
Abstract
The green sulfur bacterium, Chlorobaculum tepidum, is an anaerobic photoautotroph that performs anoxygenic photosynthesis. Although genes encoding rubredoxin (Rd) and a putative flavodiiron protein (FDP) were reported in the genome, a gene encoding putative NADH-Rd oxidoreductase is not identified. In this work, we expressed and purified the recombinant Rd and FDP and confirmed dioxygen reductase activity in the presence of ferredoxin-NAD(P)+ oxidoreductase (FNR). FNR from C. tepidum and Bacillus subtilis catalyzed the reduction of Rd at rates comparable to those reported for NADH-Rd oxidoreductases. Also, we observed substrate inhibition at high concentrations of NADPH similar to that observed with ferredoxins. In the presence of NADPH, B. subtilis FNR and Rd, FDP promoted dioxygen reduction at rates comparable to those reported for other bacterial FDPs. Taken together, our results suggest that Rd and FDP participate in the reduction of dioxygen in C. tepidum and that FNR can promote the reduction of Rd in this bacterium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The green sulfur bacterium (GSB), Chlorobaculum tepidum, (formerly identified as Chlorobium tepidum) is an obligate anaerobic photoautotroph. C. tepidum performs anoxygenic photosynthesis using various sulfur compounds, including sulfide, thiosulfate, and elemental sulfur as the electron sources for assimilation of carbon dioxide (Sakurai et al. 2010; Eddie and Hanson 2013; Gregersen et al. 2011). GSB exhibit only Type I photosynthetic reaction centers (RCs), which can promote the reduction of ferredoxin (Fd) (Miller et al. 1992; Kjær and Sheller 1996; Seo et al. 2001; Tsukatani et al. 2004; Vassiliev et al. 2000). Photosynthetic carbon dioxide assimilation is performed via the reverse tricarboxylic acid (rTCA) cycle (also known as the Arnon-Buchanan cycle) (Evans et al. 1966; Buchanan and Arnon 1990; Tang and Blankenship 2010; Buchanan et al. 2017), in which reduced Fd promotes the reduction of acetyl CoA to pyruvate via a reaction catalyzed by pyruvate-Fd oxidoreductase (PFOR; acetyl CoA + CO2 + 2Fdred + 2H+ → pyruvate + 2Fdox + CoA). The annotated genome sequence of C. tepidum includes three 2[4Fe-4S]-type Fds [accession numbers CT1260, CT1261, CT1736 (Eisen et al. 2002)]; each was purified to homogeneity from cell-free extracts (Buchanan and Arnon 1971; Tanaka et al. 1974, 1975; Seo et al. 2000; Yoon et al. 2001). Recent studies report that flavodoxin, a potential electron carrier from the marine GSB, Chlorobium phaeovibrioides, may be expressed under iron-limiting conditions and transport electrons as part of the photosynthetic electron transfer cascade (Bertsova et al. 2019a, b). Among these, the role of Fds was investigated extensively; Fds are proposed to function as electron donors to enzymes such as PFOR, the key enzymes of the reverse TCA cycle (Yoon et al. 2001), Fd-NAD(P)+ reductase[(FNR, (Seo and Sakurai 2002)], nitrogenase (Wahlund et al. 1993) and 8-vinyl-protochlorophyllide reductase (Saunders et al. 2013). Although Fds also participate in the corresponding photosynthetic electron transfer reactions in oxygenic photosynthesis in plants and cyanobacteria (Knaff and Hirasawa 1991; Medina and Gómesz-Moreno 2004; Fukuyama 2004; Sétif 2001), some iron sulfur clusters characteristic of GSB such as Fds, PFOR and RCs undergo rapid degradation in the presence of dioxygen (Vassiliev et al. 2000; Yoon et al. 2001; Kusumoto et al. 1994). In order to survive, cells require a means for rapid and efficient scavenging of dioxygen and reactive oxygen species (ROS). Accordingly, the C. tepidum genome includes many putative genes that encode proteins capable of protecting cells from dioxygen and ROS; expression of these genes may be critical for cell survival in microoxic conditions (Li et al. 2009), as is the case of oxygenic phototrophs.
In obligate and facultative heterotrophic anaerobes such as Clostridia, Desulfovibrio, methanogenic archaea, and also in some apicoplast eukaryotes, flavodiiron protein (FDP) plays a critical role as a scavenger of dioxygen, ROS and/or nitric oxide (Martins et al. 2019; Folgosa et al. 2018; Romão et al. 2016a and references therein). Some reports suggest that the earliest ROS scavenging enzymes existed before the emergence of oxygen-utilizing organisms; these systems may have evolved to provide protection against small quantities of oxygen in the primordial environment that mainly originated from the photolysis of water (Anbar et al. 2007). In oxygenic phototrophic organisms such as cyanobacteria and the green alga Chlamydomonas reinhardtii, FDPs may play important roles in conferring tolerance against oxidative stress (Alboresi et al. 2019; Jokel et al. 2015). FDP is a soluble protein that includes a flavin cofactor and one diiron cluster as a minimal catalytic unit. FDPs are classified into several subgroups based on their domain architecture (Martins et al. 2019). In C. tepidum genome, one putative Class A FDP gene (CT2285) was annotated as rubredoxin-oxygen oxidoreductase (Eisen et al. 2002; Li et al. 2009); this gene encodes a protein comprised of a β-lactamase-like domain with a bound diiron cluster and a flavin mononucleotide (FMN) binding domain with a flavodoxin-like fold (Martins et al. 2019; Folgosa et al. 2018; Romão et al. 2016a). The gene disruption analyses carried out in C. tepidum revealed that an intact CT2285 gene is indispensable for the photoautotrophic growth under microoxic conditions (Li et al. 2009). However, no further biochemical or metabolic characterization of the CT2285 gene product have been investigated. Soluble electron transfer proteins such as rubredoxin (Rd), coenzyme F420, or Fd would be required as electron donors for reduction of Class A FDPs (Martins et al. 2019; Folgosa et al. 2018; Romão et al. 2016a and references therein). Toward that end, substantial amounts of Rd as well as Fds were detected in the soluble fraction of cell-free extracts of C. tepidum (Yoon et al. 1999).
In this study, we generated and purified recombinant Rd and FDP from C. tepidum (CtRd and CtFDP, respectively) and assessed their reactivity utilizing FNRs from C. tepidum (CtFNR (Seo and Sakurai 2002)) and Bacillus subtilis [BsFNR (YumC) (Seo et al. 2004)]. Our results indicate that Rd functions efficiently as an electron donor to CtFDP.
Materials and methods
Expression and purification of recombinant C. tepidum rubredoxin (CT2024)
The open reading frame (ORF) region of the C. tepidum rubredoxin (CtRd) gene (NCBI GeneID: 1,007,537, CT2024) was amplified by PCR with primers including fwd: 5′-GAAGGAGATATAGATATGCAGAAATGGGTTTGTGT-3′ and rev: 5′-GGGCAGGAATTCGATGAAATCGGTAAGGGGTCAGG-3′ using genomic DNA extracted from the C. tepidum TLS strain as a template. An expression vector including the CtRd gene was prepared by inserting the PCR amplicons into a linearized pETBlue-1 vector that was digested at the EcoRV restriction site (Novagen, Merck KGaA, Darmstadt, Germany) using the Seamless Ligation Cloning Extract method (Okegawa and Motohashi 2015). The ORF inserted into the vector was verified by DNA sequencing at the Institute for Gene Research, Kanazawa University, and it was confirmed to be identical to the database sequence.
E. coli Tuner™ (DE3) pLacI cells (Novagen) transformed with the recombinant expression vector were cultured overnight in Luria–Bertani broth containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. A 100th volume of the primary culture was transferred into terrific broth medium containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol (typically, 8 ml pre-culture into 800 ml terrific broth medium in a 2 L volume Erlenmeyer Flask). The cells were incubated at 37 °C for several hours in a rotary shaker at 100 rpm. When OD600 reached approximately 1.0, isopropyl β- d-1-thiogalactopyranoside (IPTG) and ferric ammonium sulfate were added to the culture to final concentrations of 0.5 mM and 0.2 mM, respectively; the temperature was decreased to 27 °C. After incubation for 12–16 h, the E. coli cells were harvested by a centrifugation at 5000 × g for 10 min, dissolved in the same volume of 50 mM tris(hydroxymethyl)aminomethane (Tris)–HCl buffer (pH 8.0) containing 200 mM NaCl and 1 mM phenylmethylsulfonyl fluoride (PMSF), and disrupted with sonication (three cycles of 2 min treatment followed by 2 min cooling on ice each; Branson Sonifier 250, USA). The solution was centrifuged at 10,000 × g for 20 min, and the supernatant was recovered.
Ammonium sulfate was added to the supernatant to 35% saturation, and the solution was stirred at 4 °C overnight. After centrifugation at 10,000 × g for 30 min, ammonium sulfate was added to the supernatant to 80% saturation, and the solution was again stirred at 4 °C overnight. The solution was centrifuged at 10,000 × g for 30 min, and the precipitate was recovered. Then, the precipitate was dissolved in 20 mM Tris–HCl buffer (pH 8.0) and dialyzed against 20 mM Tris–HCl buffer (pH 8.0) at 4 °C. The dialysis buffer was changed twice with 3-h intervals. After centrifugation of the solution at 10,000 × g for 30 min, the supernatant was applied to a DEAE Sepharose A-25 column (26 × 200 mm, GE Healthcare, UK) equilibrated with 20 mM Tris–HCl buffer (pH 8.0). After washing the column with one bed volume of the same buffer, CtRd was eluted with a linear gradient of NaCl concentrations from 0 to 1 M (720 ml), and fractions with purple coloration were pooled. Ammonium sulfate powder was added to the pooled fractions to a final concentration of 2 M. After standing for 4 h at 4 °C, the solution was centrifuged at 10,000 × g for 30 min. Then, the supernatant was applied to the Phenyl Sepharose 6FF gel column (26 × 200 mm, GE Healthcare) pre-equilibrated with the 50 mM Tris–HCl buffer (pH 8.0) containing 2 M ammonium sulfate. After washing the column with the same solution, CtRd was eluted with a reverse gradient of ammonium sulfate concentration from 2 to 0 M (720 ml) in the same buffer. The fractions with purple coloration were retained and dialyzed against 20 mM Tris–HCl buffer (pH 8.0), with three changes of dialysis buffer every 3 h at 4 °C. After centrifugation at 10,000 × g for 20 min and filtration with 0.22 μm pore filer, the supernatant was applied to a Source 30Q column (18 mm × 200 mm, GE Healthcare) pre-equilibrated with the same buffer at room temperature. After washing with two column volume of the same buffer, CtRd was eluted with a liner gradient of sodium chloride concentration from 0 to 0.4 M (180 ml). The fractions with purple coloration were collected and applied to a hydroxyl apatite column (CHT I, Bio Rad Laboratories Inc., Hercules, CA, USA) that was equilibrated with 20 mM 2-[4-(2-Hydroxyethyl)-1-piperazinyl] ethanesulfonic acid (HEPES)-NaOH buffer (pH 7.0); CtRd was eluted with the same buffer. The fractions with a A492/A280 ratio of over 0.3 were collected, concentrated by ultrafiltration [Vivaspin Turbo 15 (5000 MWCO), Sartorius, Germany], and applied to a Superdex 75 column (10/300, GE Healthcare) equilibrated with 20 mM Tris–HCl buffer (pH 8.0) containing 200 mM NaCl. CtRd was eluted using same solution, and the fractions with purple coloration were collected, dialyzed against 20 mM HEPES–NaOH buffer (pH 7.0), and stored at − 80 °C until use.
Expression and purification of recombinant C. tepidum flavodiiron protein (CT2285)
FDP from C. tepidum (CtFDP) was prepared in recombinant form expressed in E. coli cells. The ORF region of the gene encoding CtFDP (CT2285, NCBI GeneID: 1,007,274) was amplified by PCR using genomic DNA from C. tepidum TLS strain as the template and primers including 5′-ATGACCGATAATAAAATTCTTCCAATTAC-3′ and 5′-GTATTCCGGGTTGTTTTACCTTC-3′. The amplified DNA fragment was ligated into the linearized pETBlue-I expression vector (Novagen). DNA sequence of the cloned ORF region of the CT2285 gene was verified by the DNA sequencing at the Institute for Genomic Research, Kanazawa University.
Expression of CtFDP gene was performed in E. coli Tuner™ (DE3) pLacI cells transformed with the recombinant expression vector. The experimental procedures from bacterial growth to the precipitation of soluble proteins with ammonium sulfate were the same as those described above for CtRd. The precipitate that emerged between the 35 and 80% saturations of ammonium sulfate was dissolved in 50 mM Tris–HCl buffer (pH 8.0) and dialyzed against the same buffer at 4 °C with three changes over 3-h intervals. After removing undissolved proteins by the centrifugation at 10,000 × g for 20 min, the supernatant was applied to a DEAE Sepharose A-25 column (26 × 200 mm, GE Healthcare). After washing the column with a one column volume of the same buffer, CtFDP was eluted with a linear gradient of sodium chloride at concentrations from 0 to 0.5 M (720 ml). The fractions containing CtFDP were detected by the presence of flavin absorption bands with maxima at ~ 380 nm and 450 nm in the UV–visible absorption spectra. The fractions with intense flavin absorption bands were collected and dialyzed against 20 mM Tris–HCl (pH 8.0) buffer, and they were applied to a Source 30Q column (18 × 200 mm, GE Healthcare) pre-equilibrated with the same buffer. After washing the column with two column volumes of the same buffer, CtFDP was eluted with a linear sodium chloride concentration gradient, from 0 to 300 mM (180 ml) in the same buffer. CtFDP containing fractions that were identified by flavin absorption as described above were collected, concentrated by ultrafiltration (Vivaspin Turbo 15 [5000 MWCO], Sartorius), and applied to a Superdex 200 10/300 column (GE Healthcare) that was pre-equilibrated with 20 mM Tris–HCl buffer (pH 8.0) containing 200 mM NaCl. The fractions with light brown coloration were collected and dialyzed against 20 mM HEPES–NaOH buffer (pH 7.0) and stored at − 80 °C for the subsequent reconstitution with cofactors, as described below.
Enrichment of iron and FMN cofactors in C. tepidum FDP
The reconstitution of CtFDP with iron and flavin mononucleotide (FMN) cofactors was performed according to the methods described for Ruegeria lacuscaerulensis DddQ (Brummett and Dey 2016) and Treponema denticola FDP (Frederick et al. 2015) with slight modifications. Briefly, freshly prepared 0.2 M solution of Mohr’s salt [(NH4)2Fe(SO4)2·6H2O] was added to the purified CtFDP solution (4.4 mg protein/ml) to a final concentration of 0.5 mM; the solution was permitted to stand for several minutes and was then dialyzed against 50 mM Tris–HCl buffer (pH 8.0) containing 1 mM ethylenediaminetetraacetate (EDTA) for 12 h followed by dialysis at 4 °C against 50 mM Tris–HCl buffer (pH 8.0) with two dialysis buffer changes in 12 h intervals. A stock solution of FMN (8.4 mM) was added to the dialyzed CtFDP solution (3.4 mg protein/ml) to a final concentration of 170 μM, followed by gentle shaking at 4 °C overnight. Unbound FMN was removed by multiple cycles of concentration and dilution with 20 mM HEPES–NaOH buffer (pH 7.0) using ultrafiltration [Vivaspin Turbo 15 (5000 MWCO), Sartorius] until the flow-through solution was nearly colorless.
Quantification of iron and FMN contents
The iron concentration of CtFDP was determined using the ferrozine assay (Dey and Frederick 2018) with ε562 = 27.9 mM−1 cm−1 for the ferrozine-iron complex.
The FMN concentration of CtFDP was evaluated using the trichloroacetic acid method (Ohnishi et al. 1994). Briefly, 50 μl of 100% TCA solution was added to the 950 μl containing ~ 10 μM CtFDP in 20 mM HEPES–NaOH buffer resulting in a final trichloroacetic acid concentration of 5%. The solution was shaken gently for several minutes and then permitted to stand for 30 min at room temperature in the dark, followed by centrifugation at 18,000 × g for 30 min. The supernatant was neutralized by an addition of 1 M Tris solution (~ 350 μl). The FMN concentration of the neutralized solution was estimated using ε447 = 12.2 mM−1 cm−1, including compensation for volume changes.
Enzymatic assays
Reduction of CtRd mediated by CtFNR and BsFNR was measured by monitoring the absorbance change at 492 nm in a 20 mM HEPES–NaOH buffer (pH 7.0) at 25 °C under the aerobic conditions. The mixture contained 10 mM glucose 6-phosphate (G6P; Oriental Yeast Co., Ltd., Tokyo, Japan), 5 U/mL glucose-6-phosphate dehydrogenase (G6PDH, Leuconostoc mesenteroides, Worthington Biochemicals Co., USA) and 1–10 nM FNR together with 0–1 mM NAD(P)+ (Oriental Yeast Co., Ltd.) and 0–0.1 mM CtRd. The reaction was initiated by the addition of FNR and followed for 1 min in a double beam spectrophotometer (V-560, JASCO Co., Tokyo, Japan). The control assays included all of the assay reagents except FNR; the results of the controls were subtracted for the estimation of turnover rate. Turnover rates are expressed as the number of CtRd molecules reduced by the FNR monomer using the formula Δε492 = 6.89 mM−1 cm−1 for CtRd (Yoon et al. 1999). The averages of triplicate measurements were used for the estimation of the Michaelis constant (KM) and turnover number (kcat) by a nonlinear regression analysis using the Michaelis–Menten equation in Igor Pro software (ver. 6.3, WaveMetrics, Portland, OR, USA). Error estimated from curve-fitting are presented as ± standard deviation.
Rd-dioxygen reductase activity was determined as the rate of dioxygen consumption monitored by an oxygen electrode system (Oxytherm+, Hansatech Instruments Ltd, Norfolk, UK). The reaction mixture included 5 mM G6P, 2 U/ml G6PDH, 10 μM NAD(P)+, 100 nM BsFNR, 100 μM CtRd, and 100 nM CtFDP in 20 mM HEPES–NaOH buffer (pH 7.0) at 25 °C. Dioxygen concentration was monitored during stepwise addition of each component. The turnover rate was estimated as the number of dioxygen molecules per FMN molecule in 1 s.
Additional methods
SDS-PAGE analysis was performed on 12% and 20% acrylamide gels with protein bands visualized with Coomassie Brilliant Blue (AE-1340 EzStain AQUA, ATTO CORP., Japan). The molecular weight marker set (Precision Plus Protein Standard) was purchased from Bio-Rad Laboratories Inc.
The molecular mass of the native form of each of the enzymes was deduced by gel-permeation chromatography on a Superdex-75 10/300 column for CtRd and Superdex-200 10/300 column for CtFDP (GE Healthcare) at a flow rate of 0.4 mL min−1 using 20 mM Tris–HCl buffer (pH 8.0) containing 200 mM NaCl as the eluent and molecular mass standards purchased from Sigma-Aldrich (Kit for Molecular Weights 12,000–200,000, MWGF200; St. Louis, MO, USA) with the addition of aprotinin (MW = 6500, Nacalai tesque, Inc, Kyoto, Japan).
N-terminal amino acid sequences were determined by Edman degradation of the proteins followed by analysis using a protein sequencer, PPSQ-51A system (Shimadzu, Kyoto, Japan).
UV–visible absorption spectra were measured with a double beam spectrophotometer (V-560, JASCO, Japan) in 20 mM HEPES–NaOH buffer (pH 7.0) at 25 °C.
Electron paramagnetic resonance (EPR) spectrum was measured with a Bruker ELEXSYS E500 spectrometer equipped with a liquid helium cryostat and temperature controller (ESR900 and ITC503, Oxford Instruments, UK). After evacuation of 200 μM CtRd in 20 mM HEPES–NaOH buffer (pH 7.0) under argon, the CtRd solution was transferred into an EPR tube in a glove box under a dinitrogen atmosphere. Reduced CtRd was prepared by an addition of approximately tenfold excess of sodium dithionite into the CtRd solution in the glovebox followed by transfer into the EPR tube.
Protein concentrations were determined using the Bicinchoninic acid (BCA) protein assay kit (Nacalai tesque, Inc) according to the manufacturer’s instructions; bovine serum albumin was used as the protein standard.
Protein and substrate concentrations were determined using the following extinction coefficients: CtRd [ε492 = 6.89 mM−1 cm−1 (Yoon et al. 1999)], BsFNR [ε457 = 12.3 mM−1 cm−1 (Seo et al. 2004)], CtFNR [ε466 = 10.3 mM−1 cm−1 (Seo and Sakurai 2002)], CtFDP [ε449 = 12.0 mM−1 cm−1 (this study)], and NAD(P)H (ε340 = 6.2 mM−1 cm−1). NAD(P)+ concentration was determined from the reduced form in the presence of excess G6P and G6PDH.
Amino acid sequence alignments of CT2078 and CT2285 represented in Figures S1 and S2 were performed on the T-coffee server (Notredame et al. 2000).
Results and discussion
Molecular mass and spectroscopic properties of recombinant CtRd and CtFDP
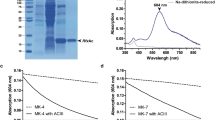
In the previous report that described the purification of native Rd from C. tepidum cells (Yoon et al. 1999), the CT2024 gene product was identified as the major Rd among the three putative Rd genes (CT1100, CT1101, and CT2024). In this work, we prepared recombinant CT2024 gene in the E. coli cells and purified the protein product to homogeneity as judged by SDS-PAGE analysis (Fig. 1a). The N-terminal amino acid sequence of purified CtRd was determined to be MQKWV(C)VP(C)GYEYDP; the two Cys residues were not detectable, in placed corresponding with those deduced from the sequence of the CT2024 gene. The molecular weight of CtRd without iron is estimated at 5869 based on its putative amino acid sequence; the apparent molecular mass of the purified protein was estimated at 13.1 kDa by gel-permeation chromatography. This result suggests that CtRd generates a homodimer under the experimental conditions reported (Yoon et al. 1999). Purified CtRd exhibits absorption bands that are typical for a [Fe-(SCys)4] type cluster protein with maxima at 492, 373, and 280 nm and a shoulder at ~ 570 nm in the UV–visible regions (Fig. 1b). The A492/A280 ratio of the purified CtRd was estimated at 0.32 (Fig. 1b). Reduction by excess sodium dithionite resulted in almost no absorbance in visible region (Fig. 1b). An EPR spectrum of oxidized CtRd at 10 K exhibited a sharp signal that is typical of high-spin rhombic Fe3+ (Biplab et al. 2017) with the g values of 4.30 and 9.58; almost no signal was observed after sodium dithionite reduction (Fig. 1c).

a SDS-PAGE analyses of CtRd and CtFDP on 20% and 12% poly acryl amid gels, respectively, with Coomassie Brilliant Blue staining. Lane 1, 3: molecular mass marker, Lane 2: CtRd (approximately 500 pmol/lane), Lane 4: CtFDP (approximately 1 μg/lane). The mass values (kDa) of the markers are indicated at the left side of each gel image. b UV–visible absorption spectra of air oxidized (continuous line) and dithionite-reduced (dotted line) CtRd. The spectra of approximately 61 μM CtRd in 20 mM HEPES–NaOH buffer (pH 7.0) were measured at 23 °C. The absorption coefficient was estimated based on the reported value at 492 nm (Yoon et al. 1999). c EPR spectra of approximately 200 μM CtRd in oxidized (continuous line) and reduced (dotted line) forms. The measurement conditions: temperature, 10 K; power, 10 mW; microwave frequency, 9.39222 GHz; modulation amplitude, 10 G at 100 kHz. d UV–visible absorption spectra of CtFDP in air oxidized forms before reconstitution (broken line) and after reconstitution (continuous line). The measurement was performed in 20 mM HEPES–NaOH buffer (pH 7.0) at 23 °C. The absorption coefficients were estimated based on the protein concentration before protein reconstitution and the absorption coefficient of FMN cofactor after reconstitution, respectively
Purified CtFDP migrated as a single major band on an SDS-PAGE gel with an apparent molecular mass of 47.3 kDa (Fig. 1a). The apparent mass was estimated at 91.8 kDa by gel-permeation chromatography. These results suggested that CtFDP was also a homodimer under the experimental conditions featured here. N-terminal amino acid sequencing of purified CtFDP resulted in the sequence (M)TDNKILPITDDVSWI; this corresponded to the gene product of the CT2085 gene both with and without the start methionine residue. The deduced amino acid sequence of the CT2285 gene product predicts a protein with a β-lactamase-like domain and conserved amino acid residues consistent with their roles as ligands of the diiron cluster as well as a flavodoxin-like domain, as found in FDPs from other bacterial species (Figure S1) (Martins et al. 2019; Folgosa et al. 2018; Romão et al. 2016a; Frazão et al. 2000; Vicente et al. 2008). Purified CtFDP maintained its brownish-yellow color after the gel-permeation chromatography, although its absorption coefficient at ~ 450 nm was low compared to those of other bacterial FDPs (Vicente et al 2012; Hillmann et al. 2009); this may be due to the release of the FMN cofactor during the purification, as previously reported (Frederick et al. 2015; Fang et al. 2012). Therefore, enrichment for FMN and ferrous ion contents was performed as described in the Materials and Methods section. UV–visible spectrum of the reconstituted CtFDP resulted in absorption bands with maxima at 449, 370, and 277 nm, which are typical for a flavin prosthetic group (Fig. 2c). The FMN and iron contents of the CtFDP were estimated at ~ 1.0 mol FMN and 2.5 ± 0.13 mol iron per mol CtFDP protomer. Reconstituted CtFDP was used in all the enzymatic assays to follow.

a NAD(P)H concentration dependency at 100 μM CtRd and b CtRd concentration dependency of CtRd reduction rate catalyzed by BsFNR with 5 μM NADPH (open circle), CtFNR with 50 μM NADPH (closed circle) and CtFNR with 100 μM NADH (closed square). The measurement was performed in 20 mM HEPES–NaOH buffer (pH7.0) at 25 °C. The observed rate indicates the generation of the number of reduced CtRd per one second per one FNR protomer. The curves are fitted using the Michaelis Menten equation with the kcat and KM values of CtFNR with NADH; 34.7 ± 0.6 s−1 and 207 ± 10 μM (broken line in a), BsFNR with CtRd at 5 μM NADPH; 255 ± 10 s−1 and 21 ± 2 μM (continuous line in b), CtFNR with CtRd at 50 μM NADPH; 24.8 ± 0.8 s−1 and 32 ± 2 μM (dotted line in b) and CtFNR with CtRd at 100 μM NADH; 16.5 ± 1.4 s−1 and 19 ± 5 μM (broken line in b), respectively
CtRd reduction by ferredoxin-NAD(P)+ oxidoreductases
In the facultative and obligate anaerobic heterotrophic bacteria NADH-Rd oxidoreductase [NROR, (EC 1.18.1.1)] catalyzes the reduction of Rd (Martins et al. 2019). Blast homology search using the amino acid sequence of Pseudomonas aeruginosa NROR (pae_pa5349, NCBI ID: 879,643) to query putative C. tepidum genes predicts CT2078 (NCBI GeneID: 1,007,483) as an NROR homolog; this is currently annotated as an NADH oxidase (Eisen et al. 2002; Li et al. 2009) (Figure S2A). Although the key residues required for NADH-binding are conserved in the deduced amino acid sequence and align with those of PaNROR (Hagelueken et al. 2007) and E. coli NROR (Romão et al. 2016b), the calculated identity and the length of the C-terminal region (Ser314 to Lys452) were low and comparatively long, respectively (Figure S2A). In the crystal structure of the PaNROR-PaRd complex, the C-terminal region includes the major area underlying interactions with Rd (Hagelueken et al. 2007). Blast homology search (Altschul et al. 1990) and structural modeling on the SWISS Model server (Waterhouse et al. 2018) revealed that CT2078-encoded peptide, notably the C-terminal region, was highly homologous (both with respect to sequence and structure) to the H2O-forming NADH oxidase [NOX, (Geueke et al. 2003; Kawasaki et al. 2004; Lountos et al. 2006)]. Likewise, Cys42 in the catalytic site of NOX from Lactobacillus sanfranciscensis was conserved in CT2078 (Lountos et al. 2006) (Figure S2B); taken together, these results suggest that CT2078 might function as a NADH oxidase rather than an NROR in C. tepidum cells. A previous study focused on a CT2078 null mutant reported that this disruption exhibited minimal impact on C. tepidum survival, while disruption of the CtFDP gene (CT2285) was fatal to cells undergoing photosynthetic growth in microoxic conditions; the existence of an NROR-independent electron transfer path to the CtFDP was suggested (Li et al. 2009). NROR is a member of NAD(P)H/FAD-dependent oxidoreductase family of the flavoprotein superfamily; this superfamily also includes FNRs (Martins et al. 2019; Hagelueken et al. 2007; Argyrou et al. 2004; Dym and Eisenberg 2001). In this study, we assessed the capacity of CtRd to undergo reduction via the actions of FNRs from both C. tepidum and B. subtilis, considering that their interactions with oxidation/reduction was carried out by the small iron sulfur protein, Fd (Seo et al. 2002,2004).
We found that BsFNR reduced CtRd at a high rate (190 s−1 at 5 μM NADPH); this was more rapid than the rate of Fd reduction catalyzed by this protein (Fig. 2a) [~ 50 s−1, (Seo et al. 2014)] and was comparable to those of NRORs from other organisms [8.4–350 s−1, (Kawasaki et al. 2009; Cabeza et al. 2015; Chen et al. 1993; Lee et al. 1998; Ma and Adams 1999)]. By contrast, the rate of CtRd reduction by CtFNR remained one-fifth to one-tenth that of BsFNR at the same concentrations of NADPH (Fig. 2a). One of the reasons underlying this difference may relate to higher ratios of reduced FNR to NADP+/H at the redox equilibria in BsFNR than in CtFNR, as reported in previous studies (Seo et al. 2016a, b). At over 10 μM NADPH for BsFNR and 100 μM NADPH for CtFNR, we observed decreased rates of reaction in response to increasing NADPH concentrations; this finding is indicative of substrate inhibition. Substrate inhibition was also observed in steady state assays with Fds and both BsFNR and CtFNR (Seo et al. 2002,2009). The KM values calculated for CtRd using both FNRs (19–32 μM, Fig. 2b) are significantly larger than that calculated for BsFd [< 5 μM, (Seo et al. 2009, 2014)]. Despite the relatively low degree of structural similarity observed between CtRd and BsFd, and the lower affinity toward CtRd, substrate inhibition was detected at similar NADPH concentrations for both FNRs; these results suggested that the reduction of CtRd proceeded using a similar binding mode as that used by BsFd. By contrast, when using NADH as an electron donor in reactions with CtFNR, the rate increased with increasing NADH concentrations; no substrate inhibition was observed at concentrations as high as 1 mM (Fig. 2a). This is likely due to the lower affinity of CtFNR for NADH (Seo and Sakurai 2002).
Oxygen reduction by CtFDP
Because BsFNR can function as a suitable NADPH-CtRd reductase in the presence of dioxygen, the NADPH-BsFNR pair was used to examine the reduction of CtRd and the measurement of the dioxygen reductase activity in the presence of CtFDP (Fig. 3). Stepwise additions of G6P, G6PDH, NADP+, BsFNR, and CtRd did not result in significant increases in the observed rate of oxygen consumption (broken line in Fig. 3); this result indicated that dioxygen reduction catalyzed by the combination of these components was very low in the absence of CtFDP under these experimental conditions. Given that the absence of CtRd resulted in a very slow rate of oxygen consumption (continuous line in Fig. 3), we can conclude that CtRd (but not BsFNR) donated electrons to CtFDP in this assay. In the presence of an NADPH regenerating system, the oxygen concentration decreased linearly with time. The observed rate was estimated to be 11.46 ± 0.07 μmol O2·s−1·μmol FMN−1 at 25 °C and pH 7.0; this is comparable to the rates reported for other FDPs [5‒28 s−1, (Vicente et al. 2012; Hillmann et al. 2009; Frederick et al. 2015; Di Matteo et al. 2008)]. An ddition of catalase after the change in O2 concentration became steady resulted in a scarce change in O2 concentration (Figure S3). These results support that CtRd can donate electrons to CtFDP and CtFDP can function as a H2O-forming dioxygen reductase.

The O2 consumption catalyzed by CtFDP. The measurement was performed in 20 mM HEPES–NaOH buffer (pH 7.0) containing 5 mM G6P, 2 U/ml G6PDH, 100 nM BsFNR, 10 μM NADP+, 100 μM CtRd and 100 nM CtFDP as final concentrations at 25 °C. The order of the additions of each component is indicated by the arrowheads at the corresponding time of the traces. The observed rate of O2 consumption after addition of CtRd in continuous line was estimated to be 11.46 ± 0.07 μmol O2·s−1·μmol FMN−1
In C. tepidum cells, the electron transfer path to CtRd was not clear. Although our results indicate that CtFNR can support NAD(P)H-dependent CtRd reduction as effectively as CtPFOR (Yoon et al. 1999), C. tepidum can grow in the photoautotrophic mode only; photosynthesis-independent NAD(P)+ reduction has not been reported. Considering the interchangeability of Rd with Fd as electron acceptors of FNR reported in this work, the RC may be among the potent candidates for CtRd-mediated reduction. Deletion of the gene encoding CtFDP exhibits profound effects on bacterial growth in the light and under microoxic conditions (Li et al. 2009). However, this deletion also demonstrates significant impact on growth in the dark (Li et al. 2009). Further investigation will be required to improve our understanding of the dioxygen and ROS response of this bacterial species.
References
Alboresi A, Storti M, Cendron L, Morosinotto T (2019) Role and regulation of class-C flavodiiron proteins in photosynthetic organisms. Biochem J 476:2487–2498
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Anbar AD, Duan Y, Lyons TW, Arnold GL, Kendall B, Creaser RA, Kaufman AJ, Gordon GW, Scott C, Garvin J, Buick R (2007) A whiff of oxygen before the great oxidation event? Science 317:1903–1906
Argyrou A, Blanchard JS (2004) Flavoprotein disulfide reductases: advances in chemistry and function. Prog Nucleic Acid Res Mol Biol 78:89–142
Bertsova YV, Kulik LV, Mamedov MD, Baykov AA, Bogachev AV (2019a) Flavodoxin with an air-stable flavin semiquinone in a green sulfur bacterium. Photosynth Res 142:127–136
Bertsova YV, Mamedov MD, Bogachev AV (2019b) Na+-Translocating Ferredoxin:NAD+ oxidoreductase is a component of photosynthetic electron transport chain in green sulfur bacteria. Biochemistry (Mosc) 84:1403–1410
Brummett AE, Dey M (2016) New Mechanistic insight from substrate- and product-bound structures of the metal-dependent dimethylsulfoniopropionate lyase DddQ. Biochemistry 55:6162–6174
Buchanan BB, Arnon DI (1971) Ferredoxins from photosynthetic bacteria, algae, and higher plants. Methods Enzymol 23(C):413–440
Buchanan BB, Arnon DI (1990) A reverse KREBS cycle in photosynthesis: consensus at last. Photosyn Res 24:47–53
Buchanan BB, Sirevåg R, Fuchs G, Ivanovsky RN, Igarashi Y, Ishii M, Tabita FR, Berg IA (2017) The Arnon-Buchanan cycle: a retrospective, 1966–2016. Photosynth Res 134117–134131
Cabeza MS, Guerrero SA, Iglesias AA, Arias DG (2015) New enzymatic pathways for the reduction of reactive oxygen species in Entamoeba histolytica. Biochim Biophys Acta 1850:1233–1244
Chen L, Liu MY, Legall J, Fareleira P, Santos H, Xavier AV (1993) Purification and characterization of an NADH-rubredoxin oxidoreductase involved in the utilization of oxygen by Desulfovibrio gigas. Eur J Biochem 216:443–448
Dey M, Brummett AE (2018) Isolation and assays of bacterial dimethylsulfoniopropionate lyases. Methods Enzymol 605:291–323
Di Matteo A, Scandurra FM, Testa F, Forte E, Sarti P, Brunori M, Giuffrè A (2008) The O2-scavenging flavodiiron protein in the human parasite Giardia intestinalis. J Biol Chem 283:4061–4068
Dym O, Eisenberg D (2001) Sequence-structure analysis of FAD-containing proteins. Protein Sci 10:1712–1728
Eddie BJ, Hanson TE (2013) Chlorobaculum tepidum TLS displays a complex transcriptional response to sulfide addition. J Bacteriol 195:399–408
Eisen JA, Nelson KE, Paulsen IT, Heidelberg JF, Wu M, Dodson RJ, Deboy R, Gwinn ML, Nelson WC, Haft DH, Hickey EK, Peterson JD, Durkin AS, Kolonay JL, Yang F, Holt I, Umayam LA, Mason T, Brenner M, Shea TP, Parksey D, Nierman WC, Feldblyum TV, Hansen CL, Craven MB, Radune D, Vamathevan J, Khouri H, White O, Gruber TM, Ketchum KA, Venter JC, Tettelin H, Bryant DA, Fraser CM (2002) The complete genome sequence of Chlorobium tepidum TLS, a photosynthetic, anaerobic, green-sulfur bacterium. Proc Natl Acad Sci U S A 99:9509–9514
Evans MC, Buchanan BB, Arnon DI (1966) A new ferredoxin-dependent carbon reduction cycle in a photosynthetic bacterium. Proc Natl Acad Sci U S A 55:928–934
Fang H, Caranto JD, Mendoza R, Taylor AB, Hart PJ, Kurtz DM Jr (2012) Histidine ligand variants of a flavo-diiron protein: effects on structure and activities. J Biol Inorg Chem 17:1231–1239
Folgosa F, Martins MC, Teixeira M (2018) Diversity and complexity of flavodiiron NO/O2 reductases. FEMS Microbiol Lett. 365–267
Frazão C, Silva G, Gomes CM, Matias P, Coelho R, Sieker L, Macedo S, Liu MY, Oliveira S, Teixeira M, Xavier AV, Rodrigues-Pousada C, Carrondo MA, Le Gall J (2000) Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nat Struct Biol 11:1041–1045
Frederick RE, Caranto JD, Masitas CA, Gebhardt LL, MacGowan CE, Limberger RJ, Kurtz DM Jr (2015) Dioxygen and nitric oxide scavenging by Treponema denticola flavodiiron protein: a mechanistic paradigm for catalysis. J Biol Inorg Chem 20:603–613
Fukuyama K (2004) Structure and function of plant-type ferredoxins. Photosyn Res 81:289–301
Geueke B, Riebel B, Hummel W (2003) NADH oxidase from Lactobacillus brevis: a new catalyst for the regeneration of NAD. Enz Microbial Tech 32:205–211
Gregersen LH, Bryant DA, Frigaard NU (2011) Mechanisms and evolution of oxidative sulfur metabolism in green sulfur bacteria. Front Microbiol 24:116
Hagelueken G, Wiehlmann L, Adams TM, Kolmar H, Heinz DW, Tümmler B, Schubert WD (2007) Crystal structure of the electron transfer complex rubredoxin rubredoxin reductase of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 104:12276–12281
Hillmann F, Riebe O, Fischer RJ, Mot A, Caranto JD, Kurtz DM Jr, Bahl H (2009) Reductive dioxygen scavenging by flavo-diiron proteins of Clostridium acetobutylicum. FEBS Lett 583:241–245
Jokel M, Kosourov S, Battchikova N, Tsygankov AA, Aro EM, Allahverdiyeva Y (2015) Chlamydomonas Flavodiiron proteins facilitate acclimation to anoxia during sulfur deprivation. Plant Cell Physiol 56:1598–1607
Kawasaki S, Ishikura J, Chiba D, Nishino T, Niimura Y (2004) Purification and characterization of an H2O-forming NADH oxidase from Clostridium aminovalericum: existence of an oxygen-detoxifying enzyme in an obligate anaerobic bacteria. Arch Microbiol 181:324–330
Kawasaki S, Sakai Y, Takahashi T, Suzuki I, Niimura Y (2009) O2 and reactive oxygen species detoxification complex, composed of O2-responsive NADH: rubredoxin oxidoreductase-flavoprotein A2-desulfoferrodoxin operon enzymes, rubperoxin, and rubredoxin, in Clostridium acetobutylicum. Appl Environ Microbiol 75:1021–1029
Kjær B, Scheller HV (1996) An isolated reaction center complex from the green sulfur bacterium Chlorobium vibrioforme can photoreduce ferredoxin at high rates. Photosyn Res 47:33–39
Knaff DB, Hirasawa M (1991) Ferredoxin-dependent chloroplast enzymes. Biochim Biophys Acta 1056:93–125
Kusumoto N, Inoue K, Nasu H, Sakurai H (1994) Preparation of a photoactive reaction center complex containing photo-reducible Fe-S centers and photooxidizable cytochrome c from the green sulfur bacterium Chlorobium tepidum. Plant Cell Phys 35:17–25
Lee HJ, Basran J, Scrutton NS (1998) Electron transfer from flavin to iron in the Pseudomonas oleovorans rubredoxin reductase-rubredoxin electron transfer complex. Biochemistry 37:15513–15522
Li H, Jubelirer S, Garcia Costas AM, Frigaard NU, Bryant DA (2009) Multiple antioxidant proteins protect Chlorobaculum tepidum against oxygen and reactive oxygen species. Arch Microbiol 191:853–867
Lountos GT, Jiang R, Wellborn WB, Thaler TL, Bommarius AS, Orville AM (2006) The crystal structure of NAD(P)H oxidase from Lactobacillus sanfranciscensis: insights into the conversion of O2 into two water molecules by the flavoenzyme. Biochemistry 45:9648–9659
Ma K, Adams MW (1999) A hyperactive NAD(P)H: rubredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol 181:5530–5533
Maiti BK, Almeida RM, Moura I, Moura JJG (2017) Rubredoxins derivatives: simple sulphur-rich coordination metal sites and its relevance for biology and chemistry. Coordination Chem Rev 352:379–397
Martins MC, Romão CV, Folgosa F, Borges PT, Frazão C, Teixeira M (2019) How superoxide reductases and flavodiiron proteins combat oxidative stress in anaerobes. Free Radic Biol Med 140:36–60
Medina M, Gómez-Moreno C (2004) Interaction of ferredoxin-NADP+ reductase with its substrates: optimal interaction for efficient electron transfer. Photosynth Res 79:113–131
Miller M, Liu X, Snyder SW, Thurnauer MC, Biggins J (1992) Photosynthetic electron-transfer reactions in the green sulfur bacterium Chlorobium vibrioforme: evidence for the functional involvement of iron-sulfur redox centers on the acceptor side of the reaction center. Biochemistry 31:4354–4363
Notredame C, Higgins DH, Heringa J (2000) T-Coffee: a novel method for multiple sequence alignments. J Mol Biol 302:205–217
Ohnishi K, Niimura Y, Yokoyama K, Hidaka M, Masaki H, Uchimura T, Suzuki H, Uozumi T, Kozaki M, Komagata K, Nishino T (1994) Purification and analysis of a flavoprotein functional as NADH oxidase from Amphibacillus xylanus overexpressed in Escherichia coli. J Biol Chem 269:31418–31423
Okegawa Y, Motohashi K (2015) A simple and ultra-low cost homemade seamless ligation cloning extract (SLiCE) as an alternative to a commercially available seamless DNA cloning kit. Biochem Biophys Rep 4:148–151
Romão CV, Vicente JB, Borges PT, Frazão C, Teixeira M (2016a) The dual function of flavodiiron proteins: oxygen and/or nitric oxide reductases. J Biol Inorg Chem 21:39–52
Romão CV, Vicente JB, Borges PT, Victor BL, Lamosa P, Silva E, Pereira L, Bandeiras TM, Soares CM, Carrondo MA, Turner D, Teixeira M, Frazão C (2016b) Structure of Escherichia coli flavodiiron nitric oxide reductase. J Mol Biol 428:4686–4707
Sakurai H, Ogawa T, Shiga M, Inoue K (2010) Inorganic sulfur oxidizing system in green sulfur bacteria. Photosynth Res 104:163–176
Saunders AH, Golbeck JH, Bryant DA (2013) Characterization of BciB: a ferredoxin-dependent 8-vinyl-protochlorophyllide reductase from the green sulfur bacterium Chloroherpeton thalassium. Biochemistry 52:8442–8451
Seo D, Sakurai H (2002) Purification and characterization of ferredoxin-NAD(P)+ reductase from the green sulfur bacterium Chlorobium tepidum. Biochim Biophys Acta 1597:123–132
Seo D, Tomioka A, Kusumoto N, Kamo M, Enami I, Sakurai H (2001) Purification of ferredoxins and their reaction with purified reaction center complex from the green sulfur bacterium Chlorobium tepidum. Biochim Biophy Acta 1503:377–384
Seo D, Kamino K, Inoue K, Sakurai H (2004) Purification and characterization of ferredoxin-NADP+ reductase encoded by Bacillus subtilis yumC. Arch Microbiol 182:80–89
Seo D, Okabe S, Yanase M, Kataoka K, Sakurai T (2009) Studies of interaction of homo-dimeric ferredoxin-NAD(P)+ oxidoreductases of Bacillus subtilis and Rhodopseudomonas palustris, that are closely related to thioredoxin reductases in amino acid sequence, with ferredoxins and pyridine nucleotide coenzymes. Biochim Biophys Acta 1794:594–601
Seo D, Asano T, Komori H, Sakurai T (2014) Role of the C-terminal extension stacked on the re-face of the isoalloxazine ring moiety of the flavin adenine dinucleotide prosthetic group in ferredoxin-NADP+ oxidoreductase from Bacillus subtilis. Plant Physiol Biochem 81:143–148
Seo D, Soeta T, Sakurai H, Sétif P, Sakurai T (2016a) Pre-steady-state kinetic studies of redox reactions catalyzed by Bacillus subtilis ferredoxin-NADP+ oxidoreductase with NADP+/NADPH and ferredoxin. Biochim Biophys Acta 1857:678–687
Seo D, Kitashima M, Sakurai T, Inoue K (2016b) Kinetics of NADP+/NADPH reduction-oxidation catalyzed by the ferredoxin-NAD(P)+ reductase from the green sulfur bacterium Chlorobaculum tepidum. Photosynth Res 130:479–489
Sétif P (2001) Ferredoxin and flavodoxin reduction by photosystem I. Biochim Biophys Acta 1507:161–179
Tanaka M, Haniu M, Evans MCW, Rao KK (1974) Amino acid sequence of ferredoxin from a photosynthetic green bacterium, Chlorobium limicola. Biochemistry 13:2953–2959
Tanaka M, Haniu M, Yasunobu KT, Evans MCW, Rao KK (1975) The amino acid sequence of ferredoxin II from Chlorobium limicola, a photosynthetic green bacterium. Biochemistry 14:1938–1943
Tang KH, Blankenship RE (2010) Both forward and reverse TCA cycles operate in green sulfur bacteria. J Biol Chem 285:35848–35854
Tsukatani Y, Miyamoto R, Itoh S, Oh-oka H (2004) Function of a PscD subunit in a homodimeric reaction center complex of the photosynthetic green sulfur bacterium Chlorobium tepidum studied by insertional gene inactivation: regulation of energy transfer and ferredoxin-mediated NADP+ reduction on the cytoplasmic side. J Biol Chem 279:51122–51130
Vassiliev IR, Ronan MT, Hauska G, Golbeck JH (2000) The bound electron acceptors in green sulfur bacteria: resolution of the g-tensor for the F(X) iron-sulfur cluster in Chlorobium tepidum. Biophys J 78:3160–3169
Vicente JB, Carrondo MA, Teixeira M, Frazão C (2008) Structural studies on flavodiiron proteins. Methods Enzymol 437:3–19
Vicente JB, Tran V, Pinto L, Teixeira M, Singh U (2012) A detoxifying oxygen reductase in the anaerobic protozoan Entamoeba histolytica. Eukaryot Cell 11:1112–1118
Wahlund TM, Madigan MT (1993) Nitrogen fixation by the thermophilic green sulfur bacterium Chlorobium tepidum. J Bacteriol 175:474–478
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modeling of protein structures and complexes. Nucleic Acids Res 46:W296–W303
Yoon KS, Hille R, Hemann C, Tabita FR (1999) Rubredoxin from the green sulfur bacterium Chlorobium tepidum functions as an electron acceptor for pyruvate ferredoxin oxidoreductase. J Biol Chem 274:29772–29778
Yoon KS, Bobst C, Hemann CF, Hille R, Tabita FR (2001) Spectroscopic and functional properties of novel 2[4Fe-4S] cluster-containing ferredoxins from the green sulfur bacterium Chlorobium tepidum. J Biol Chem 276:44027–44036
Acknowledgements
The authors would like to thank ENAGO for the English language review.
Funding
This work was partly supported by Japan Society for the Promotion of Science KAKENHI Grant Number JP17K07304 (to DS) and JP18K06296 (to KI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Erko stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ittarat, W., Sato, T., Kitashima, M. et al. Rubredoxin from the green sulfur bacterium Chlorobaculum tepidum donates a redox equivalent to the flavodiiron protein in an NAD(P)H dependent manner via ferredoxin-NAD(P)+ oxidoreductase. Arch Microbiol 203, 799–808 (2021). https://doi.org/10.1007/s00203-020-02079-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-020-02079-4