
Abstract
The nuclear factor erythroid 2(NFE-2)-related factor 2 (Nrf2), a transcription factor, is a major player in antioxidant and detoxification system implied by the cells in normal physiological condition as well as during intrinsic or extrinsic cellular stress. During tumorigenesis, activation of Nrf2 could be tumor suppressive or oncogenic. Aberrant Nrf2 signaling makes tumor cells resistant to chemotherapy and radiation. However, Nrf2 can also be cytoprotective in nature where it acts independently or in crosstalk with other cellular signaling pathways to maintain cellular homeostasis. In this book chapter, we highlight the role of Nrf2 in various types of cancers describing Nrf2 signaling in inflammation and oxidative stress to support either cancer cell survival or death. Current status of Nrf2 as a target in cancer therapy is also discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Reactive oxygen species (ROS) are continuously generated in our body due to internal metabolism as well as external environmental exposure. In physiological conditions, cells produce ROS for useful purposes and ROS signaling protects cells during inflammation and stress response. Moreover, it plays crucial role during cell division and autophagy to propagate and maintain homeostasis [1]. However, ROS generation and its accumulation in excess result in oxidative stress which is harmful for many cellular processes. Oxidative stress leads to inflammation, immune disorders, and many diseases like cancer and aging [2].
Nrf2 is a transcription factor that belongs to cap ‘n’ collar (CNC) subfamily of basic region leucine zipper (bZip) transcription factors. Nrf2 was first cloned as a factor bound to NFE2-binding motif in β-globin gene responsible for erythropoiesis development and platelet development [3]. Although Nrf2 is not necessary for differentiation of hematopoetic cells, it was found to induce a group of drug-metabolizing enzymes (DMEs), such as NAD(P)H:quinone oxidoreductase 1 (NQO1) and glutathione S-transferase (GST) by means of antioxidants and electrophiles [4, 5]. Induction of such proteins promotes detoxification and elimination of several endogenous as well exogenous chemicals and thereby Nrf2 regulates oxidative stress response caused by inflammation [6].
Over past few decades, from several studies, the major role of Nrf2 has been shown to produce resistance to oxidant stress. While searching for genome-wide targets of Nrf2, many antioxidant response element (ARE) containing genes responsible for antioxidant homeostasis and drug detoxification have been identified [7]. Cells maintain concentration of Nrf2 in a very controlled manner, where Nrf2 is repressed by Keap1 (Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1). Under normal cellular condition, Keap1 mediates ubiquitination and subsequent proteosomal degradation of Nrf2 [8]. However, the protective nature of Nrf2 can also be exploited by tumor cells to build a prosurvival niche for further tumor progression and drug resistance [9]. In this context, the emerging molecular mechanism and function of Nrf2 in the regulation of inflammation-mediated cancer are described below.
The function of Nrf2 signaling in cancer cells, in response to toxic chemicals and radiations (ionizing or ultraviolet radiation) has been well established [10, 11]. Cancer progression has been found to be prevented by Nrf2 through several pathways such as by generation of many antioxidants and detoxifying enzymes to suppress the reactive oxygen species (ROS). Recently, it has been found that cancer cells overexpress Nrf2 which ultimately leads to cancer progression [4, 12] and metastasis [13]. With ongoing research, the functions of Nrf2 in various phases of cancer progression have been highlighted deeply.
Although Nrf2 activity is majorly regulated by its inhibitor Keap1, many other regulatory mechanisms are also in existence. Nrf2 activity is regulated at the level of transcription, translation, and post-translational phase independent of Keap1-mediated regulation. Nrf2 is regulated by protein kinases like JNK, GSK3β, PKC, and Akt [14]. Nrf2 binding to caveolin-1 or p21 also alters its functioning. Moreover, Nrf2 could be regulated at epigenetic level as well by various microRNAs like miRNA-28, 144, and -200a [15]. The above-mentioned processes are equally vital factors for the activation of Nrf2 to keep the cellular redox potential in a balanced state to reduce the possibility of inflammation and thereby progression to cancer development.
2 Regulation of Nrf2 and Keap1
2.1 Keap1-Dependent Regulation
Keap1 plays an important role as redox sensor and involved in cellular defense by regulating the Nrf2 protein under normal as well as stressed conditions. In inactive state, Keap1 forms a homodimer to sequester Nrf2 in the cytosol and prevents Nrf2 translocation in the nucleus in association with the actin cytoskeleton [16]. In addition, Keap1 mediates the Cul3-based poly-ubiquitination and proteasomal degradation of Nrf2.
2.2 Keap1-Dependent Proteasomal Degradation of Nrf2
The continuous activation of oxidative stress could be prevented by maintaining Nrf2 at very low level [17, 18]. Proteasomal degradation of Nrf2 promoted by Keap1 protein makes Nrf2 a high turnover protein with a half-life of 10–20 min only. Nrf2-Keap1 binding is based on double glycine repeat domain (DLG domain) in Keap1 and Neh2 domain in Nrf2 [19]. Two motifs within the Neh2 domain exhibit different affinity for Keap1; one binds strongly whereas, the other binds with low affinity. In normal cellular conditions, Nrf2 concentration is kept lowered through weak binding of Nrf2-Keap1 and thus targeting Nrf2 for ubiquitination by Cul3-E3 ligase and thereby degradation by 26S proteasomal pathway [20]. (Fig. 1).

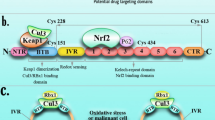
Domain structure of Nrf2. The Nrf2 protein contains seven conserved Nrf2–ECH homology (Neh) domains. Neh1 domain binds to DNA and dimerizes with transcription factors; Neh2 binds to Keap1 through DLG and ETGE motif and contains seven lysine residues for ubiquitin ligation; Neh3 is needed for transcriptional activation; Neh4 and Neh5 are transactivation domains; Neh6 regulates Nrf2 stability; Neh7 is responsible for RXR/binding
2.3 Keap1 Degradation by Autophagy
The proteasome is a subcellular structure that is utilized for the degradation of specific proteins targeted for destruction. However, autophagy is generally considered as degradation pathway for nonspecific proteins, unfolded proteins, and cellular organelles. Findings from past few decades suggest that the autophagic degradation pathway is also capable of degrading specific, targeted proteins [21,22,23]. By binding to poly-ubiquitinated protein marked for degradation and to autophagosome, sequestosome1 (p62), a substrate adaptor via its multiple protein–protein interaction capability targets specific cellular proteins to undergo degradation via autophagosome [23, 24]. From some studies it has been elucidated that between Keap1 and p62 there is a significant relationship, where p62 regulates degradation of Keap1 by autophagy. A reduced expression of Keap1 has been observed in a group of cell line where p62 was overexpressed. On the other hand, when p62 was knocked out by siRNA, a high level of Keap1 protein expression with decreased protein level of Nrf2- and Ner2-regulated gene were observed [25]. When p62 is absent in a cell, the cellular Keap1 concentration is maintained twice as compared to normal level. When p62–Keap1 interaction mechanism was thoroughly studied, a concept has arrived which states that, under oxidative stress, Keap1 bound with Nrf2 undergoes conformational change, which allows release of Nrf2, making an empty site in Keap1 protein where p62 can bind by STGE motif which can be compared with ETGE motif of Nrf2. Binding of p62 with Keap1 allows LC3 to bind which was already bound to autophagosome membrane and allow Keap1 for degradation [26].
3 Keap1-Independent Regulation
From many studies, it has been found that Keap1 is not only one candidate for regulation of Nf2. Sulforaphane, an Nrf2 inducer, overexpressed without hampering the binding between Nrf2 and Keap1. This explains the presence of alternative pathways for Nrf2 activation [27]. The expression and activity of a protein can be regulated at various time scale including transcriptional, post-transcriptional, protein abundance, post-translational modification, and subcellular localization. The phosphorylation of Nrf2 by several signaling machinery and the involvement of epigenetic factors such as microRNAs may also play a role in Nrf2 activation. In this section, alternative processes of Nrf2 regulation are thoroughly described.
3.1 Auto-Regulation and Transcriptional Regulation
Nrf2 acts as a transcription factor by up regulating the expression of genes that contain antioxidant response element (ARE) sequence in the promoter region. Likewise, the aryl hydrocarbon receptor [28] up regulates production of a number of phase I antioxidant enzymes such as Cytochrome p450 which contains xenobiotic response element (XRE) at their promoter site [29] which mediates the group of reactive intermediates, and these reactive intermediates can deliberately activate the antioxidant signaling pathway through the ARE. In another study, a compound TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) an inducer for both Nrf2 and AHR can induce all those genes which contain both ARE and XRE on their promoter region [30]. For some genes, which contain promoter where both Nrf2 and AHR can bind and induce gene expression. Interestingly, in Nrf2 promoter the ARE/XRE sequence has been found suggesting the possibility of auto-regulation of Nrf2 at transcriptional level. The XRE and ARE regions are present in close vicinity within the promoter region of Nrf2 suggesting the possibility of Nrf2 to control its own transcription that is auto-regulation. In support of this another group showed that the D3T (3H-1, 2-dithiole-3-thione), an inducer of ARE, increases Nrf2 at both the RNA and protein levels, and this overexpression can be inhibited by cyclohexamide, a transcriptional inhibitor. When ARE sequence is inserted at the promoter region of luciferase enzyme gene, its overexpression in the presence of Nrf2 is the evidence of the auto-regulation of Nrf2, allowing a positive feedback loop providing cellular defense against oxidative stress [31]. Many chemo-preventive agents are capable of trigger Nrf2 gene expression suppressing NF-κB activity. NF-κB can repress transcription of genes which contain ARE at their promoter region. In the promoter sequence of Nrf2, there is a region where NF-κB can bind and down regulate those genes which are expressed by Nrf2. This finding suggests that a crosstalk between NF-κB and Nrf2 regulate cellular response against oxidative stress [32].
3.2 Post-Transcriptional Modulator: microRNAs
Over the past decades, microRNAs acquire attention for their function to control the modification of many signaling pathways, as well as the Nrf2 pathway. microRNAs including miR-28 [33], miR-144 [34], miR-200 [15], and miR-34 [35] play a crucial part for Nrf2 signaling in response to oxidative stress. In erythroid cells, an inhibitory effect of miR-144 on Nrf2 signaling pathway has been found. When expression of miR-144 is activated in erythroid cells, a significant loss of Nrf2 protein level with subsequent decrease of restoration of glutathione and modification of antioxidant activity proves the inverse effect of miR-144 on Nrf2 pathway [34]. In breast epithelial cells, a similar relationship was observed within miR-28 and Nrf2. MiR-28 has been seen in various cancers, e.g., lymphoma, glioma, squamous carcinoma, etc. and it is found to bind with Nrf2 mRNA resulting in its degradation along with induction of Nrf2 protein degradation. Neither the induction of Keap1 protein nor the binding between Nrf2 and Keap1 has been affected in the presence of miR-28 explaining that miR-28 function as Nrf2 regulator which is independent of Keap1 [33].
3.3 Post-Translational Modifications: Phosphorylation/Acetylation
In response to oxidative stress, Nrf2 as transcription factor, move in the nucleus to activate various proteins to reduce cellular ROS. After maintaining cellular redox state when the activation of Nrf2 as transcription factor is no longer needed, the nuclear Nrf2 is phosphorylated which leads Nrf2 for nuclear export thereby degradation. Various pathways like extracellular signal-regulated kinase (ERK), glycogen synthase kinase 3 beta (GSK3β), protein kinase C (PKC), phosphatidylinositol 3-kinase (PI3K/AKT), mitogen-activated protein kinase (MAPK) signaling pathways, etc. play crucial roles in this regulation. In response to oxidative stress, protein kinase C (PKC) phosphorylate at Ser40 of Neh2 domain of Nrf2 interrupts the binding of Keap1 with Nrf2; the free Nrf2 then moves into the nucleus [14]. PKC belongs to a family of serine/threonine kinases which can be subdivided into three classes: classical, novel, and atypical. In a study, the atypical class of PKC phosphorylates Nrf2 which causes Nrf2 to transport into the nucleus. The translocated Nrf2 then acts as transcription factor to allow the expression of phase II antioxidant enzymes in response to oxidative stress [36]. The nuclear localization sequences (NLSs) and nuclear export sequences [27] in Nrf2 have been identified and confirmed that after released from Keap1, the free Nrf2 cannot move into the nucleus by its own. For translocation of Nrf2 into the nucleus, certain adaptor proteins like importin forms Nrf2-Importin complex and moves it through nuclear envelope [37]. Casein kinase 2 (CK2) targets approximately 13 potential phosphorylation sites within Nrf2 sequence [38] These phosphorylations by CK2 help Nrf2 for nuclear translocation, which can be repressed by inhibitors of CK2 [39]. The interplay between many signaling pathways regulating post-translational modification of Nrf2 will help to understand cell defense mechanism against oxidative stress.
3.4 Maintaining Cellular Homeostasis by Nrf2
To maintain normal cellular homeostasis, the function of Nrf2 is regulated by several ways. To bind with ARE/EpRE sequence, Nrf2 heterodimerization occurs with musculoaponeurotic fibrosarcoma oncogene homolog (Maf) protein. This Maf protein, which is bZIP type transcription factors, is also required for transactivation of Nrf2 [40,41,42]. In human Nrf2 protein, seven highly conserved regions known as Nrf2-ECH homology (Neh) domains have been found. Among the family of Neh domains, the Neh1 domain contains a CNC–bZIP structure which mediates heterodimerization with Maf [43]. Bach which is an ARE/EpRE-binding protein acts as a transcriptional repressor of Nrf2 especially in heme oxygenase-1 gene expression involved in oxidative stress response [44]. Neh4 and Neh5 domains play an important role in the transactivation activity of Nrf2. After binding with CREB-binding protein (CBP), the potency of Nrf2 as transcriptional activator increases by nearly 100-fold [45,46,47].
Nrf2 can be stabilized by phase II inducers. Till now, nine structurally diverse chemical groups have been identified as inducers those are involved in activation of phase II genes by Nrf2 [48]. Within a cell, primary sensors are found and they have certain cysteine residues in the polypeptide chain which actively participate in redox sensing. These phase 2 inducers can all modify sulfhydryl groups of reactive cysteine residues by alkylation, oxidation, or reduction. Interestingly, Keap1 has 27 cysteine residues which are highly reactive; as a result, phase II inducers sense those reactive cysteine residues of Keap1 and target Keap1 and allow the release of Nrf2. Dexamethasone mesylate, a sulfhydryl reactive inducer, directly reacts with cysteine amino acids of Keap1 and triggers dissociation of Keap1 from Nrf2 [49]. The phase 2 inducer 15-Deoxy-delta-12, 14-prostaglandin J2 (15d- PGJ2) interacts directly with Keap1 inducing Nrf2 [50]. Among the most reactive cysteine residues, mutations in Cys273 or Cys288 make Keap1 incapable of suppressing Nrf2 activity [51, 52]. Phase 2 inducers are also found to be activating some protein kinases as mediators of oxidative stress, for example, extracellular signal-regulated kinases (ERK) [53,54,55], MAPK/ERK kinase-1 [56], MEK kinase 1 [57], p38 mitogen-activated protein kinase (MAPK) [54, 55], PKR-like endoplasmic reticulum kinase (PERK) [58], phosphatidylinositol 3-kinase (PI3K) [59, 60], and protein kinase C (PKC) [36, 61, 62]. Furthermore, specific kinase inhibitors are capable of blocking the phase II antioxidant enzymes and ARE/XRE-triggered gene induction [14, 62]. It is possible that PKC and PERK and also their upstream signaling molecules may be playing some role as sensors for oxidative stress (Fig. 2).

Maintaining cellular homeostasis by Nrf2. Reactive oxygen species (ROS), either from external sources or generated within cell during inflammation, causes cancer. Nrf2 in response to ROS induces a group of antioxidants phase 2 scavengers, detoxifying enzymes which play an important role in fighting against cancer. Many other pathways like NF-κB, MAPK, and PKC also regulate Nrf2 signaling
4 Nrf2 Signaling
Nrf2 regulates expression of an array of antioxidant genes upon oxidative stress. Of many oxidative stress-induced pathways, Nrf2-Keap1 signaling plays a pivotal protective role for normal cells. Moreover, since Nrf2-deficient mice are more prone to carcinogenesis [63, 64] and Nrf2 loss leads to enhanced metastasis, it is thought to act as a tumor suppressor [65, 66]. However, recent studies have indicated an oncogenic function of Nrf2. Cancer cells are consistently under oxidative stress and to overcome this problem, Nrf2 has been found to highly express in these cells thereby enhancing their therapeutic resistance. In the following section, we will elaborate signaling of Nrf2 in normal and cancer cells as well.
4.1 Nrf2 Signaling in Normal Cells
In physiological conditions, Nrf2 remains in cytosol and binds to Kelch/DGR domain of its inhibitor Keap1 with ETGE and DLG motifs present in Neh2 domain of Nrf2. Upon binding to these two sites, Keap1 targets Nrf2 for degradation. Keap1-dependent degradation is Culin3 mediated which is E3-ubiquitin ligase and conjugates ubiquitin moieties at the seven lysine residues present in the Neh2 domain of Nrf2. As a result, in the absence of any stress, Nrf2 is degraded in the cytosol and unable to translocate to the nucleus [67, 68].
However, cellular stress alters Nrf2 binding to Keap1 by its DLG domain. Stress changes Keap1 conformation by oxidizing cysteine residues present in its intervening region (IVR). Conformationally modified Keap1 is unable to bind Nrf2 at DLG domain. Now Keap1 remains bind to that Nrf2 which is not further subjected to poly-ubiquitination and degradation. Thereby newly translated Nrf2 translocates to the nucleus. In nucleus, Nrf2 binds to antioxidant response element (ARE) of a number of genes which are involved in cytoprotection. Nrf2 binding to ARE is mediated by masculoaponeurotic fibrosarcoma (Maf) family proteins. By dimerizing with Nrf2, Maf family proteins have two functions: (1) to facilitate Nrf2 binding to ARE, (2) to recruit co-activator for transcriptional activation of genes. Genes whose transcription is activated by Neh2 binding are related to antioxidant response, phase II detoxifying enzymes, glutathione synthesis, drug metabolism, drug resistance, HIPK2 signaling, etc. (Fig. 3) [67, 68].

Nrf2 signaling in normal and stressed conditions. In physiological conditions, Nrf2 undergoes Keap1-mediated proteasomal degradation by Cul3-ubiquitin ligase. In cellular stress, it dissociates with Keap1 and translocates into the nucleus and induce expression of various genes that are involved in maintaining cellular homeostasis
4.2 Nrf2 Signaling in Tumor Suppression
It is a known that Nrf2 regulates expression level of genes important for antioxidant functions. These genes exert cytoprotective effects by their anti-inflammatory role but if inflammation persists, it results into carcinogenesis via formation of tumor-supportive microenvironment [69]. During carcinogenesis when a normal cell changes into a transformed cell, Nrf2 prevents this process by its antioxidant and anti-inflammatory functions [70]. Specific mutations in Nrf2 or its suppression during this process leads to carcinogenesis. Hence, it is thought that Nrf2 acts as tumor suppressor and tumor suppressive role of Nrf2 has been investigated thoroughly. By studying mutations in Nrf2 or knocking down Nrf2 gene, several reports have shown that Nrf2 is a tumor suppressor and its deletion/suppression leads to cervical, colorectal, oral, head and neck, lung, pancreatic, hepatic, and skin carcinogenesis [71,72,73]. In Nrf2 null mice, there is decreased expression of HO-1, NQO1, GST, GCL (glutamate-cysteine ligase), and UGT (UDP-glucuronosyltransferase) in comparison to Nrf2 wild type mice. Moreover, a single nucleotide polymorphism at rs6721961 locus in human Nrf2 promoter results in its decreased levels which enhances the risk for non-small-cell lung cancer (NSCLC) [50]. In the first stage of metal-induced carcinogenesis, Nrf2 acts as anti-oncogenic protein and inhibits malignant cell transformation. Nrf2 is present at higher level in lung adenocarcinoma tissues. However, its knockdown enhances arsenic-induced transformation in BEASE-2B cell in the process of lung carcinogenesis [74]. Nrf2 knockdown results in decreased level of ROS which increases cell survival and proliferation during transformation and leads to tumorigenesis. Therefore, antioncogenic function of Nrf2 is due to its antioxidant gene regulation which reduces ROS level and thereby cell transformation. Likewise, exposure of BEAS-2BR cells with cadmium also leads to lung carcinogenesis. In this process, cadmium exposure leads to ROS generation in untransformed cells. ROS generation leads to increased level of TNF-α which activates NF-κB and COX-2 ultimately resulting in tumor forming inflammatory microenvironment. Overexpression of Nrf2 downstream targets resulted in reduced ROS level and inhibition of cadmium induced malignant cell transformation [70]. Additionally, Nrf2 null mice are highly prone to chemical (DMBA or TPA)-induced skin carcinogenesis. Mice having dominant-negative mutant of Nrf2 (dnNrf2) in basal keratinocytes of the epidermis show a prolonged inflammatory response following skin injury and develop skin carcinomas. Here, the basal level of Nrf2 target genes is needed to prevent skin tumorigenesis. Hence, reduced chemical-induced detoxification as well as increased oxidative damage increases susceptibility of dnNrf2 mice for skin tumorigenesis [73]. Nrf 2 has an important role in colon carcinogenesis also. Nrf2 knockout results in chronic inflammation with higher expression of inflammatory marker genes such as COX-2, cPLA (cytosolic phospholipase A2), and LTB4 (leukotriene B4), along with it there is a higher level of PCNA and cMyc in intestinal tissue of respective mice. These Nrf2 knockout mice develop higher number of tumors upon DSS treatment [75]. Collectively, Nrf2 knockout inhibits oxidative stress response pathway, augments inflammation and also increases proliferation of intestinal crypt cells resulting in intestinal carcinogenesis. In significant number of breast cancers cell lines, Nrf2 is present in low level. This is due to increased Cul3-mediated proteasomal degradation of Nrf2.
4.3 Nrf2 Signaling in Tumor Promotion
In 2004, Ikeda et al. highlighted the oncogenic function of Nrf2 in liver carcinoma for the first time [76]. In subsequent years, increasing pieces of evidence suggested that Nrf2 could act as oncogene as well. Constitutive Nrf2 activation results in many types of cancers.
During carcinogenesis, as a cell gets transformed it has higher proliferation capacity. Nrf2 has been found to be involved in this increased proliferation capacity in many cell types. Nrf2 does so by up regulating expression of genes involved in NADPH formation such as transaldolase 1 (TALDO1), phosphogluconate dehydrogenase (PGD), transketolase (TKT), and glucose-6-phosphate dehydrogenase (G6PD). In addition, transcription of some genes of metabolic pathway like isocitrate dehydrogenase 1 (IDH1), phosphoribosyl pyrophosphate amidotransferase (PPAT), methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), and malic enzyme 1 (ME1) is also enhanced. This leads to increased cellular level of purines which is a raw material for DNA and RNA synthesis. Hence, transformed cells proliferate at a higher rate than their normal counterparts [68, 77].
In some tumors, Nrf2 is hyperactive and protects cancer cells from oxidative stress. This hyperactivation of Nrf2 could be due to mutation, increased transcription, or post-translational modification. Hyperactive Nrf2 increases tumorigenesis by positive feedback loop between Nrf2 signaling and histone deacetylase 4 (HDAC4) and pentose phosphate pathway (PPP). Nrf2 directly regulates expression of apoptotic genes like Bcl-2 and Bcl-xL [68].
During arsenic-induced carcinogenesis, Nrf2 is responsible for protection of transformed cells by lowering the level of ROS and apoptosis in BEAS-2B cells. This lowered ROS level is due to increased expression of antioxidant genes like catalase and superoxide dismutase-2 and apoptosis resistance was due to direct regulation of Bcl-2 and Bcl-xL gene expression by Nrf2 [74]. Thus in lung adenocarcinoma, increased level of Nrf2 and its downstream targets helps in malignant cell transformation and tumorigenesis.
Likewise, cadmium also induces lung carcinogenesis. In cadmium-transformed BEAS-2BR cells, there is inhibition of autophagy which leads to increased level of p62. P62 is a competitive inhibitor of Nrf2 for Keap1 binding. Hence, it binds to Keap1 thereby increasing the level of Nrf2 into nucleus. In nucleus, Nrf2 increases expression of catalase and superoxide dismutase resulting in decreased ROS level. It also increases expression of anti-apoptotic genes like Bcl-2 and Bcl-xL, promoting resistance to cell death [70].
Overexpression of Nrf2 is also related to breast carcinogenesis. In MCF7 and MDA-MB-231 cell lines, NRF2 knockdown by siRNA results in decreased cell proliferation and cell migration during metastasis process [78]. Nrf2 regulates breast cancer metastasis by positively regulating RhoA GTPase which is important in cell migration [79]. RhoA along with its downstream effector proteins enhances stress fiber and focal adhesion formation which is necessary in breast cancer metastasis. Additionally, Nrf2 binds and silences estrogen-related receptor-α (ERR1) expression which is a negative regulator of Rho protein. Moreover, Nrf2 enhances breast cancer progression by activating HIF1-α expression which is a key transcription factor sensing hypoxia stress and regulating genes involved in angiogenesis, apoptosis, cell proliferation, and survival [78].
In cervical cancers, nuclear expression of Nrf2 is increased. Higher Nrf2 level is associated with reduced Keap1 expression due to hypermethylation of its gene. Moreover, increased Nrf2 correlates with decreased apoptosis, enhanced cell proliferation, induced cell migration, and invasion of SiHa (cervical cancer cells) cells [71].
Nrf2 protein level constantly remains high in head and neck squamous cell carcinoma (HNSCC) due to gain-of-function mutation in Nrf2 gene [80]. Likewise, Nrf2 expression is higher in oral squamous cell carcinoma (OSCC) in comparison to normal squamous mucosa [81].
5 Nrf2 Signaling in Maintaining Genomic Stability
Since genomic alterations are one of the basic needs for a cell to get transformed into cancerous cell, any lesion that causes genomic instability leads to cancer. Reactive oxygen species, ultraviolet radiation, X-rays, ionizing radiation, and most of the chemotherapeutic drugs tend to deregulate genomic stability either to develop cancer or to treat it. Various checkpoints act at specific places in a cell as surveillance to inhibit progression of this instability. Along with checkpoints, many repair pathways are also present to rectify genomic alterations. Up regulation of repair genes or down regulation of checkpoint proteins is the mechanism deployed by cancer cells to show drug or chemo-resistance or to maintain their growth. Cancer cells override the barrier of checkpoints or enhance repair capacity for their unperturbed cell cycle progression or to develop drug resistance. They do so by incorporating mutations in genes of DNA repair pathway and checkpoint pathway proteins. Nrf2 alters expression of genes involved in base excision repair, non-homologous end joining, and homologous recombination. One of the enzymes in base excision repair pathway, 8-oxoguanine DNA glycosylase (OGG1), rectifies oxidized lesions at guanine bases induced by reactive oxygen species. Nrf2 regulates expression of OGG1 in mitochondria as well as in nucleus to enhance base excision repair capacity of cells during oxidative stress and thereby protects them from programmed cells death due to unrepaired bases [82]. During radiation therapy, cancer cells activate expression of Nrf2 which in turn up regulates p53-binding protein 1 (53BP1) expression. Elevated level of 53BP1 enhances non-homologous end joining capacity of cancer cells and thereby helps to develop radiation resistance [83]. Moreover, many genes in DNA repair pathway contain AREs, their regulation by Nrf2 is yet to be resolved clearly. However, Nrf2 regulates ionizing radiation-induced Rad51 foci formation which is involved in homologous recombination repair. It does so by increasing mRNA level of Rad51 and thereby increasing radiation resistance in cancer cells [84]. To increase homologous recombination repair capacity of cancer cells, BRCA1, whose gene is up regulated in breast and ovarian cancers, binds to Nrf2 and protects it by Keap1-dependent degradation. This enhances survival of cancer cells in oxidative stress conditions (Fig. 4) [85].

Nrf2 signaling in maintaining genomic stability. Following DNA damage either by tumor oxidative stress microenvironment or by chemo- or radiotherapy, cells activate Nrf2 expression which thereby increases cells’ ability to repair DNA damage that results in their survival and resistance to therapy
6 Nrf2 Signaling in Autophagy
Autophagy is a vital process for cells to maintain cellular homeostasis. It is necessary for nucleic acid synthesis, degradation of aggregated proteins, unfolded proteins, and removal of damaged mitochondria in physiological conditions [86]. However, autophagy could be a result of cellular stress like starvation, oxidative stress, and metabolic stress also [87]. P62 protein is a key player in autophagic signaling as it localizes to autophagosome and acts as a receptor for molecules to be undergoing autophagy. Nrf2 remains in positive feedback loop with p62. In mitochondrial stress, Ser403 residue of p62 gets phosphorylated by CK2 (casein kinase 2) or TBK1 (TANK-binding kinase 1). S403 phosphorylated targets p62 to the ubiquitinated cargos. Then, mTORC1-dependent phosphorylation of p62 at S351 takes place which enhances binding of p62 with Keap1. Keap1 binding to p62 located at autophagic cargos sequesters it at autophagosome and thereby targets it for degradation [88]. Resulting Nrf2 translocates to the nucleus and exerts its protective effects to the cells. This mechanism is utilized by hepatocellular carcinoma cells to protect them against oxidative stress and also to develop chemo-resistance.
K67 (the acetonyl naphthalene derivative) inhibits protein–protein interaction between S349 phosphorylated p62 and Keap1. It binds to DC pocket of Keap1 that is the site responsible for p62 binding and for Nrf2 binding as well. Treatment of hepatocellular carcinoma with inhibitor K67 reduces proliferation of HCC cells and also confers resistance to cisplatin and sorafenib [89]. Quercetin also activates Keap1-Nrf2-p62 axis and thereby reduces hepatic toxicity by various toxicants [89].
Moreover, during oxidative stress conditions, trehalose activates autophagy-mediated Nrf2- Keap1 regulation. Overexpression of p62 mimics Nrf2 activation [90].
Curcumin reduces inflammation, oxidative stress, and cytotoxic to cancer cells. Dietary supplementation of curcumin reduces AFB1-mediated inflammation by decreasing the level of inflammatory cytokines (NF-қB, IL-6, IL-8, IL-10, etc.) and increasing pro-inflammatory cytokines. This results into decreased AFB1 mediated inflammation. Curcumin also does so by up regulating Nrf2 and thereby its downstream gene, HO-1 in broiler hepatocytes [91].
In breast cancer MCF7 and T47D cells, Nrf2 signaling augments antioxidant response in glucose deprivation and increased autophagy conditions. This confers protection to breast cancer cells even in low glucose means in metabolic stressed conditions [92].
During protein synthesis, formation of misfolded protein is a general phenomenon and about 30% of total synthesized proteins become misfolded. These misfolded proteins do not have any function in cells; therefore, they are degraded by either proteasome or by autophagy-mediated processes. If these proteins remain in cells being unprocessed, it induces cytotoxicity. So for cells to increase their life span, it is very important to remove un/misfolded proteins as they form to inhibit their accumulation. These protein aggregates could be formed inside the cells. One could be due to inhibition of degradation pathway, leading to protein aggregate accumulation which is called aggresomes. Second types of aggregates are generated during cellular stress like oxidative stress, heat shock named as aggresome-like induced structures (ALIS). Both types of aggregates are degraded by selective autophagy called as aggrephagy. TRIM (Triplicate motif protein), during oxidative stress conditions, forms a positive feedback loop having Nrf2, Keap1, p62, and TRIM16 proteins by regulating expression level of these proteins [93, 94].
Upon traumatic brain injury (TBI), inflammation and microglial activation takes place to reduce cytotoxicity. Valproic acid (VPA) up regulates p62-Keap1-Nrf2 signaling axis following brain injury and causes increased acetylation of histones. These results in VPA-mediated up regulation in antioxidant and autophagy response in TBI and it protects from neurodegeneration [95].
7 Nrf2 Signaling in Inflammation
Inflammasomes are the complexes that are made up of upstream caspase-1, NLRP3 (nucleotide-binding oligomerization domain (NOD)-like receptor containing pyrin domain 3) and ASC (apoptosis-associated speck-like protein containing a CARD). Here, NLRP3 acts as a sensor protein; ASC acts as adaptor protein and caspase-1 acts as effector protein. In addition to NLRP3, AIM2 (absent in melanoma 2), NLRP1, and NLRC4 (NLR family CARD domain containing protein 4) also act as a sensor for inflammosome. Like Nrf2 signaling, inflammosome pathway is also activated upon cellular stress. Inflammosome activation induces inflammation which causes cell death. Nrf2 and inflammosome pathway are activated in acute as well as in chronic inflammation, upon generation of ROS, following induction of autophagy. Since both of these pathways are activated upon same stress conditions, there is an obvious crosstalk between these two pathways [96]. Nrf2 regulates expression of genes that are present in inflammosome pathway as well. Following stress, sensor proteins in inflammosome pathway sense the stress and induce ASC oligomerization forming ASC specks. Then, caspase-1 binding activates proteases which ultimately induce pro-inflammatory cytokines such as IL-1 and IL-18. Pro-inflammatory cytokines initiate inflammation. TXNIP (thioredoxin-interacting protein), generated by ROS, also interacts inflammosome sensor proteins and activates inflammosome assembly. Nrf2 activation by ROS induction augments inflammosome activation in mouse cells and in human skin cells as well. Several studies suggest that Nrf2 activation reduces NF-қB activation which is required for NLRP3 priming to inflammasomes and thereby inhibits inflammasomes activation. This occurs due to the fact that Nrf2 activation causes cytoprotection whereas inflammosome activation results in cell death. Hence, Nrf2-inflammosome crosstalk occurs antagonistically. Both pathways are involved in inflammatory stress and nrf2 is also involved in cancer, it is relevant to study crosstalk and its significance in cancer cells also [96].
Moreover, in inflammatory conditions, lung injury is induced which is related to a high rate of mortality. This inflammation-induced lung injury could be a result of intestinal ischemia-reperfusion (I/R). In such conditions, Nrf2 down regulates TLR4, inflammation, apoptosis, and regulates Akt activation. In murine lung epithelial cells, Nrf2 knockdown up regulates TLR4, HO-1, autophagy, and apoptosis in glucose-deprived conditions. In addition, Akt signaling is also down regulated [97].
8 Nrf2 Signaling in Chemo-Resistance
Moreover, cancer stem cells (CSCs) are population of cancer cells which has self-renewal properties like other stem cells. Cancer stem cells are responsible for tumor relapse after treatment of various types of cancers like lung, brain, and breast. They have low level of ROS like that of adult stem cells. Due to low ROS level, CSCs develop resistance to oxidative stress. In comparison to normal cells, CSCs present in breast cancer express increased levels of antioxidant proteins like catalase, superoxide dismutase, and glutathione peroxidase. This creates lower level of cellular-free radicals by increasing ROS metabolism in oxidative environment during radiation-induced DNA damage. In breast cancer cell lines like MCF7, there is a high level of cluster of differentiation 44 (CD44) proteins. CD44 is a glycoprotein and acts as receptor for proteins of extracellular matrix, hyaluronic acid which is a well-known marker of CSCs in various cancers. Increased expression of CD44 correlates with tumor initiation and progression due to increased motility and invasion capacity of cancer cells. Higher CD44 levels are also found to correlate with higher Nrf2 level in breast CSCs. This ultimately contributes to maintenance of lower ROS level upon radiation therapy and leads to radiation resistance against anticancer therapy through high CD44-high Nrf2 axis in breast tumors [98].
In gastric cancer, Nrf2-P-glycoprotein axis is responsible for multidrug resistance. During chemotherapy, some cancers develop drug resistance. This can be due to reduction in drug accumulation by membrane transporters. P-glycoprotein (P-gp), which is an efflux pump, plays a crucial role in drug resistance via decreasing drug concentration by causing drug efflux out of the cancer cells. There is a positive correlation between Nrf2 and p-gp in gastric cancer cells. Cumulative results of higher expression of Nrf2-dependent phase II detoxifying enzymes, antioxidant enzymes, and P-gp-dependent efflux of anticancer drugs creates tumor-supportive environment and leads to tumor drug resistance in gastric cancer [99].
Moreover, increased Nrf2 expression in K-RasG12V (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) mutated cells confers drug resistance. Majority of cells undergo cancerous transformation due to K-rasG12V mutation. Nrf2 levels increase in such transformed cells which is responsible for cytoprotection and drug resistance through up regulation of its downstream target gene-NQO1. Nrf2 knockout in Ras transformed cells leads to increased apoptosis, lower cellular proliferation, and less tumor growth. This is due in part by lowed activation of antioxidant genes in Nrf2 knockout cells and increased ROS generation [100].
In case of ovarian cancer stem cell like cells (CSCs), aldehyde dehydrogenase (ALDH)-dependent up regulation of Nrf2 results in drug resistance. Highly active ALDH, an enzyme that causes aldehyde oxidation to their respective acids, is one of the hallmarks of CSC. In 1258 ovarian cancer meta-analysis, ALDH has been found to be up regulated and it correlates with tumor survival and drug resistance. However, ALDH up regulation accompanies an increase in expression of a multidrug resistance protein which induces efflux of drug. ALDH-dependent Nrf2 signaling activation leads to Nrf2 translocation into the nucleus, increased transcription of NQO-1 and AKR1C1 genes. Also the total cellular Nrf2 level increases along with its nuclear localization. Higher nuclear localization is a result of increase in p62 level which causes dissociation of Nrf2 with Keap1, its release, and translocation into the nucleus. Therefore, ALDH-dependent Nrf2 activation through p62-related signaling in ovarian cells leads to CSC promoting properties and anticancer drug resistance [23].
In colon cancer, drug resistance is developed by Her2-Nrf2 signaling pathway. Somatic mutation in Her2, a member of receptor tyrosine kinase family of proteins, results in ovarian cancer like that of breast cancer. Resulting Her2 overexpression leads to resistance against anticancer drug like oxaliplatin in many cancers including colon cancer. Nrf2 inhibition in Her2-mutated colon cancer increases oxaliplatin-induced apoptosis and drug sensitivity in LS174T colon cancer cell line. Moreover, HCT116 colon cancer cells spheres, Nrf2 is highly expressed giving rise to doxorubicin treatment resistance in these cells [101].
In hepatocellular carcinoma (HCC), cisplatin resistance is induced by NRAL/miR-340-5p/Nrf2 signaling. NRAL (Nrf2 regulation-associated lncRNA) is a 495 nucleotide long non-coding RNA (lncRNA-ENST00000412153) present near 2722 kb from Nrf2 gene. CDDP depletes antioxidant ability of tumor cells thereby causing their apoptosis. NRAL develops resistance to CDDP in hepatocellular carcinoma by regulating Nrf2. It does so by directly interacting miR-340-5p which is an endogenous miRNA and targets Nrf2 mRNA for degradation. Therefore, NRAL acts as competing endogenous RNA (ceRNA) for miR-340-5p and thereby increasing Nrf2 level which ultimately results antioxidant capacity of cells and develops in CDDP, cisplatin resistance in HCC [102].
Nrf2 also confers drug resistance in bladder cancers. In bladder cancer, there is a positive feedback loop between Nrf2 and YAP (Yes-associated protein) that protects tumor cell against oxidative stress and apoptosis. YAP regulates expression of FOXM1 which is a transcription factor involved in Nrf2 transcription. Moreover, expression of YAP is also regulated by Nrf2 which creates a positive loop between Nrf2 and YAP. This regulates cellular oxidative homeostasis via GSH and develops cisplatin resistance in bladder cancers [103].
9 Aberrant Activation of Nrf2
Aberrant activation of Nrf2 in tumor cells confers malignancy. Many immuno-histochemical and clinicopathological studies show strong correlations between Nrf2 activation and tumor progression [104,105,106,107,108]. These evidences make Nrf2 an important prognostic factor in a wide range of cancers. From many case studies, it has been found that Nrf2 have been constitutively activated due to changes in DNA structure, RNA expression, or due to alteration in protein–protein interactions. Somatic mutations of Keap1 and Nrf2 genes generally found in the head and neck cancer, lung cancer, and bladder cancer. From the crystal structure, it has been found that most mutations of Keap1 are in the coding region and in case of Nrf2 the DLG/ETGE motifs are found to be the mutation-prone sites [109, 110]. Mutated Nrf2, resulted from alternative splicing of Nrf2 with exon2 skipping, is not able to bind to Keap1. This type of mutations are commonly found in lung, head and neck squamous cell carcinoma, and hepatocellular cancer with permanent localization of Nrf2 in the nucleus [111]. An adaptor protein p62 selects ubiquitinated Keap1 for autophagy resulting in accumulation of free Nrf2. In hepatocellular carcinoma, constitutive activation of Nrf2 is often observed due to the overexpression of phosphorylated p62 [88]. In fumarate hydratase deficiency, accumulated fumarate alkylates some thiol groups of Keap1 thereby stabilizing Nrf2 [28]. K-ras mutant mice induce overexpression of Myc through activation of Ras signaling. This overexpressed Myc then binds to the promoter of Nrf2 up regulating its transcription and thereby induces tumorigenesis [112].
10 Nrf2 Activators
Among many plant-derived photochemicals, curcumin, epigallocatechin-3-gallate, resveratrol, cafestol, lycopene, kahweol, cinnamonyl-based compounds, sulforaphane (SF), garlic organosulfur compounds, zerumbone, and carnosol are found to act as Nrf2 activators. Sulforaphane, an isothiocyanate, found in broccoli shows protection not only against skin tumor and tobacco-derived lung cancer but also shows reduction of abnormal crypt foci following exposure of N-nitrosobis (2 oxopropyl) amine (BOP) [113, 114]. Curcumin, another chemo-preventive natural product, also has been identified to activate Nrf2. In benzo(a)pyrene-induced murine lung and liver carcinoma, curcumin has been shown to increase Nrf2 expression with hampered binding to DNA, increased oxidative stress, and inflammation [115]. Dimethyl fumarate (DMF) causes alkylation of Keap1 on critical cysteine residue to inhibit Nrf2 ubiquitination, and thereby increases its stabilization and expression of various Nrf2 target genes [116]. DMF which is approved by Food and Drug Administration (FDA) for multiple sclerosis (MS), also shows some potential as an anticancer drug in various types of cancers like glioblastoma, head and neck cancer, melanoma, and colon cancer [117].
11 Nrf2 Inhibitors
In case of cancers with consecutive Nrf2 expression, inhibition of Nrf2 signaling by pharmacological modulators has been appeared as a potential therapeutic intervention. Molecules like ascorbic acid, luteolin, trigonelline, ochratoxin A, and all-trans retinoic acid (ATRA) are able to suppress the Nrf2 pathway either by inhibiting Nrf2 binding to DNA or decreasing Nrf2 mRNA and protein expression [118]. Retinoic acid receptor α (RARα) and ATRA mimetic hamper both basal and inducible Nrf2 activity both in in vitro and in vivo [119]. In the presence of ATRA, Nrf2 forms a complex with RARα. This Nrf2 and RARα complex inhibits binding of Nrf2 on the ARE thereby makes Nrf2 unable to induce expression of its target genes.
12 Conclusion
From a number of studies, it has been cleared that inflammation provides a cell, an environment for tumor development. Aberrant production of ROS causes inflammation in our body. During inflammation, various types of chemokines and cytokines are produced that give rise to an ideal condition for tumor generation. In these cells, a constant increase in ROS generally observed which maintains tumor progression. If not controlled, the excessive ROS causes more mutations and ultimately drives tumor cell migration and invasion. In response to ROS-mediated oxidative stress, a group of cytoprotective enzymes are expressed by transcription factor Nrf2, thereby reducing ROS production. For cancer prevention in which oxidative stress contributes to the pathogenesis, elevating Nrf2 activity remains an important approach.
References
Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15.
Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol Ther. 2010;125:376–93.
Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–30.
Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22.
Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93:14960–5.
Ma Q. Xenobiotic-activated receptors: from transcription to drug metabolism to disease. Chem Res Toxicol. 2008;21:1651–71.
Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal. 2010;13:1713–48.
Hamada S, Taguchi K, Masamune A, Yamamoto M, Shimosegawa T. Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis. 2018;38:661–70.
Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–9.
Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, et al. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol. 2013;33:2436–46.
Long M, Tao S, Rojo de la Vega M, Jiang T, Wen Q, Park SL, et al. Nrf2-dependent suppression of azoxymethane/dextran sulfate sodium-induced colon carcinogenesis by the cinnamon-derived dietary factor cinnamaldehyde. Cancer Prev Res. 2015;8:444–54.
Tao S, Rojo de la Vega M, Chapman E, Ooi A, Zhang DD. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol Carcinog. 2018;57:182–92.
Wang H, Liu X, Long M, Huang Y, Zhang L, Zhang R, et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med. 2016;8:334ra51.
Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–74.
Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011;286:40725–33.
Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A. 2004;101:2046–51.
Alam J, Killeen E, Gong P, Naquin R, Hu B, Stewart D, et al. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am J Physiol Renal Physiol. 2003;284:F743–52.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–91.
Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–20.
Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9.
Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol. 2006;18:375–82.
Kaniuk NA, Kiraly M, Bates H, Vranic M, Volchuk A, Brumell JH. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes. 2007;56:930–9.
Kim D, Choi BH, Ryoo IG, Kwak MK. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018;9:896.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45.
Copple IM, Lister A, Obeng AD, Kitteringham NR, Jenkins RE, Layfield R, et al. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem. 2010;285:16782–8.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23.
Li Y, Paonessa JD, Zhang Y. Mechanism of chemical activation of Nrf2. PLoS One. 2012;7:e35122.
Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–37.
Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab. 2002;3:481–90.
Loignon M, Miao W, Hu L, Bier A, Bismar TA, Scrivens PJ, et al. Cul3 overexpression depletes Nrf2 in breast cancer and is associated with sensitivity to carcinogens, to oxidative stress, and to chemotherapy. Mol Cancer Ther. 2009;8:2432–40.
Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–92.
Nair S, Doh ST, Chan JY, Kong AN, Cai L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br J Cancer. 2008;99:2070–82.
Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res Treat. 2011;129:983–91.
Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116:4338–48.
Li N, Muthusamy S, Liang R, Sarojini H, Wang E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech Ageing Dev. 2011;132:75–85.
Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am J Physiol. 2003;285:C334–42.
Theodore M, Kawai Y, Yang J, Kleshchenko Y, Reddy SP, Villalta F, et al. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008;283:8984–94.
Pi J, Bai Y, Reece JM, Williams J, Liu D, Freeman ML, et al. Molecular mechanism of human Nrf2 activation and degradation: role of sequential phosphorylation by protein kinase CK2. Free Radic Biol Med. 2007;42:1797–806.
Apopa PL, He X, Ma Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J Biochem Mol Toxicol. 2008;22:63–76.
Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. 1995;15:4184–93.
Kataoka K, Igarashi K, Itoh K, Fujiwara KT, Noda M, Yamamoto M, et al. Small Maf proteins heterodimerize with Fos and may act as competitive repressors of the NF-E2 transcription factor. Mol Cell Biol. 1995;15:2180–90.
Takagi Y, Kobayashi M, Li L, Suzuki T, Nishikawa K, Yamamoto M. MafT, a new member of the small Maf protein family in zebrafish. Biochem Biophys Res Commun. 2004;320:62–9.
Motohashi H, Katsuoka F, Engel JD, Yamamoto M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci U S A. 2004;101:6379–84.
Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21:5216–24.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86.
Kobayashi A, Ito E, Toki T, Kogame K, Takahashi S, Igarashi K, et al. Molecular cloning and functional characterization of a new Cap‘n’ collar family transcription factor Nrf3. J Biol Chem. 1999;274:6443–52.
Kobayashi M, Itoh K, Suzuki T, Osanai H, Nishikawa K, Katoh Y, et al. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells. 2002;7:807–20.
Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci U S A. 2001;98:3404–9.
Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–13.
Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, et al. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol Cell Biol. 2004;24:36–45.
Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–5.
Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–51.
Buckley BJ, Marshall ZM, Whorton AR. Nitric oxide stimulates Nrf2 nuclear translocation in vascular endothelium. Biochem Biophys Res Commun. 2003;307:973–9.
Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278:484–92.
Zipper LM, Mulcahy RT. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol Sci. 2003;73:124–34.
Stacy DR, Ely K, Massion PP, Yarbrough WG, Hallahan DE, Sekhar KR, et al. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck. 2006;28:813–8.
Yu R, Chen C, Mo YY, Hebbar V, Owuor ED, Tan TH, et al. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J Biol Chem. 2000;275:39907–13.
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–209.
Kang KW, Choi SH, Kim SG. Peroxynitrite activates NF-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide. 2002;7:244–53.
Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003;546:181–4.
Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–82.
Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–80.
Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004;64:6424–31.
Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–5.
Rachakonda G, Sekhar KR, Jowhar D, Samson PC, Wikswo JP, Beauchamp RD, et al. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene. 2010;29:3703–14.
Satoh H, Moriguchi T, Taguchi K, Takai J, Maher JM, Suzuki T, et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis. 2010;31:1833–43.
Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–91.
Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in Cancer. Trends Mol Med. 2016;22:578–93.
Okada F. Inflammation-related carcinogenesis: current findings in epidemiological trends, causes and mechanisms. Yonago Acta Med. 2014;57:65–72.
Wang Y, Mandal AK, Son YO, Pratheeshkumar P, Wise JTF, Wang L, et al. Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol Appl Pharmacol. 2018;353:23–30.
Ma JQ, Tuersun H, Jiao SJ, Zheng JH, Xiao JB, Hasim A. Functional role of NRF2 in cervical carcinogenesis. PLoS One. 2015;10:e0133876.
Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420.
auf dem Keller U, Huber M, Beyer TA, Kumin A, Siemes C, Braun S, et al. Nrf transcription factors in keratinocytes are essential for skin tumor prevention but not for wound healing. Mol Cell Biol. 2006;26:3773–84.
Son YO, Pratheeshkumar P, Roy RV, Hitron JA, Wang L, Divya SP, et al. Antioncogenic and oncogenic properties of Nrf2 in arsenic-induced carcinogenesis. J Biol Chem. 2015;290:27090–100.
Cheung KL, Lee JH, Khor TO, Wu TY, Li GX, Chan J, et al. Nrf2 knockout enhances intestinal tumorigenesis in Apc(min/+) mice due to attenuation of anti-oxidative stress pathway while potentiates inflammation. Mol Carcinog. 2014;53:77–84.
Ikeda H, Nishi S, Sakai M. Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis. Biochem J. 2004;380:515–21.
Basak P, Sadhukhan P, Sarkar P, Sil PC. Perspectives of the Nrf-2 signaling pathway in cancer progression and therapy. Toxicol Rep. 2017;4:306–18.
Zhang HS, Du GY, Zhang ZG, Zhou Z, Sun HL, Yu XY, et al. NRF2 facilitates breast cancer cell growth via HIF1a-mediated metabolic reprogramming. Int J Biochem Cell Biol. 2017;95:85–92.
Zhang C, Wang HJ, Bao QC, Wang L, Guo TK, Chen WL, et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget. 2016;7:73593–606.
Martinez VD, Vucic EA, Thu KL, Pikor LA, Lam S, Lam WL. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: association with poor prognosis in head and neck cancer. Head Neck. 2014;37:727–34.
Huang CF, Zhang L, Ma SR, Zhao ZL, Wang WM, He KF, et al. Clinical significance of Keap1 and Nrf2 in oral squamous cell carcinoma. PLoS One. 2013;8:e83479.
Singh B, Chatterjee A, Ronghe AM, Bhat NK, Bhat HK. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer. 2013;13:253.
Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev. 2014;15:7–18.
Jayakumar S, Pal D, Sandur SK. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat Res. 2015;779:33–45.
Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12:68–78.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–80.
Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–31.
Ichimura Y, Komatsu M. Activation of p62/SQSTM1-Keap1-nuclear factor Erythroid 2-related factor 2 pathway in Cancer. Front Oncol. 2018;8:210.
Mizunoe Y, Kobayashi M, Sudo Y, Watanabe S, Yasukawa H, Natori D, et al. Trehalose protects against oxidative stress by regulating the Keap1-Nrf2 and autophagy pathways. Redox Biol. 2018;15:115–24.
Muhammad I, Wang X, Li S, Li R, Zhang X. Curcumin confers hepatoprotection against AFB1-induced toxicity via activating autophagy and ameliorating inflammation involving Nrf2/HO-1 signaling pathway. Mol Biol Rep 2018
Walker A, Singh A, Tully E, Woo J, Le A, Nguyen T, et al. Nrf2 signaling and autophagy are complementary in protecting breast cancer cells during glucose deprivation. Free Radic Biol Med. 2018;120:407–13.
Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108:1427–32.
Jena KK, Kolapalli SP, Mehto S, Nath P, Das B, Sahoo PK, et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018;37
Cheng KW, Cheng SC, Chen WY, Lin MH, Chuang SJ, Cheng IH, et al. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antivir Res. 2018;115:9–16.
Hennig P, Garstkiewicz M, Grossi S, Di Filippo M, French LE, Beer HD. The crosstalk between Nrf2 and Inflammasomes. Int J Mol Sci. 2018;19
Yan J, Li J, Zhang L, Sun Y, Jiang J, Huang Y, et al. Nrf2 protects against acute lung injury and inflammation by modulating TLR4 and Akt signaling. Free Radic Biol Med. 2018;121:78–85.
Ryoo IG, Choi BH, Ku SK, Kwak MK. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: implications for cancer stem cell resistance. Redox Biol. 2018;17:246–58.
Jeddi F, Soozangar N, Sadeghi MR, Somi MH, Shirmohamadi M, Eftekhar-Sadat AT, et al. Nrf2 overexpression is associated with P-glycoprotein upregulation in gastric cancer. Biomed Pharmacother. 2018;97:286–92.
Shao J, Glorieux C, Liao J, Chen P, Lu W, Liang Z, et al. Impact of Nrf2 on tumour growth and drug sensitivity in oncogenic K-ras-transformed cells in vitro and in vivo. Free Radic Res. 2018;52:661–71.
Pirpour Tazehkand A, Akbarzadeh M, Velaie K, Sadeghi MR, Samadi N. The role of Her2-Nrf2 axis in induction of oxaliplatin resistance in colon cancer cells. Biomed Pharmacother. 2018;103:755–66.
Wu LL, Cai WP, Lei X, Shi KQ, Lin XY, Shi L. NRAL mediates cisplatin resistance in hepatocellular carcinoma via miR-340-5p/Nrf2 axis. J Cell Commun Signal 2018
Ciamporcero E, Daga M, Pizzimenti S, Roetto A, Dianzani C, Compagnone A, et al. Crosstalk between Nrf2 and YAP contributes to maintaining the antioxidant potential and chemoresistance in bladder cancer. Free Radic Biol Med. 2018;115:447–57.
Kawasaki Y, Okumura H, Uchikado Y, Kita Y, Sasaki K, Owaki T, et al. Nrf2 is useful for predicting the effect of chemoradiation therapy on esophageal squamous cell carcinoma. Ann Surg Oncol. 2014;21:2347–52.
Onodera Y, Motohashi H, Takagi K, Miki Y, Shibahara Y, Watanabe M, et al. NRF2 immunolocalization in human breast cancer patients as a prognostic factor. Endocr Relat Cancer. 2014;21:241–52.
Shibata T, Saito S, Kokubu A, Suzuki T, Yamamoto M, Hirohashi S. Global downstream pathway analysis reveals a dependence of oncogenic NF-E2-related factor 2 mutation on the mTOR growth signaling pathway. Cancer Res. 2010;70:9095–105.
Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16:3743–53.
Zhao M, Xu H, Zhang B, Hong B, Yan W, Zhang J. Impact of nuclear factor erythroid-derived 2-like 2 and p62/sequestosome expression on prognosis of patients with gliomas. Hum Pathol. 2015;46:843–9.
Fukutomi T, Takagi K, Mizushima T, Ohuchi N, Yamamoto M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol Cell Biol. 2014;34:832–46.
Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700.
Goldstein LD, Lee J, Gnad F, Klijn C, Schaub A, Reeder J, et al. Recurrent loss of NFE2L2 exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Rep. 2016;16:2605–17.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9.
Kensler TW, Curphey TJ, Maxiutenko Y, Roebuck BD. Chemoprotection by organosulfur inducers of phase 2 enzymes: dithiolethiones and dithiins. Drug Metabol Drug Interact. 2000;17:3–22.
Kuroiwa Y, Nishikawa A, Kitamura Y, Kanki K, Ishii Y, Umemura T, et al. Protective effects of benzyl isothiocyanate and sulforaphane but not resveratrol against initiation of pancreatic carcinogenesis in hamsters. Cancer Lett. 2006;241:275–80.
Garg R, Gupta S, Maru GB. Dietary curcumin modulates transcriptional regulators of phase I and phase II enzymes in benzo[a]pyrene-treated mice: mechanism of its anti-initiating action. Carcinogenesis. 2008;29:1022–32.
Phillips JT, Fox RJ. BG-12 in multiple sclerosis. Semin Neurol. 2013;33:56–65.
Chen AF, Kirsner RS. Mechanisms of drug action: the potential of dimethylfumarate for the treatment of neoplasms. J Invest Dermatol. 2011;131:1181.
Magesh S, Chen Y, Hu L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med Res Rev. 2012;32:687–726.
Wang XJ, Hayes JD, Henderson CJ, Wolf CR. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc Natl Acad Sci U S A. 2007;104:19589–94.
Conflicts of Interest
There are no conflicts to declare.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sarkar, S., Ghosh, N., Kundu, M., Sil, P.C. (2020). Nrf2 and Inflammation-Triggered Carcinogenesis. In: Deng, H. (eds) Nrf2 and its Modulation in Inflammation. Progress in Inflammation Research, vol 85. Springer, Cham. https://doi.org/10.1007/978-3-030-44599-7_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-44599-7_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-44597-3
Online ISBN: 978-3-030-44599-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)