
Abstract
A pool of myoblasts available for myogenesis is important for skeletal muscle size. The decreased number of skeletal muscle fibers could be due to the decreased myoblast proliferation or cytotoxicity. Identification of toxicants that regulate myoblast apoptosis is important in skeletal muscle development or regeneration. Here, we investigate the cytotoxic effect and its possible mechanisms of arsenic trioxide (As2O3) on myoblasts. C2C12 myoblasts underwent apoptosis in response to As2O3 (1–10 μM), accompanied by increased Bax/Bcl-2 ratio, decreased mitochondria membrane potential, increased cytochrome c release, increased caspase-3/-9 activity, and increased poly (ADP-ribose) polymerase (PARP) cleavage. Moreover, As2O3 triggered the endoplasmic reticulum (ER) stress indentified through several key molecules of the unfolded protein response, including glucose-regulated protein (GRP)-78, GRP-94, PERK, eIF2α, ATF6, and caspase-12. Pretreatment with antioxidant N-acetylcysteine (NAC, 0.5 mM) dramatically suppressed the increases in reactive oxygen species (ROS), lipid peroxidation, ER stress, caspase cascade activity, and apoptosis in As2O3 (10 μM)-treated myoblasts. Furthermore, As2O3 (10 μM) effectively decreased the phosphorylation of Akt, which could be reversed by NAC. Over-expression of constitutive activation of Akt (c.a. Akt) also significantly attenuated As2O3-induced myoblast apoptosis. Taken together, these results suggest that As2O3 may exert its cytotoxicity on myoblasts by inducing apoptosis through a ROS-induced mitochondrial dysfunction, ER stress, and Akt inactivation signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In adults, repair of degenerated muscles relies on a small population of skeletal muscle stem cells known as satellite cells (Mauro 1961). Satellite cells, a popular of quiescent muscle precursor cells that reside beneath the basal lamina, provide the predominant source of additional myonuclei for muscle growth (Mitchell and Pavlath 2001; Rosenblatt et al. 1994). Once activated, satellite cells give rise to myoblasts that proliferate, differentiate, and fuse to form new muscle fibers or to repair damaged muscle fibers (Charge and Rudnicki 2004; Hawke and Garry 2001). A pool of myoblasts available for myogenesis is important for muscle size in vivo and in vitro (Miyake et al. 2011). The decreased number of muscle fibers could be due to the decreased myoblast proliferation or cytotoxicity. Moreover, in most cases, the apoptosis of myoblasts serves as a physiological behavior to remove excess myoblasts during myogenesis or muscle regeneration, while inappropriate apoptosis will pathologically lead to degeneration that associated with various muscular dystrophies and atrophies (Tews and Goebel 1997; Tidball et al. 1995). Therefore, identification of toxicants that regulate myoblast cell cytotoxicity is important in understanding skeletal muscle growth, disease, and regeneration.
Inorganic arsenic is an environmental toxicant and carcinogen. Humans can be exposed to arsenic primarily from air, food, and water. Chronic exposure to arsenic via drinking water has known to be associated with numerous cancers (e.g., skin, lung, and bladder) and non-cancer harmful health (e.g., skin lesions, peripheral vascular disease, cardiovascular disease, and diabetes mellitus) (Navas-Acien et al. 2005; Yoshida et al. 2004). Previous studies have demonstrated that low dose of inorganic arsenic is capable of inhibiting myoblast differentiation (Steffens et al. 2011). Arsenic trioxide is known to be used to treat acute promyelocytic leukemia and can induce apoptosis in various cancer and normal cell lines (Cai et al. 2010; Florea et al. 2007; Li et al. 2010; Lu et al. 2011; Tang et al. 2009). Intracellular reactive oxygen species (ROS) mediates multiple cellular responses, including protein kinase activation (Torres and Forman 2003), cell cycle progression (Boonstra and Post 2004), cell differentiation (Yen et al. 2010), and apoptotic cell death (Orrenius et al. 2007). ROS has been reported to regulate As2O3-induced apoptosis (Chen et al. 1998). It has been observed that As2O3 activates caspase cascade signaling and induces various cell apoptosis by alteration in the mitochondria membrane potential (MMP), mitochondria function protein (Bcl-2, Bax, and cytochrome c), and caspase activity resulting in oxidative stress (Chen et al. 1998; Lu et al. 2011). Moreover, arsenic-induced oxidative stress can effectively induce cell apoptosis through activation of the endoplasmic reticulum (ER) stress pathway (Lu et al. 2011; Yen et al. 2011). The serine/threonine protein kinase Akt, also known as protein kinase B, mediates effects of extracellular signals on various cellular processes including growth, differentiation, survival, and metabolism (Manning and Cantley 2007). It has also been shown that As2O3 treatment leads cell apoptosis by inactivating the Akt-related cell survival pathway in U937 cells (Choi et al. 2002). However, the cytotoxic effect and mechanism of arsenic on myoblasts still remain unclear. The aim of this study is to test the potential cytotoxicity of As2O3 on myoblasts and to evaluate the cellular mechanism involved in the As2O3-induced myoblast apoptosis.
Materials and methods
Cell culture
C2C12 myoblasts were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured in growth medium consisting of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin) in 5 % carbon dioxide (CO2) at 37 °C.
Drugs
0.1 M As2O3 (Sigma) was prepared in 1 N NaOH, diluted to 10−3 M in PBS, and adjusted to pH 7.2 using HCl. As2O3 was diluted further with PBS, and the solutions were kept at 4 °C.
Cell viability assay
Cell proliferation was determined by a colorimetric assay using 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Sigma). Cell viability was conducted in C2C12 cultures per well in 12-well dishes. C2C12 myoblast cultures were seeded at 2 × 105 cells per well. After 16-h attachment period, arsenic test media (0–30 μM) was added. Plates were then incubated in an atmosphere of 37 °C, 5 % CO2 for a further 24 h. This assay measures the activity of living cells via mitochondrial dehydrogenase activity that reduces MTT to purple formazan. The formazan was solubilized by DMSO, and its absorbance at 570 nm was measured. Results were representative of at least three independent experiments.
Measurement of mitochondria membrane potential (MMP)
Measurement of mitochondria membrane potential was determined as described earlier (Chen et al. 2006). Briefly, C2C12 cells were treated with 10 μM As2O3 and incubated for 6, 12, and 24 h. Cells were then incubated with 40 nM of 3-3′-dihexyloxacarbo-cyanine (DiOC6) for 30 min at 37 °C and then pelleted by centrifugation at 1,000 rpm for 5 min. The pellets were resuspended and washed twice with PBS. The mitochondria membrane potential was determined by a FACScan flow cytometer (Becton–Dickinson).
Cytochrome c release from mitochondria
C2C12 myoblasts were washed once with ice-cold PBS. To isolated mitochondria and cytosol fractions, the cells were lysed in buffer containing 10 mM Tris–HCl pH 7.4, 10 mM NaCl and 12.5 mM EDTA, and the cell extract was centrifuged at 600×g for 5 min to pellet the nuclei. The supernatant was centrifuged at 16,000×g for 20 min to pellet mitochondria fraction. The supernatant was as the cytosol fraction.
Annexin V-FITC apoptosis detection
C2C12 cells (2 × 105) were plated in 6-well plate. After overnight incubation at 37 °C, cells were treated with As2O3 (0, 1, 3, and 10 μM) for 24 h with or without 0.5 mM NAC (Sigma) pretreatment for 30 min, and then apoptosis was assessed by using annexin V-FITC apoptosis detection kit (Becton–Dickinson) in accordance with the manufacture’s instructions. In brief, cells were dissociated using 0.05 % trypsin/EDTA for 1 min (keeping floating cells) then centrifuged, re-suspended in 100 μl binding buffer, transferred in a 5 ml FACS tube, combined with 5 μl Annexin V-FITC (conjugated with fluorescein isothiocyanate), and 10 μl propidium iodide. After incubation for 30 min at RT in dark, 400 μl of binding buffer was added to each tube and the samples were immediately analyzed using a FACS flow cytometer.
Protein extraction and immunoblotting
Whole-cell lysates were prepared from C2C12 myoblast cells by incubation in RIPA buffer [10 mM Tris (pH 7.4), 150 mM NaCl, 1 mM ethylene glycol tetraacetic acid, 0.1 % sodium dodecyl sulfate, w 1 mM NaF, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/mL aprotinin, and 1 μg/mL leupeptin]. The cell suspension was left on ice for 20 min and then centrifuged at 10,000×g for 20 min at 4 °C. We used the supernatant for the experiments. An equal amount of protein was separated by 8–12 % SDS-PAGE and electrotransferred onto polyvinylidene difluoride membrane (0.2 μm) using transfer buffer (192 mM glycine, 25 mM Tris, 20 % methanol, pH 8.3) followed by blocking in TBST (Tris-buffered saline/Tween-20) buffer (20 mM Tris, 150 mM NaCl, 0.01 % Tween-20, pH 7.5) supplemented with 5 % non-fat powdered milk. The membranes were then probed with the primary antibodies for Bcl-2, Bax, cytochrome c, caspase-3, caspase-9, caspase-12, glucose-regulated protein (GRP)-78, GRP-94, C/EBP homologous protein (CHOP), poly(ADP-ribose) polymerase (PARP), Akt, β-actin, and α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA); phospho-PKR (double-stranded RNA-activated protein kinase)-like ER kinase (PERK) and phospho-eukaryotic translation initiation factor α (eIF2α) (cell Signaling Technology, Danvers, MA, USA); activating transcription factor (ATF)-6 (IMGENEX, San Diego, CA, USA); and phospho-Akt (EPITOMICS, Burlingame, CA, USA) overnight at 4 °C. The blots were washed with TBST (three times for 10 min, room temperature), incubated with the secondary goat anti-rabbit or anti-mouse antibodies conjugated with horseradish peroxidase (1:7,500 dilution in TBST for 1 h at room temperature), and then washed again in TBST (three times for 10 min, room temperature). The blots were developed using an enhanced chemiluminescence reagent and exposed to X-ray film. Densitometric analysis was carried out using Molecular Analyst software (version 1.3; BioRad, Hercules, CA, USA).
Measurement of intracellular ROS formation
The generation of intracellular ROS was determined using a fluorescein-labeled dye, 2′,7′-dichlorofluorescin diacetate (DCFH-DA). The non-fluorescent dye permeated cells easily and was hydrolyzed to 2′,7′-dichlorofluorescin (DCF) upon interaction with intracellular ROS. After treatment, cells were incubated with 20 μM DCFH-DA foe 30 min at 37 °C. Then, cells were washed twice with ice-cold PBS and harvested by trypsin. Then, the cells were immediately analyzed by a FACScan flow cytometer (Becton–Dickinson) to determine the ROS generation. Each group collected 10,000 individual cells.
Lipid peroxidation (LPO) assay
Malondiadehyde (MDA) concentrations were determined using the TBARS Assay Kit (Cayman Chemical) according to the manufacturer`s recommendations for colorimetric measurement of MDA. In brief, cells were sonicated on iced lysis buffer. Samples were then centrifuged at 1,600×g for 10 min at 4 °C, and the supernatant was preserved for analysis, To 100 μl of each sample and standard curve solutions, 100 μl of provided SDS solution was added followed by 4 ml of color reagent. Samples and standard curve solutions were boiled for 1 h, and the reaction was terminated by incubation on ice for 10 min followed by centrifugation at 1,600×g for 10 min at 4 °C. Then, 150 μl of samples and standard curve solutions were loaded in duplicate onto a 96-well plate, and absorbance was read at 540 nm in a Powerwave HT (BioTek Instruments, Winooski, VT, USA) spectrophotometer. The protein concentration was determined using the BCA kit (Pierce, Rockford, IL, USA) with an absorption band of 570. LPO levels was expressed as nanomoles (nmol) MDA per milligram protein and estimated from the standard curve.
Transient transfection of c.a. Akt
A control pcDNA 3.1 empty vector and a constitutively active form of Akt [myristoylated (myr) Akt] were gift from Dr. M.L. Kuo (Kuo et al. 2001). C2C12 cells were transfected with myr-Akt or pcDNA3.1 using Lipofectamine 2000 reagent (Invitrogen) and preformed as previously described (Yen et al. 2010).
Statistics
Data are expressed as mean ± SE. We assessed the significant difference from the respective controls for each experimental test condition using analysis of variance and the t test, with p < 0.05 considered significant.
Results
Effects of As2O3 on cell viability and apoptosis in C2C12 myoblasts
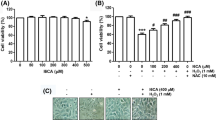
We first examined the effect of As2O3 on the viability of C2C12 by MTT assay. As shown in Fig. 1a, As2O3 (1–30 μM) significantly reduced C2C12 viability after 24-h exposure in a concentration-dependent manner (p < 0.05). To examine whether As2O3 causes cell apoptosis, myoblasts were stained with Annexin V-PI and analyzed by flow cytometry. As shown in Fig. 1b, treatment with As2O3 (3 and 10 μM) for 24 h showed a significantly higher apoptosis rate (3 μM, 21 ± 0.7 %; 10 μM, 47 ± 6.6 %) over the untreated C2C12 cells (p < 0.05). However, low concentration of As2O3 (1 μM) did not stimulate significant cell apoptosis. Because 10 μM As2O3 led to greater apoptosis, this concentration was used in the subsequent experiments.

As2O3 decreases cell viability and induces apoptosis in myoblasts. C2C12 myoblasts were cultured in the presence or absence of As2O3 (0.5–30 μM) for 24 h. A C2C12 myoblast viability was determined by MTT assay. B Myoblast apoptosis was determined by annexin V/PI and analyzed by flow cytometry. The cells in the bottom right quadrant (FITC+ and PI−) and the upper right quadrant (FITC+ and PI+) represent early apoptotic and late apoptotic cells, respectively (a). The percentage of annexin V-positive cells was measured (b). All data are presented as mean ± SE of three independent experiments. *p < 0.05 as compared with control
Caspase-3 plays an essential role as executor in apoptosis (Porter and Janicke 1999). In addition, the cleavage of PARP by caspases provides one of the most recognizable markers in apoptosis (Lazebnik et al. 1994). We assessed the cleavages of PARP and caspase-3 proteins in myoblasts after exposure to As2O3 for various time intervals. Western blot analysis showed that the cleaved forms of caspase-3 (17 kDa fragments) and PARP (89 kDa fragments) were markedly increased after 8 h of treatment with 10 μM As2O3 and maintained to 24 h (Fig. 2a). Moreover, pro-caspase 9 was also markedly decreased in As2O3-treated myoblasts (Fig. 2b).

As2O3 triggers caspases activation, mitochondrial dysfunction, and PARP cleavage in myoblasts. C2C12 cells were treated with As2O3 (10 μM) for 0.5–24 h. The cleavages of caspase-9/3 and PARP (A) and the expressions of Bax and Bcl-2 (B) in cell lysates were detected by Western blotting. α-tubulin was used as an internal control. Representative images of three independent experiments are shown. C Flow cytograms of MMP in C2C12 myoblasts exposure to As2O3. The cells were treated with 10 μM As2O3 for 0, 6, 12, and 24 h, then stained with DiOC6, and analyzed by flow cytometry. Data are presented as mean ± SE of three independent experiments. *p < 0.05 as compared with control. D The release of cytochrome c from mitochondria to cytosol in C2C12 cells after exposure to As2O3 was determined by Western blotting. β-actin was used as an internal control. Representative images of three independent experiments are shown
As2O3 triggers apoptosis through the caspase-dependent intrinsic mitochondrial pathway
The proteins of Bcl-2 family play a major role in regulation of apoptosis by functioning as promoters (e.g., Bax) or inhibitors (Bcl-2 or Bcl-xl) of cell death (Chao and Korsmeyer 1998; Hockenbery et al. 1990; Reed 1995). To explore the involvement of Bcl-2 family members in apoptosis induced by As2O3, the expression levels of both Bcl-2 and Bax were analyzed by Western blotting. We found that treatment of C2C12 with As2O3 for 24 h resulted in a reduction in the expression of anti-apoptotic protein Bcl-2 and an increase in the expression of pro-apoptotic protein Bax in a time-dependent manner (Fig. 2b). A key step in the intrinsic apoptotic pathway is the damage of mitochondria and release of cytochrome c, which in terns on the caspase cascade (Garrido et al. 2006). To determine whether As2O3-induced apoptosis in myoblasts was through a mitochondria pathway, we examined whether As2O3 could modulate MMP and cytochrome c. As shown in Fig. 2c, treatment with As2O3 for 6, 12, and 24 h induced the decrease of the MMP in a time-dependent manner. Western blot analysis also revealed that treatment with 10 μM As2O3 to C2C12 cells effectively increased the release of cytochrome c from mitochondria into the cytosol fraction (Fig. 2d). These results indicate that the mitochondria pathway plays an important role in As2O3-induced apoptosis.
As2O3 induces the ER stress response of C2C12 myoblasts
ER stress has been shown to be involved in inorganic arsenic-induced cell apoptosis of cultured osteoblasts and pancreatic β-cells (Lu et al. 2011; Tang et al. 2009). To test whether As2O3 induces ER stress in myoblast, we examined the ER stress markers expression after treatment with As2O3 in C2C12 myoblasts. As shown in Fig. 3, As2O3 (10 μM) effectively triggered the expressions of ER stress-related molecules including phosphorylated PERK, phosphorylated eIF2α, GRP-78, GRP-94, CHOP, cleaved ATF6, and cleaved caspase 12 in myoblasts in a time-dependent manner. These results indicate that As2O3 is capable of inducing the ER stress in C2C12 myoblasts.

As2O3 induces the expressions of ER stress-related molecules in myoblasts. C2C12 cells were treated with 10 μM As2O3 for indicated time courses. The expressions of p-PERK, peIF2α, GRP78, GRP94, cleaved ATF6, CHOP, caspase 12, and β-actin were analyzed by Western blotting. Results shown are representative of at last three independent experiments
As2O3 induces the intracellular ROS production in C2C12 cells
Next, we tested whether intracellular ROS is associated with As2O3-induced apoptosis in myoblasts. As shown in Fig. 4a, the intracellular ROS levels were increased at 30 min and significantly increased as early as 1 h following exposure to 10 μM As2O3, and peak production of ROS was after 4 h (p < 0.05). As expected, pretreatment with antioxidant NAC (0.5 mM) significantly reduced the intracellular ROS level induced by As2O3 (Fig. 4a). As2O3 also significantly increased the lipid peroxidation product MDA levels in myoblasts, which could be reversed by NAC (Fig. 4b). We further examined whether NAC could rescue the As2O3-induced ER stress and apoptosis. As shown in Fig. 4c and d, pretreatment with NAC could effectively decrease the increased expression of CHOP, cleavages of caspase-12, caspase-9, caspase-3, and PARP, and release of cytochrome c from mitochondria in As2O3-treated myoblasts. Moreover, NAC also significantly attenuated the increased cell apoptosis (Fig. 5a) and decreased cell survival in As2O3-treated myoblasts (Fig. 5b). These results indicate that ROS play an important role in As2O3-induced myoblast apoptosis.

The role of ROS in As2O3-induced apoptosis and ER stress in myoblasts. C2C12 cells were treated with 10 μM As2O3 for various time courses in the presence or absence of 0.5 mM NAC. A The levels of ROS were measured by flow cytometry using a fluorescein-labeled dye (2′,7′-dichlorofluorescein diacetate). B MDA production was determined by a commercial assay as described in “Materials and methods”. Data are presented as mean ± SE of three independent experiments. *p < 0.05 as compared with control. # p < 0.05 as compared with As2O3 alone. In C and D, the expressions of CHOP, cytochrome c, caspase-12, caspase-9, caspase-3, and PARP in myoblasts were determined by Western blot analysis. C2C12 cells were treated with 10 μM As2O3 for 4 h (C) or 24 h (D) in the present or absence of 0.5 mM NAC. Representative images of three independent experiments are shown

NAC attenuates the As2O3-induced apoptosis and decreased cell viability. C2C12 myoblasts were pretreated with 0.5 mM NAC for 30 min and then treated with or without 10 μM As2O3 for 24 h. A The percentage of annexin V-positive cells were measured. B Cell viability was determined by MTT assay. Data are presented as mean ± SE of three independent experiments. *p < 0.05 as compared with control. # p < 0.05 as compared with As2O3 alone
Akt pathway plays a role in regulating As2O3-induced myoblast apoptosis
It has been reported that the activation of Akt is critical for cell survival (Manning and Cantley 2007). We next investigated the effect of As2O3 on the phosphorylation of Akt protein by Western blotting analysis. As shown in Fig. 6a, As2O3 (10 μM) effectively decreased the phosphorylation of Akt in myoblasts in a time-dependent manner. Furthermore, we evaluated the relationship between As2O3-induced apoptosis and Akt signaling. A myr-Akt plasmid coding for an active form of Akt was transiently transfected into C2C12 myoblasts. A significant reduction in the As2O3-enhanced cleaved PARP expression was shown in the myr-Akt-transfected myoblasts as compared with control pcDNA-transfected cells (Fig. 6b). In addition, pretreatment with NAC markedly prevented the inhibition of Akt phosphorylation in As2O3-treated myoblasts (Fig. 6c). These results indicate that ROS may contribute to the suppression of Akt phosphorylation and then lead to the myoblast apoptosis induced by As2O3.

Akt activation is involved in As2O3-induced apoptosis. C2C12 myoblasts were treated with 10 μM As2O3 for indicated time courses. A As2O3 down-regulated the phosphorylation of Akt in a time-dependent manner. B Control pcDNA and c.a. Akt were transfected into cells for 24 h. Over-expression of myr-Akt reduced As2O3-induced PARP cleavage. Data are presented as mean ± SE of three independent experiments. *p < 0.05 as compared with control. # p < 0.05 as compared with As2O3 alone. C C2C12 cells were treated with 10 μM As2O3 in the presence or absence of 0.5 mM NAC. The phosphorylation of Akt was determined by Western blot analysis. D Schematic diagram of the signaling pathways involved in As2O3-induced cell apoptosis in cultured C2C12 myoblasts
Discussion
Myoblasts dictate skeletal muscle size, with adequate numbers of myoblasts being required for precise muscle regeneration (Deponti et al. 2007; Jansen and Pavlath 2008). It has been shown that lower concentrations of inorganic arsenic (0.02–0.5 μM) inhibit the myoblast differentiation (Steffens et al. 2011; Yen et al. 2010). Li et al. (2010) reported that higher concentrations of As2O3 (4–32 μM) obviously reduced smooth muscle cell viability in a concentration-dependent manner (Li et al. 2010). As2O3 (3–10 μM) could also induce apoptosis in cultured bone marrow mesenchymal stem cells (Cai et al. 2010; Yadav et al. 2010). Treatment with high concentrations of As2O3 (30, 60, and 90 μM) for various periods (24, 48, and 72 h) caused primary cardiomyocyte apoptosis in a dose- and time-dependent manner (Raghu and Cherian 2009). A previous clinic study has indicated that the plasma levels of arsenic are 5.54–7.30 μM in the patients of acute promyelocytic leukemia treated with As2O3 (Shen et al. 1997). In the present study, we found that treatment with As2O3 (3–10 μM) in C2C12 myoblasts significantly decreases cell viability and increases cell apoptosis in a dose-dependent manner.
The cytotoxic effects of As2O3 on various cell types are apparently mediated by apoptosis. Mitochondria play a crucial role in regulating apoptotic cell death (Green and Reed 1998). The mitochondria function is regulated by Bcl-2 family proteins, which can be subdivided into anti-apoptotic and pro-apoptotic members (Adams and Cory 2007). It has been reported that high ratio of Bax to Bcl-2 can cause a loss of MMP, resulting in the release of cytochrome c from the intermembrane space of mitochondria to the cytosol (Tophkhane et al. 2007). Then, the released cytochrome c triggers the formation of apoptosome-containing apoptotic protease activating factor 1 (Apaf-1) and caspase-9. Caspase-9 activates the effector procaspases, including procaspase-3, to carry out the process of apoptosis (Zou et al. 2003). In this study, a significant increase in Bax and a decrease in Bcl-2 expressions were observed in As2O3-treated myoblasts. We also found that As2O3 induces a decrease in MMP, an increase in cytochrome c release, and an increase in caspases (caspase-3/-9) activation and subsequently causes the PARP cleavage and apoptosis in myoblasts. These results suggest that a mitochondria-dependent pathway is involved in As2O3-induced apoptosis in myoblasts.
ER is the site of synthesis and folding of secretory proteins. The protein folding in the ER is impaired under various physiology and pathology conditions, collectively called ER stress (Kaufman 1999). In general, ER stress triggers three major branches of unfolded protein response (UPR) including the PERK/eIF2α, ATF6, and inositol-requiring enzyme (IRE)-1, which serve as proximal sensors of protein folding status in the ER (Ron and Walter 2007; Todd et al. 2008). Under ER stress, ER-localized chaperons are induced (GRP78 and GRP94), protein synthesis is slowed, and a protein degrading system is initiated (Szegezdi et al. 2003). However, when the ER functions are impaired beyond restoration, apoptosis occurs to protect the organism by eliminating damaged cells (Oyadomari and Mori 2004). CHOP plays a role in ER-induced apoptosis (Szegezdi et al. 2006). During ER stress, three arms of UPR induce the transcription of CHOP (Ma et al. 2002; Wang et al. 1998; Yoshida et al. 2000). CHOP is a proapoptotic transcription factor and can down-regulation of Bcl-2 protein and translocation of Bax protein from cytosol to mitochondria (Oyadomari and Mori 2004). Caspase-12 is an ER-resident caspase that is activated under ER stress conditions and can mediate apoptosis (Nakagawa and Yuan 2000). Once activated, caspase-12 causes cytochrome c-independent caspase-9 activation, followed by caspase-3 activation (Morishima et al. 2002). Recent studies have shown that the ER stress is involved in As2O3-induced apoptosis in human lens epithelial cells (Zhang et al. 2007), MC3T3-E1 osteoblasts (Tang et al. 2009), pancreatic β-cell-derived RIN-m5F cells (Lu et al. 2011). In the present study, the Western blot analysis revealed that exposed As2O3 in C2C12 myoblasts increases the expressions of p-PERK, p-eIF2α, GRP-78, GRP-94, and CHOP and the cleavages of ATF6 and caspase-12 proteins. These results indicate that ER stress participates in the As2O3-induced C2C12 myoblast apoptosis.
The generation of ROS is also one of the common responses to cellular injury and apoptotic cell death (Jimi et al. 2004). Previous studies demonstrated that treatment with hydrogen peroxide can induce C2C12 myoblast apoptosis, at least in part, through a mechanism involving the intrinsic mitochondrial cell death pathway (Jiang et al. 2005a, b). The accumulation of ROS is associated with the collapse of MMP and the subsequent oxidative damage to the mitochondrial membranes that impairs the membrane integrity, leading to disruption of MMP, cytochrome c release, caspase-9/-3 activation, and apoptosis (Antonsson 2001). A recent report has shown that As2O3 causes ER stress in pancreatic β-cell through the generation of excess ROS (Lu et al. 2011). In the present work, a significant increase in the levels of intracellular ROS was quantitatively recognized in C2C12 myoblasts after exposure to As2O3. Moreover, we also found that NAC, an antioxidant, significantly suppresses As2O3-induced ER stress and apoptotic events (CHOP expression, depolarization of MMP, release of cytochrome c, caspases-12/-9/-3 activation, and PARP cleavage). These results suggest that the generation of ROS is an early event that initiates and activates the ER stress- and mitochondria-related apoptotic pathways in C2C12 myoblasts exposed to As2O3.
Akt is an important mediator of growth factors and involved in cell growth and survival (Manning and Cantley 2007). Over-expression of Akt has been found to prevent apoptosis in many cell types and resulting in a resistance to or delay cell death (Fresno Vara et al. 2004). It has been reported that Akt regulates the apoptotic mechanism by phosphorylating and inactivating the Bcl-2 family member BAD, which controls the release of cytochrome c from mitochondria (Datta et al. 1997; Osaki et al. 2004). A modulating role of PI3k/Akt signaling in the expression of Bim, a Bcl-2 interacting mediator of cell death, has been demonstrated. LY294002, a PI3K inhibitor, could increase the Bim expression in cells concomitant with an increase in cell death (Qi et al. 2006). Furthermore, Akt mediated the cell survival through the inactivation of caspase-9 and FKHRL1, a member of the Forkhead family of transcription factors (Brunet et al. 1999). H2O2 induction of ROS has also been shown to inhibit the phosphorylation of Akt and phosphoinositide-dependent kinase 1 (PDK1) in neuronal cells (Chen et al. 2010). In the present study, we observed that As2O3 markedly decreases the phosphorylation of Akt in C2C12 myoblasts without changes in total Akt proteins. Pretreatment with NAC dramatically prevented the As2O3-inhibited Akt phosphorylation. We also found that over-expression of c.a. Akt significantly suppressed As2O3-promoted apoptotic cleavage of PARP. These results indicate that Akt may be as a protective (anti-apoptotic) role in As2O3-induced C2C12 myoblast apoptosis.
In conclusion, we demonstrated that apoptosis is involved in As2O3-induced myoblast cytotoxicity that is associated with intracellular ROS-regulated mitochondrial dysfunction, ER stress response, or Akt inactivation (Fig. 6d). In addition, pretreatment with NAC inhibited As2O3-induced myoblast apoptosis. These findings suggest that As2O3 may be an important environmental risk factor for skeletal muscle cell development/growth.
References
Adams JM, Cory S (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26:1324–1337
Antonsson B (2001) Bax and other pro-apoptotic Bcl-2 family “killer-proteins” and their victim the mitochondrion. Cell Tissue Res 306:347–361
Boonstra J, Post JA (2004) Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 337:1–13
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868
Cai BZ, Meng FY, Zhu SL, Zhao J, Liu JQ, Liu CJ, Chen N, Ye ML, Li ZY, Ai J, Lu YJ, Yang BF (2010) Arsenic trioxide induces the apoptosis in bone marrow mesenchymal stem cells by intracellular calcium signal and caspase-3 pathways. Toxicol Lett 193:173–178
Chao DT, Korsmeyer SJ (1998) BCL-2 family: regulators of cell death. Annu Rev Immunol 16:395–419
Charge SB, Rudnicki MA (2004) Cellular and molecular regulation of muscle regeneration. Physiol Rev 84:209–238
Chen YC, Lin-Shiau SY, Lin JK (1998) Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis. J Cell Physiol 177:324–333
Chen YW, Huang CF, Tsai KS, Yang RS, Yen CC, Yang CY, Lin-Shiau SY, Liu SH (2006) Methylmercury induces pancreatic beta-cell apoptosis and dysfunction. Chem Res Toxicol 19:1080–1085
Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S (2010) Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest 90:762–773
Choi YJ, Park JW, Suh SI, Mun KC, Bae JH, Song DK, Kim SP, Kwon TK (2002) Arsenic trioxide-induced apoptosis in U937 cells involve generation of reactive oxygen species and inhibition of Akt. Int J Oncol 21:603–610
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241
Deponti D, Francois S, Baesso S, Sciorati C, Innocenzi A, Broccoli V, Muscatelli F, Meneveri R, Clementi E, Cossu G, Brunelli S (2007) Necdin mediates skeletal muscle regeneration by promoting myoblast survival and differentiation. J Cell Biol 179:305–319
Florea AM, Splettstoesser F, Busselberg D (2007) Arsenic trioxide (As2O3) induced calcium signals and cytotoxicity in two human cell lines: SY-5Y neuroblastoma and 293 embryonic kidney (HEK). Toxicol Appl Pharmacol 220:292–301
Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M (2004) PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 30:193–204
Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G (2006) Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 13:1423–1433
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312
Hawke TJ, Garry DJ (2001) Myogenic satellite cells: physiology to molecular biology. J Appl Physiol 91:534–551
Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ (1990) Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348:334–336
Jansen KM, Pavlath GK (2008) Prostaglandin F2alpha promotes muscle cell survival and growth through upregulation of the inhibitor of apoptosis protein BRUCE. Cell Death Differ 15:1619–1628
Jiang B, Xiao W, Shi Y, Liu M, Xiao X (2005a) Heat shock pretreatment inhibited the release of Smac/DIABLO from mitochondria and apoptosis induced by hydrogen peroxide in cardiomyocytes and C2C12 myogenic cells. Cell Stress Chaperones 10:252–262
Jiang B, Xiao W, Shi Y, Liu M, Xiao X (2005b) Role of Smac/DIABLO in hydrogen peroxide-induced apoptosis in C2C12 myogenic cells. Free Radic Biol Med 39:658–667
Jimi S, Uchiyama M, Takaki A, Suzumiya J, Hara S (2004) Mechanisms of cell death induced by cadmium and arsenic. Ann NY Acad Sci 1011:325–331
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233
Kuo ML, Chuang SE, Lin MT, Yang SY (2001) The involvement of PI 3-K/Akt-dependent up-regulation of Mcl-1 in the prevention of apoptosis of Hep3B cells by interleukin-6. Oncogene 20:677–685
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371:346–347
Li JX, Shen YQ, Cai BZ, Zhao J, Bai X, Lu YJ, Li XQ (2010) Arsenic trioxide induces the apoptosis in vascular smooth muscle cells via increasing intracellular calcium and ROS formation. Mol Biol Rep 37:1569–1576
Lu TH, Su CC, Chen YW, Yang CY, Wu CC, Hung DZ, Chen CH, Cheng PW, Liu SH, Huang CF (2011) Arsenic induces pancreatic beta-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways. Toxicol Lett 201:15–26
Ma Y, Brewer JW, Diehl JA, Hendershot LM (2002) Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol 318:1351–1365
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129:1261–1274
Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9:493–495
Mitchell PO, Pavlath GK (2001) A muscle precursor cell-dependent pathway contributes to muscle growth after atrophy. Am J Physiol Cell Physiol 281:C1706–C1715
Miyake M, Hayashi S, Iwasaki S, Uchida T, Watanabe K, Ohwada S, Aso H, Yamaguchi T (2011) TIEG1 negatively controls the myoblast pool indispensable for fusion during myogenic differentiation of C2C12 cells. J Cell Physiol 226:1128–1136
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y (2002) An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277:34287–34294
Nakagawa T, Yuan J (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 150:887–894
Navas-Acien A, Sharrett AR, Silbergeld EK, Schwartz BS, Nachman KE, Burke TA, Guallar E (2005) Arsenic exposure and cardiovascular disease: a systematic review of the epidemiologic evidence. Am J Epidemiol 162:1037–1049
Orrenius S, Gogvadze V, Zhivotovsky B (2007) Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol 47:143–183
Osaki M, Oshimura M, Ito H (2004) PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 9:667–676
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389
Porter AG, Janicke RU (1999) Emerging roles of caspase-3 in apoptosis. Cell Death Differ 6:99–104
Qi XJ, Wildey GM, Howe PH (2006) Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J Biol Chem 281:813–823
Raghu KG, Cherian OL (2009) Characterization of cytotoxicity induced by arsenic trioxide (a potent anti-APL drug) in rat cardiac myocytes. J Trace Elem Med Biol 23:61–68
Reed JC (1995) Regulation of apoptosis by bcl-2 family proteins and its role in cancer and chemoresistance. Curr Opin Oncol 7:541–546
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529
Rosenblatt JD, Yong D, Parry DJ (1994) Satellite cell activity is required for hypertrophy of overloaded adult rat muscle. Muscle Nerve 17:608–613
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY (1997) Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89:3354–3360
Steffens AA, Hong GM, Bain LJ (2011) Sodium arsenite delays the differentiation of C2C12 mouse myoblast cells and alters methylation patterns on the transcription factor myogenin. Toxicol Appl Pharmacol 250:154–161
Szegezdi E, Fitzgerald U, Samali A (2003) Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann NY Acad Sci 1010:186–194
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7:880–885
Tang CH, Chiu YC, Huang CF, Chen YW, Chen PC (2009) Arsenic induces cell apoptosis in cultured osteoblasts through endoplasmic reticulum stress. Toxicol Appl Pharmacol 241:173–181
Tews DS, Goebel HH (1997) DNA-fragmentation and expression of apoptosis-related proteins in muscular dystrophies. Neuropathol Appl Neurobiol 23:331–338
Tidball JG, Albrecht DE, Lokensgard BE, Spencer MJ (1995) Apoptosis precedes necrosis of dystrophin-deficient muscle. J Cell Sci 108:2197–2204
Todd DJ, Lee AH, Glimcher LH (2008) The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol 8:663–674
Tophkhane C, Yang S, Bales W, Archer L, Osunkoya A, Thor AD, Yang X (2007) Bcl-2 overexpression sensitizes MCF-7 cells to genistein by multiple mechanisms. Int J Oncol 31:867–874
Torres M, Forman HJ (2003) Redox signaling and the MAP kinase pathways. BioFactors 17:287–296
Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D (1998) Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J 17:5708–5717
Yadav S, Shi Y, Wang F, Wang H (2010) Arsenite induces apoptosis in human mesenchymal stem cells by altering Bcl-2 family proteins and by activating intrinsic pathway. Toxicol Appl Pharmacol 244:263–272
Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH (2010) Arsenic inhibits myogenic differentiation and muscle regeneration. Environ Health Perspect 118:949–956
Yen CC, Ho TJ, Wu CC, Chang CF, Su CC, Chen YW, Jinn TR, Lu TH, Cheng PW, Su YC, Liu SH, Huang CF (2011) Inorganic arsenic causes cell apoptosis in mouse cerebrum through an oxidative stress-regulated signaling pathway. Arch Toxicol 85:565–575
Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K (2000) ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol 20:6755–6767
Yoshida T, Yamauchi H, Fan Sun G (2004) Chronic health effects in people exposed to arsenic via the drinking water: dose-response relationships in review. Toxicol Appl Pharmacol 198:243–252
Zhang H, Duncan G, Wang L, Liu P, Cui H, Reddan JR, Yang BF, Wormstone IM (2007) Arsenic trioxide initiates ER stress responses, perturbs calcium signalling and promotes apoptosis in human lens epithelial cells. Exp Eye Res 85:825–835
Zou H, Yang R, Hao J, Wang J, Sun C, Fesik SW, Wu JC, Tomaselli KJ, Armstrong RC (2003) Regulation of the Apaf-1/caspase-9 apoptosome by caspase-3 and XIAP. J Biol Chem 278:8091–8098
Acknowledgments
This study was supported by grant from the National Science Council of Taiwan (NSC97-2314-B-002-052-MY3).
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yuan-Peng Yen and Keh-Sung Tsai contributed equally to this work.
Rights and permissions
About this article
Cite this article
Yen, YP., Tsai, KS., Chen, YW. et al. Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Arch Toxicol 86, 923–933 (2012). https://doi.org/10.1007/s00204-012-0864-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-012-0864-9