N, Gnecchi M, Lopez-Ilasaca M et al (2006) Mesenchymal stem cells overexpressing Akt dramatically repair infarcted myocardium and improve cardiac function despite infrequent cellular fusion or differentiation. Mol Ther J Am Soc Gene Ther 14(6):840–850. doi:10.1016/j.ymthe.2006.05.016
Nouspikel T (2013) Genetic instability in human embryonic stem cells: prospects and caveats. Future Oncol 9(6):867–877. doi:10.2217/fon.13.22
Nuttall ME, Shah F, Singh V, Thomas-Porch C, Frazier T, Gimble JM (2014) Adipocytes and the regulation of bone remodeling: a balancing act. Calcif Tissue Int 94(1):78–87. doi:10.1007/s00223-013-9807-6
Oh CD, Maity SN, Lu JF et al (2010) Identification of SOX9 interaction sites in the genome of chondrocytes. PLoS One 5(4):e10113. doi:10.1371/journal.pone.0010113
Olivier E, Bouhassira E (2011) Differentiation of human embryonic stem cells into mesenchymal stem cells by the raclure method. In: Nieden NI (ed) Embryonic stem cell therapy for Osteo-degenerative diseases. Methods in molecular biology, vol 690. Humana, Clifton, pp 183–193
Orbay H, Tobita M, Mizuno H (2012) Mesenchymal stem cells isolated from adipose and other tissues: basic biological properties and clinical applications. Stem cells Int 461718. doi: 10.1155/2012/461718
Orozco L, Munar A, Soler R et al (2013) Treatment of knee osteoarthritis with autologous mesenchymal stem cells: a pilot study. Transplantation 95(12):1535–1541. doi:10.1097/TP.0b013e318291a2da
Ortuno MJ, Ruiz-Gaspa S, Rodriguez-Carballo E et al (2010) p38 regulates expression of osteoblast-specific genes by phosphorylation of osterix. J Biol Chem 285(42):31985–31994. doi:10.1074/jbc.M110.123612
Pal P, Lochab S, Kanaujiya JK et al (2013) E3 ubiquitin ligase E6AP negatively regulates adipogenesis by downregulating proadipogenic factor C/EBPalpha. PLoS One 8(6):e65330. doi:10.1371/journal.pone.0065330
Pan S, Yang X, Jia Y, Li R, Zhao R (2013) Microvesicle-shuttled miR-130b reduces fat deposition in recipient primary cultured porcine adipocytes by inhibiting PPAR-gamma expression. J Cell Physiol. doi:10.1002/jcp.24486
Park BO, Ahrends R, Teruel MN (2012) Consecutive positive feedback loops create a bistable switch that controls preadipocyte-to-adipocyte conversion. Cell Rep 2(4):976–990. doi:10.1016/j.celrep.2012.08.038
Paul G, Ozen I, Christophersen NS et al (2012) The adult human brain harbors multipotent perivascular mesenchymal stem cells. PLoS One 7(4):e35577. doi:10.1371/journal.pone.0035577
Peng Y, Shi K, Wang L et al (2013) Characterization of Osterix protein stability and physiological role in osteoblast differentiation. PLoS One 8(2):e56451. doi:10.1371/journal.pone.0056451
Perez-Ilzarbe M, Agbulut O, Pelacho B et al (2008) Characterization of the paracrine effects of human skeletal myoblasts transplanted in infarcted myocardium. Eur J Heart Fail 10(11):1065–1072. doi:10.1016/j.ejheart.2008.08.002
Porada CD, Almeida-Porada G (2010) Mesenchymal stem cells as therapeutics and vehicles for gene and drug delivery. Adv Drug Deliv Rev 62(12):1156–1166. doi:10.1016/j.addr.2010.08.010
Pratap J, Galindo M, Zaidi SK et al (2003) Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res 63(17):5357–5362
Quesenberry PJ, Becker PS (1998) Stem cell homing: rolling, crawling, and nesting. Proc Natl Acad Sci USA 95(26):15155–15157
Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ (2008) Efficient tumour formation by single human melanoma cells. Nature 456(7222):593–598
Ringden O, Uzunel M, Rasmusson I et al (2006) Mesenchymal stem cells for treatment of therapy-resistant graft-versus-host disease. Transplantation 81(10):1390–1397
Sacchetti B, Funari A, Michienzi S et al (2007) Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131(2):324–336. doi:10.1016/j.cell.2007.08.025
Samuelsson L, Stromberg K, Vikman K, Bjursell G, Enerback S (1991) The CCAAT/enhancer binding protein and its role in adipocyte differentiation: evidence for direct involvement in terminal adipocyte development. EMBO J 10(12):3787–3793
Sarkar D, Vemula PK, Zhao W, Gupta A, Karnik R, Karp JM (2010) Engineered mesenchymal stem cells with self-assembled vesicles for systemic cell targeting. Biomaterials 31(19):5266–5274. doi:10.1016/j.biomaterials.2010.03.006
Schaap-Oziemlak AM, Raymakers RA, Bergevoet SM et al (2010) MicroRNA hsa-miR-135b regulates mineralization in osteogenic differentiation of human unrestricted somatic stem cells. Stem Cells Dev 19(6):877–885. doi:10.1089/scd.2009.0112
Schrepfer S, Deuse T, Reichenspurner H, Fischbein MP, Robbins RC, Pelletier MP (2007) Stem cell transplantation: the lung barrier. Transpl Proc 39(2):573–576. doi:10.1016/j.transproceed.2006.12.019
Schriebl K, Satianegara G, Hwang A et al (2012) Selective removal of undifferentiated human embryonic stem cells using magnetic activated cell sorting followed by a cytotoxic antibody. Tissue Eng Part A 18(9–10):899–909
Schuldiner M, Yanuka O, Itskovitz-Eldor J, Melton DA, Benvenisty N (2000) Effects of eight growth factors on the differentiation of cells derived from human embryonic stem cells. Proc Natl Acad Sci USA 97(21):11307–11312
Shi M, Li J, Liao L et al (2007) Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: role in homing efficiency in NOD/SCID mice. Haematologica 92(7):897–904
Shi K, Lu J, Zhao Y et al (2013) MicroRNA-214 suppresses osteogenic differentiation of C2C12 myoblast cells by targeting Osterix. Bone 55(2):487–494. doi:10.1016/j.bone.2013.04.002
Shiojima I, Walsh K (2006) Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev 20(24):3347–3365. doi:10.1101/gad.1492806
Shujia J, Haider HK, Idris NM, Lu G, Ashraf M (2008) Stable therapeutic effects of mesenchymal stem cell-based multiple gene delivery for cardiac repair. Cardiovasc Res 77(3):525–533
Simonsen JL, Rosada C, Serakinci N et al (2002) Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat Biotechnol 20(6):592–596
Sinha KM, Zhou X (2013) Genetic and molecular control of osterix in skeletal formation. J Cell Biochem 114(5):975–984. doi:10.1002/jcb.24439
Sinha KM, Yasuda H, Zhou X, Decrombrugghe B (2013) Osterix and NO66 histone demethylase control the chromatin architecture of Osterix target genes during osteoblast differentiation. J Bone Miner Res Off J Am Soc Bone Miner Res. doi:10.1002/jbmr.2103
Sordi V, Malosio ML, Marchesi F et al (2005) Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood 106(2):419–427
Sottile V, Thomson A, McWhir J (2003) In Vitro Osteogenic Differentiation of Human ES Cells. Cloning Stem Cells 5(2):149–155
Stenderup K, Justesen J, Clausen C, Kassem M (2003) Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 33(6):919–926. doi:10.1016/j.bone.2003.07.005
Takada I, Kouzmenko AP, Kato S (2009a) Molecular switching of osteoblastogenesis versus adipogenesis: implications for targeted therapies. Expert Opin Ther Targets 13(5):593–603. doi:10.1517/14728220902915310
Takada I, Kouzmenko AP, Kato S (2009b) Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol 5(8):442–447. doi:10.1038/nrrheum.2009.137
Tang QQ, Lane MD (2000) Role of C/EBP homologous protein (CHOP-10) in the programmed activation of CCAAT/enhancer-binding protein-beta during adipogenesis. Proc Natl Acad Sci USA 97(23):12446–12450. doi:10.1073/pnas.220425597
Tang C, Weissman IL, Drukker M (2012) The safety of embryonic stem cell therapy relies on teratoma removal. Oncotarget 3(1):7–8. PMID 22294556
Tao Z, Chen B, Tan X et al (2011) Coexpression of VEGF and angiopoietin-1 promotes angiogenesis and cardiomyocyte proliferation reduces apoptosis in porcine myocardial infarction (MI) heart. Proc Natl Acad Sci USA 108(5):2064–2069. doi:10.1073/pnas.1018925108
Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS (2000) Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 290(5489):134–138
Tong Q, Tsai J, Tan G, Dalgin G, Hotamisligil GS (2005) Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol Cell Biol 25(2):706–715. doi:10.1128/MCB.25.2.706-715.2005
Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM (1994) mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8(10):1224–1234
Tormin A, Li O, Brune JC et al (2011) CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood 117(19):5067–5077. doi:10.1182/blood-2010-08-304287
Tornvig L, Mosekilde LI, Justesen J, Falk E, Kassem M (2001) Troglitazone treatment increases bone marrow adipose tissue volume but does not affect trabecular bone volume in mice. Calcif Tissue Int 69(1):46–50. doi:10.1007/s002230020018
Tremoleda JL, Forsyth NR, Khan NS et al (2008) Bone Tissue Formation from Human Embryonic Stem Cells In Vivo. Cloning Stem Cells 10(1):119–132
Trivedi P, Hematti P (2008) Derivation and immunological characterization of mesenchymal stromal cells from human embryonic stem cells. Exp Hematol 36(3):350–359
Tu Q, Valverde P, Li S, Zhang J, Yang P, Chen J (2007) Osterix overexpression in mesenchymal stem cells stimulates healing of critical-sized defects in murine calvarial bone. Tissue Eng 13(10):2431–2440
Ueta C, Iwamoto M, Kanatani N et al (2001) Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol 153(1):87–100
Vimalraj S, Partridge NC, Selvamurugan N (2014) A positive role of microRNA-15b on regulation of osteoblast differentiation. J Cell Physiol. doi:10.1002/jcp.24557
Wakitani S, Nawata M, Tensho K, Okabe T, Machida H, Ohgushi H (2007) Repair of articular cartilage defects in the patello-femoral joint with autologous bone marrow mesenchymal cell transplantation: three case reports involving nine defects in five knees. J Tissue Eng Regen Med 1(1):74–79. doi:10.1002/term.8
Wang YH, Han Z-B; Song Y-P; Han ZC (2012) Safety of mesenchymal stem cells for clinical application. Stem Cells Int 2012(Article ID 652034):4. doi: 10.1155/2012/652034
Wang CY, Yang SF, Wang Z et al (2013) PCAF acetylates Runx2 and promotes osteoblast differentiation. J Bone Miner Metab 31(4):381–389. doi:10.1007/s00774-013-0428-y
Weng JY, Du X, Geng SX et al (2010) Mesenchymal stem cell as salvage treatment for refractory chronic GVHD. Bone Marrow Transpl 45(12):1732–1740. doi:10.1038/bmt.2010.195
Wu Y, Zhao RC (2012) The role of chemokines in mesenchymal stem cell homing to myocardium. Stem Cell Rev 8(1):243–250
Xiao G, Jiang D, Thomas P et al (2000) MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J Biol Chem 275(6):4453–4459
Yamashita S, Miyaki S, Kato Y et al (2012) L-Sox5 and Sox6 proteins enhance chondrogenic miR-140 microRNA expression by strengthening dimeric Sox9 activity. J Biol Chem 287(26):22206–22215. doi:10.1074/jbc.M112.343194
Yang L, Cheng P, Chen C et al (2012a) miR-93/Sp7 function loop mediates osteoblast mineralization. J Bone Miner Res Off J Am Soc Bone Miner Res 27(7):1598–1606. doi:10.1002/jbmr.1621
Yang X, Balakrishnan I, Torok-Storb B, Pillai MM (2012b) Marrow stromal cell infusion rescues hematopoiesis in lethally irradiated mice despite rapid clearance after infusion. Adv Hematol 2012:142530
Yang D, Okamura H, Nakashima Y, Haneji T (2013) Histone demethylase Jmjd3 regulates osteoblast differentiation via transcription factors Runx2 and osterix. J Biol Chem 288(47):33530–33541. doi:10.1074/jbc.M113.497040
Zhang JF, Fu WM, He ML et al (2011a) MiR-637 maintains the balance between adipocytes and osteoblasts by directly targeting Osterix. Mol Biol Cell 22(21):3955–3961. doi:10.1091/mbc.E11-04-0356
Zhang JF, Fu WM, He ML et al (2011b) MiRNA-20a promotes osteogenic differentiation of human mesenchymal stem cells by co-regulating BMP signaling. RNA Biol 8(5):829–838. doi:10.4161/rna.8.5.16043
Zhao S, Wehner R, Bornhauser M, Wassmuth R, Bachmann M, Schmitz M (2010) Immunomodulatory properties of mesenchymal stromal cells and their therapeutic consequences for immune-mediated disorders. Stem Cells Dev 19(5):607–614. doi:10.1089/scd.2009.0345
Zhou M, Ma J, Chen S, Chen X, Yu X (2013) MicroRNA-17-92 cluster regulates osteoblast proliferation and differentiation. Endocrine. doi:10.1007/s12020-013-9986-y
Zimmet H, Porapakkham P, Sata Y et al (2012) Short- and long-term outcomes of intracoronary and endogenously mobilized bone marrow stem cells in the treatment of ST-segment elevation myocardial infarction: a meta-analysis of randomized control trials. Eur J Heart Fail 14(1):91–105. doi:10.1093/eurjhf/hfr148
Acknowledgments
The work was supported by a grant from the University hospital of Odense, Odense, Denmark and KACST, (Project Code: 10-BIO1308-02) (KSA). The funders had no role in preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zaher, W., Harkness, L., Jafari, A. et al. An update of human mesenchymal stem cell biology and their clinical uses. Arch Toxicol 88, 1069–1082 (2014). https://doi.org/10.1007/s00204-014-1232-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-014-1232-8