
Abstract
Plant domestication was a pivotal accomplishment in human history, which led to a reduction in genetic diversity of crop species; however, there was less research focus on how this reduced genetic diversity of crops in affecting rhizosphere microbial communities during crop domestication process. Here, we used high-throughput sequencing to explore the different effects of crops domestication on rhizosphere microbial community structure of rice (Oryza sativa L. and Oryza rufipogon Griff.) and soybean (Glycine max L. and Glycine soja Sieb. et Zucc.). Results indicated that rhizosphere fungal communities are more strongly influenced by crop domestication than bacterial communities. There was a stronger relationship for fungi and bacteria in the cultivated crops than in the wild relatives. Results also showed that the wild varieties had a higher abundance of beneficial symbionts and a lower abundance of pathogens comparing with the cultivated varieties. There was a similar tendency for both rice and soybean in rhizosphere microbial communities by comparing wild crops and their cultivated varieties. In conclusion, crop domestication had a stronger effect on the fungal communities than on the bacterial communities and had improved the microbial relationship in rhizosphere of cultivated crops.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
When wild plants were domesticated, a substantial alteration in the composition and function of the rhizomicrobiome may have occurred (Shenton et al. 2016; Leff et al. 2017). As we know modern cultivars are bred for improved yield and biomass production and a better quality for food and fodder when growing under the conditions of relatively high nutrient availability, protection against pathogens by application of pesticides, and optimal moisture by irrigation (Shi et al. 2018a, b; Tian et al. 2018). However, wild crops grow and survive naturally without these optimal growing conditions which have resulted in significant differences in the structure and functioning of the rhizosphere communities between modern crops and their wild relatives (Bulgarelli et al. 2015; Tian et al. 2017). The rhizosphere microbial community structure was affected by a variety of abiotic and biotic factors (Barbosa Lima et al. 2015; Rodríguez-Blanco et al. 2015; Wagner et al. 2016). Thus, it is reasonable to assume that the rhizomicrobiome of wild crops may contribute to host plants’ growth and survival under biotic and abiotic stress conditions more effectively than that of their cultivated relatives (Philippot et al. 2013; Mendes and Raaijmakers 2015; Perez-Jaramillo et al. 2016).
At present, many studies reported that the bacterial and fungal community structure of the plant species rhizomicrobiome specific in Arabidopsis (Schlaeppi et al. 2014; Bulgarelli et al. 2015), maize (Zea mays L.) (Bouffaud et al. 2014), rice (Oryza sativa) (Edwards et al. 2015), beet (Beta vulgaris ssp.) (Zachow et al. 2014), and sunflower (Helianthus annuus) (Leff et al. 2017). However, the drivers of microbial community assemblages in the rhizosphere are still rudimentary and the mechanisms associated with the effects of crop cultivar on the structure of the rhizosphere community remain largely unknown (Hartmann et al. 2014; Yan et al. 2015). Moreover, whether the differences that occur in the rhizosphere community assembly of cultivated versus their wild crops are true for different crops has not been determined. Therefore, we utilize different crops like wild versus cultivated rice and soybean, which are grown in the same experimental conditions to find out whether the rhizosphere microorganisms developed from wild crops to cultivated crops have the same trend.
In recent years, the concept of the rhizomicrobiome has evolved (Mendes et al. 2011) and advances in DNA-sequencing technology have allowed researchers to investigate the structure of the rhizomicrobiome in more detail than before (Hua et al. 2014). Metabarcoding and next-generation-sequencing have provided the possibilities to make detailed assessments of differences between the rhizomicrobiome of wild and their cultivated crops. In addition, high-throughput sequencing is also possible to identify the predominant microbial groups in the rhizomicrobiome and their functions that have a positive impact on wild plants’ growth and stress tolerance, which may be no longer present or present to a lesser extent in the rhizomicrobiome of cultivated relatives (Zhang et al. 2017; Mendes et al. 2018). This knowledge may eventually be useful for the development of sustainable crop management strategies that ensures sufficient crop yields with high quality, at the conditions of reduced chemical and physical inputs.
Over the past decades, plant breeders have exploited genes from wild relatives of modern crop species to improve plant growth and health. For instance, wild relatives have been used as sources of alleles to improve the ability of modern cultivars to withstand biotic and abiotic stresses in wheat (Agropyron elongatum) (Placido et al. 2013), barley (Hordeum vulgare ssp. spontaneum) (Schmalenbach et al. 2008), and lettuce (Lactuca sativa L.) (Simko et al. 2013). Similarly, entomologists have explored native habitats to identify the natural enemies of insect pests. However, in the area of crop microbial research, relatively few studies have been made to illustrate about biodiversity and functions of beneficial microbial community composition present in the native habitats of ancestors of modern crop species. Limited efforts have been made to study the bacteria of cultivated crops and their wild relatives (Shenton et al. 2016; Pérez-Jaramillo et al. 2017), and the rhizosphere fungal community across the degree of domestication of sunflower (Helianthus annuus) (Leff et al. 2017). In a previous study, the rhizosphere bacterial community structure of domesticated corn cultivars and its relative wild corn (Balsas teosinte) were compared by using terminal restriction fragment length polymorphism analysis of the 16S rRNA gene (Szoboszlay et al. 2015). However, no study has evaluated simultaneously the bacterial and fungal communities in cultivated crop versus its wild relatives using high-throughput sequencing.
In this study, we have a hypothesis that the rhizosphere bacterial and fungal communities assembly differ in cultivated rice and soybean species versus their wild relatives. The hypothesis was tested by high-throughput sequencing of bacterial 16S rRNA and fungal internal transcribed spacer (ITS) regions.
Materials and methods
Soil and plant materials
The soils were collected from the Changchun Agricultural Station of the Northeast Institute of Geography and Agroecology, located in Jilin Province, China (43°59′N, 125°23′E). The main soil physiochemical characteristics are described in Table S1. The soil type is belonging to black soil.
Two different cultivated crops and their wild relatives were chosen to assess the microbial community composition with the rhizosphere of Oryza sativa L. ssp. Japonica cv. Dongdao-4 (OsL), Oryza rufipogon Griff. (OrG), Glycine max (L.) Merr. cv. Zhonghuang 39 (GmL), and Glycine soja Sieb. et Zucc. (GsS). The varieties of cultivated rice and soybean species used represented common cultivars grown in local regions of China.
The seeds of wild rice were obtained from the Dongxiang Natural Conservation Region (28°14′N, 116°30′E, 45.8 m a.s.1.) and the seeds of wild soybean were from Northeastern China (47°47′N, 126°97′E, 238 m a.s.1.). The morphological comparison between cultivated and wild crops is shown in Figure S1.
Plant growth and rhizospheric soil collection
Seeds were surface-sterilized with 1% sodium hypochlorite for 8 min and germinated on filter paper wetted. After 5 days, germinated seeds were transferred to a 10 cm diameter × 10 cm high pots filled with described agricultural soil and incubated for 3 weeks in a growth chamber (25 °C, 16 h daylight). To synchronize the development of wild and cultivated plants, plants were incubated at 4 °C. Six seedlings growing uniformly per plant species were then transplanted into 20 cm diameter × 20 cm high plastic pots containing the described soil. The plantlets were trimmed to three seedlings per pot. The pots were subsequently kept under controlled temperature (26 °C/28 °C), photoperiod (16/8 h), and relative humidity of 70%. Three pots with the same amount of soil but without plants were used as control.
Rhizosphere soils were sampled at flowering stages in 60 and 55 days for rice and soybean, respectively. The rhizosphere soils were sampled removing the whole plant and brushing the soil adhered to the seminal roots. Bulk soil samples were taken from control.
DNA extraction for high-throughput sequencing
Soil DNA was extracted from 0.5 g (wet weight) of each sample using Fast DNA™ Spin Kit for Soil (MP Biomedicals, Santa Ana, CA, USA). The amplification of the V3–V4 hypervariable regions of 16S rRNA was performed using the primer set 341 F (5′-CCTACGGGNGGCWGCAG-3′) and 785 R (5′-GACTACHVGGGTATCTAATCC-3′) (Pérez-Jaramillo et al. 2017; Thijs et al. 2017). For fungal amplification, a fungal primer set ITS3 F (5′-GATGAAGAACGYAGYRAA-3′) and ITS4 R (5′-TCCTCCGCTTATTGATATGC-3′) (Zhou et al. 2017) was used to amplify. PCR was carried out using 0.25 uL forward primer, 0.25 µL reverse primer, and 1 µL sample DNA template, 12.5 µL 2 × KAPA HiFi HotStart Ready Mix, and 11 µL PCR Grade Water. Total volume was 25 µL. We used following PCR program on a LifePro PCR thermocycler: 94 °C for 3 min, followed by 25 cycles of 95 °C for 30 s, 53 °C for 30 s, and 72 °C for 30 s. The final extension at 72 °C was used for 8 min and the reactions were held at 4 °C. As negative control, PCR Grade Water was used instead of DNA template. The PCR products were visualized on a 1.2% agarose gel. PCR products were purified using QIAquick Gel Extraction Kit (Qiagen, USA, code No. 28704), according to the technical specification. The DNA was quantified using Qubit® Fluorometers (Thermo Fisher Scientific). Samples were subjected to 250-bp paired-end sequencing on an Illumina MiSeq instrument (San Diego, CA, USA).
16S and ITS sequences’ processing
Raw sequences were merged using FLASH v1.2.11 (Magoč and Salzberg 2011), and then, the merged sequences were filtrated by mothur v1.35.1 (Schloss et al. 2009) as follows. First, reads containing any sequence that the average quality score was less than or equal 20 bp, including ambiguous bases and homopolymer, were more than 10 bp and then were removed by mothur. Second, reads containing any mismatch sequence that was more than 4 bp were removed and primer sequences were also removed. Third, reads containing any sequence that were less than 200 bp and more than 500 bp were discarded. Chimeric sequences were removed using USEARCH v7.0 (Edgar et al. 2011) based on the SILVA database (Pruesse et al. 2007) for 16S rRNA and the UNITE database (Kõljalg et al. 2013) for ITS. All of the remaining sequences from each sample were then clustered into operational taxonomic units (OTUs) by USEARCH v6.0 (Edgar 2013) based on 97% sequence similarity. OTU taxonomic classification was conducted by BLAST searching the representative sequences set against the SILVA database and the UNITE v6.0 database (Kõljalg et al. 2013) for bacterial and fungal sequences, respectively. FunGuild v1.1, which is a flat database hosted by GitHub (https://github.com/UMNFuN/FUNGuild), was used to assign fungal phylotypes from the rarefied data to one of the three trophic modes (pathotroph and symbiotroph) where possible (Nguyen et al. 2016).
All of the Illumina raw sequence data were deposited into the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) database (http://www.ncbi.nlm.nih.gov/sra) under accession number NO. SRP079341.
Statistical analyses
The OTU table was rarefied for downstream analysis. To determine the overall structural changes in microbial community composition, a principal coordinate analysis (PCoA) was performed based on Bray–Curtis dissimilarities generated from rarefied OTU, using R package ape version 4.1 (Jari Oksanen et al. 2018). Bray–Curtis dissimilarities were used to perform permutational multivariate analysis of variance (PERMANOVA) analysis with 9999 random permutations (P < 0.05) (Schlemper et al. 2018). Differences in mean proportion were conducted using the Statistical Analysis of Metagenomics Profiles (STAMP) v2.1.3 program (P < 0.05) (Parks et al. 2014). Benjamini–Hochberg algorithm was applied to control false discovery rates (FDR). Spearman correlations were performed based on the Shannon index, Simpson index, and Sobs to determine the level of significance of differences in diversity as a result of crop cultivar, and whether these levels differed for bacteria and fungi. Correlation coefficients were calculated using the corr.test function in the stats package of R based on Spearman’s test (Halpin et al. 2016). Co-inertia analysis (COIA) was performed using the “ade4” package. Monte Carlo test was carried out for COIA using 999 random permutations. The statistical analyses were performed using SPSS version 20.0.
Results
A total of 249,549 bacterial reads after filtering out of 300,038 raw reads (ranging from 9028 to 31,654) were obtained, resulting in 3233 OTUs. A total of 144,126 effective fungal reads out of 168,266 raw sequences were obtained (ranging from 6398 to 16,973), resulting in 1350 OTUs. The result indicated that the sequencing coverage was sufficient to detect most of the species and the OTU number produced by the Illumina sequencing is reasonable (Fig. S2).
Bacterial community structure of cultivated rice and soybean and their wild relatives
PCoA showed that the dissimilarity of rhizosphere bacterial communities (57.29% explained by axis 1) (Fig. 1a) was mainly due to plant species, i.e., soybeans versus rice. Results showed that the bacterial communities were not significantly different in the rhizosphere soils of rice (F = 21.38; P = 0.1) (Fig. S3a) and soybean (F = 1.66; P = 0.2) (Fig. S3b).

Principal coordinate analysis (PCoA) showing community similarity between samples and the reliability of the data for bacteria (a) and fungi (b). Open squares are the cultivated species and solid squares are their wild relatives. Red squares are rice species and blue squares are soybean species. OrG, Oryza rufipogon Griff.; OsL, Oryza sativa L.; GsS, Glycine soja Sieb. et Zucc.; GmL, Glycine max L. (Color figure online)
PCoA showed that the rhizosphere bacterial communities were dissimilar between the rhizosphere soils and bulk soils of rice and soybean (Fig. S4). Results test showed that the bacterial communities were significantly different between the rhizosphere soils and bulk soils of rice (F = 62.71; P = 0.005) (Fig. S4a) and soybean (F = 29.505; P = 0.008) (Fig. S4b).
Fungal community structure of cultivated rice and soybean and their wild relatives
PCoA showed that the dissimilarity of rhizosphere fungal communities (70.98% explained by axis 1) (Fig. 1b) was mainly due to plant species, i.e., soybeans versus rice. Results showed that the fungal communities were significantly different in the rhizosphere soils of rice (F = 15.17; P = 0.031) (Fig. S5a) and soybean (F = 27.96; P = 0.027) (Fig. S5b).
Between cultivated rice and its wild relative, results revealed that these differences were mainly caused by unclassified Pleosporales, Nectriaceae, Trichocomaceae, Xylariaceae family—incertae sedis, and Pleosporaceae with mean proportion significant higher in OsL (cultivated rice) (Fig. S5c). For soybean, Welch’s test indicated the only family responsible for this dissimilarity was Xylariaceae family—incertae sedis with a significant higher abundance at GmL rhizosphere soils (Fig. S5d).
PCoA showed that the rhizosphere fungal communities were dissimilarity between the rhizosphere soils and bulk soils of rice and soybean (Fig. S6). Results test showed that the fungal communities were significantly different between the rhizosphere soils and bulk soils of rice (F = 31.005; P = 0.007) (Fig. S6a) and soybean (F = 67.60; P = 0.003) (Fig. S6b).
Effect of crops domestication on bacterial and fungal community structure
To illuminate the influence of crops cultivar on rhizosphere bacterial and fungal community from cultivated crop and their wild relative, result indicated that the fungal communities were significantly different in rice (P = 0.024) (Table S2), whereas the same analysis indicated that the bacterial communities were not significantly different (P = 0.114) (Table S2). For soybean, results indicated the fungal communities were significantly different (P = 0.041) (Table S3), whereas the same analysis indicated that the bacterial communities were not significantly different (P = 0.219) (Table S3).
Alpha diversity
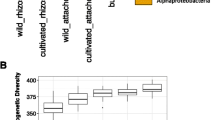
The alpha diversities of both bacteria and fungi were assessed by determining the Shannon index, Simpson index, and the number of species (OTUs) for species richness. No significant differences in diversity and species richness were observed in bacterial communities of cultivated crops versus their wild relatives (P > 0.05) (Fig. 2). In contrast, the diversity and species richness of the fungal communities of wild versus cultivated plants of both rice and soybean differed significantly (Fig. 2). For Shannon index, cultivated rice decreased compared to its wild relative (Fig. 2a), whereas cultivated soybean increased compared to its wild relative (Fig. 2b). The fungal diversity (Simpson index) of cultivated rice increased compared to its wild type (Fig. 2c), while the fungal diversity (Simpson index) of cultivated soybean decreased compared to its wide type (Fig. 2d). For Sobs, cultivated crops (rice and soybean) decreased compared to their wild relative (Fig. 2e, f).

Alpha diversity and number of species observations (sobs) (abundance) based on operational taxonomic units (OTUs) derived from bacterial 16S rRNA and fungal ITS amplicons for rice (a, c, e) and soybean (b, d, f), respectively. The data represent the mean ± SD based on three biological replicates. *Statistically significant differences at P < 0.05 were determined using the Mann–Whitney U two-tailed test (*P < 0.05; **P < 0.01; ***P < 0.001). OrG, Oryza rufipogon Griff.; OsL, Oryza sativa L.; GsS, Glycine soja Sieb. et Zucc.; GmL, Glycine max L.
Correlation analyses and co-inertia analyses
To assess the effect of crop cultivar on bacterial and fungal diversity indices of cultivated crops and their wild relatives, a linear model analysis was conducted. Results indicated that the diversity indices of rhizosphere fungal community were more significantly influenced by crop cultivar than that of the rhizosphere bacterial community of both rice (Fig. 3a) and soybean (Fig. 3b).

Correlation analyses of the effect of crop cultivar on alpha diversity. The analysis was performed using the bacterial and fungal Shannon index, Simpson index, and Sobs (OTUs richness) of rice (a) and soybean (b). Spearman correlations were performed based on the Shannon index, Simpson index, and Sobs. Correlation coefficients were calculated using the corr.test function in the stats package of R based on Spearman’s test. *Statistically significant differences at P < 0.05 were determined using Spearman correlations. Values represent the correlation coefficient
Co-inertia analysis showed that shorter arrows were in cultivated crops (OsL and GmL) than in wild relatives (OrG and GsS), indicating stronger relationship between bacterial and fungal rhizosphere communities in the cultivated crops’ rhizosphere than in the wild relatives. In addition, the projection of arrows of cultivated crops in the opposite direction of wild relatives indicated that cultivated crops had a weak similarity on the variation of bacterial–fungal communities compared with wild crops (Fig. 4).

Co-inertia analysis (COIA) of bacterial and fungal communities of rice and soybean. The back of the arrow represents the location of bacterial community and the tip of arrow represents the location of fungal community. The strength of the relationship between both communities is inversely related to the length of the arrow. Arrows represent the co-variation of both communities: OrG, Oryza rufipogon Griff.; OsL, Oryza sativa L.; GsS, Glycine soja Sieb. et Zucc.; GmL, Glycine max L.
Differences in the putative functionality between cultivated crops and their wild relatives
To determine the impact of crop cultivar on the putative functional properties of the fungal communities in the rhizosphere of wild and cultivated rice and soybean, we assessed the abundance of potential symbiotic and pathogenic fungal species. Most notably, the rhizomicrobiome of wild crops contained significantly higher abundance of putative symbiotic fungi (primarily Glomeromycota) (Fig. 5a, b), while cultivated crops exhibited a higher abundance of putative fungal pathogens (Fig. 5c, d), as indicated by the relative abundances of OTU’s assigned to Alternaria, Acremonium, Periconia, and Thanatephorus.

Percentage of potential fungal symbiotrophs (a, b) and putative fungal pathogens (c, d) in the rhizosphere communities of wild and cultivated crop species. The relative abundance of Glomeromycota as an indication of putative fungal symbionts and the relative abundances of Alternaria, Acremonium, Perciconia, and Thanatephorus OTU’s as an indication of putative fungal pathogens. The data represent the mean ± SD based on three replicates. Statistically significant differences at P < 0.05(*); P < 0.01 (**); and P < 0.001(***) as determined by the Mann–Whitney U one-tailed test. OrG, Oryza rufipogon Griff.; OsL, Oryza sativa L.; GsS, Glycine soja Sieb. et Zucc.; GmL, Glycine max L.
Discussion
To understand whether crop-associated microbial community composition has shifted during crop domestication process and whether potentially beneficial interactions between crops and their rhizomicrobiome have been lost. Although we are aware of the limitations to any conclusion due to the design of this study in which we used one wild and one cultivated plant species of both crops, the results of this study suggest that, indeed, with the development of crop, the rhizosphere communities may have substantial alterations in both rice and soybean. The results from a few other studies which showed that crop evolution resulted in a number of changes in the interactions between plants and other organisms (Prasifka et al. 2015; Turcotte et al. 2015; Leff et al. 2017). Although crop domestication affecting the rhizosphere microbial diversity is often reported, providing a general description of the rhizosphere microbiome is difficult owing to large discrepancies between different studies, which might not only be due to biological variability, but also to the practical issues related to the actual sampling of the rhizosphere (Berg and Smalla 2009). In addition, we should also consider that the molecular work and the bioinformatics pipelines were highly variable and also may strongly affect the outcome of analyses.
One of the main objectives of this study was to clarify whether cultivated crops and their wild relatives had different rhizosphere bacterial and fungal structure. Our results clearly indicate that both the diversity and the structure of the fungal rhizosphere community significantly differed between cultivated crops and their wild relatives, but not the bacterial community. This assumption holds, especially for leguminous plants, such as soybean, due to their specific relationship with symbiotic bacteria. Based on this assumption, we expected that, if there were differences between cultivated species and their wild relatives, these would be larger for bacteria than for fungi. The rhizosphere bacterial communities of wild barley cultivars have been reported to be more diverse than that of their modern counterparts (Bulgarelli et al. 2015). Our results, however, indicated that the rhizobacterial communities did not significantly differ between cultivated crop and their wild relatives for both rice and soybean. Marques et al. (2015) also found that plant domestication may not lead to alterations in the rhizobacterial community. Yet, we found remarkable differences between cultivated crops and their wild relatives of fungal rhizosphere communities. Furthermore, several reports have shown that soil has an important influence in the assembly of rhizosphere microbial community structure (Santos-González et al. 2011; Bulgarelli et al. 2012; Schreiter et al. 2014). Our results indicated that microbial community showed significant difference between rhizosphere and bulk soil of both rice and soybean. Thus, differences between the cultivated and their wild relatives may not be due to one shifting the soil microbiome. The similarity of actinomycete communities in rhizosphere of strawberry plants growing in different soils is greater than that in the bulk soil communities, which indicates that plants are the determinants of microbial community composition stronger than soil types (Costa et al. 2006). Plant species can strongly influence the composition and activity of the rhizosphere microbiota, and differences in root morphology, as well as in the amount and type of rhizo-deposits, between plants contribute greatly to this species-specific effect (Bressan et al. 2009; Ladygina and Hedlund 2010). Many studies have shown that the plant species and the cultivar can affect the composition of the rhizosphere microbiota (İnceoğlu et al. 2012; Bokulich et al. 2014; Nallanchakravarthula et al. 2014). The differences and similarities across studies can perhaps be best understood by considering the assembly of the rhizosphere microbiota as resulting from a hierarchy of events.
Another objective of this study was to determine whether the response of the rhizomicrobiome to crop domestication differed among crop species. We used crops, rice and soybean, that have fundamentally different below-ground traits, and that may, therefore, show plant-specific differences in their respective rhizomicrobiomes when comparing wild and cultivated species. Such differences and an understanding of the basis of these differences may lead to specific or common treatments of different crops in developing sustainable crop management systems. Our results demonstrated that rice and soybean clearly select distinct rhizosphere bacterial and fungal taxa (Fig. 1), which validates the previous studies. For instance, Bulgarelli et al. (2012) and Ofek et al. (2014) also reported that different crops harbor distinct rhizobacterial communities. Interestingly, the same trend was observed in rice and soybean regarding the responses of their bacterial and fungal communities to crop cultivar (Fig. 3), although the direction of the response was not similar (Figs. 1, 2).
Wild plant species support certain beneficial microbes to larger extent than their cultivated relatives. This finding is in agreement with results from the other studies, suggesting that domestication of plant species may have affected the ability of modern crop cultivars to establish the beneficial associations with rhizosphere microbiomes (Perez-Jaramillo et al. 2016). Likewise, our results also validate the previous work, indicating that crop domestication may have stimulated the prevalence of pathogens (Keesing et al. 2010). In contrast, however, Leff et al. (2017) demonstrated that the domestication of sunflowers increased the prevalence of symbionts associated with cultivated plants and decreased the prevalence of pathogens. Our results should be interpreted with great care, and should be considered as a preliminary indication, since it is rather speculative to deduce functionality from taxonomic data, except perhaps in the case of the symbiotic Glomeromycota.
Cultivated crops are phenotypically different compared to their ancestral relatives (Fig. S1). It is highly un-certain, however, if these phenotypic differences had any effect on the rhizosphere microbial community structure of wild versus cultivated relatives. Leff et al. (2017) did not detect any relationship between the structure of the microbial community in the rhizosphere and phenotypic traits such as height, number of nodes, number of branches, most recent fully expanded leaf length, most recent fully expanded leaf width, and stem diameter. Thus, we hypothesize that the patterns in rhizosphere community structure and diversity observed in the present study were probably driven by other, below-ground plant traits, such as root exudate production and root physiology. Cultivated crops have been mainly selected for faster growth and higher yield, which could have resulted in the exudation of different quantities and types of organic compounds from their roots, and, thus, in different below-ground microbial community structure (Perez-Jaramillo et al. 2016). Differences in organic compound production by domestication processes may be the result of trade-offs between growth rates and defence against biotic and abiotic stressors (Mayrose et al. 2011). Some of these interactions are mediated by key secondary metabolites or defence compounds, such as sesquiterpene lactones (Prasifka et al. 2015), which could stimulate or hinder the growth of different fungal taxa.
Overall, our study indicated that wild crops harbor different rhizomicrobiomes in comparison with their cultivated relatives. Although large differences in the structure of the rhizosphere communities of rice and soybean were detected, there was clear evidence that fungal communities were much more affected by crop domestication than bacterial communities. In this study, we also speculated on the functional consequences of these community changes. Once more, we realize that conclusions on the impact of crop domestication on the rhizomicrobiome based on the results of this study must be drawn with great caution due to the limited number of each crop species used, carrying out in very small pots and the focus on taxonomic rather than on functional analyses. Future studies, therefore, including a diverse panel of varieties are crucial to analyse the effect of domestication on rhizospheric microbiomes. In addition, we will focus on elucidating the basic processes that result in the differences in the rhizomicrobiome between a variety of presently used crops and their wild relatives. The functional consequences of these differences will also be explored, with a particular emphasis on the resistance of cultivated crops and their ancestral progenitors to biotic (such as pathogens) and abiotic stresses (such as fungicide). Furthermore, in addition to descriptive analyses of the rhizospheric microbiome, there is a strong need to elucidate the mechanisms underlying the selection of specific populations of microorganisms among the soil-borne communities. We consider the learning from nature as “going back to the roots” of non-cultivated plant species, for which rhizosphere processes and microbial interactions might be more evolved than for most agricultural crops, which are under strong anthropogenic control.
References
Barbosa Lima A, Cannavan FS, Navarrete AA, Teixeira WG, Kuramae EE, Tsai SM (2015) Amazonian dark earth and plant species from the Amazon region contribute to shape rhizosphere bacterial communities. Microb Ecol 69:855–866. https://doi.org/10.1007/s00248-014-0472-8
Berg G, Smalla K (2009) Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol 68:1–13. https://doi.org/10.1111/j.1574-6941.2009.00654.x
Bokulich NA, Thorngate JH, Richardson PM, Mills DA (2014) Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc Natl Acad Sci 111:E139–E148. https://doi.org/10.1073/pnas.1317377110
Bouffaud ML, Poirier MA, Muller D, Moenne-Loccoz Y (2014) Root microbiome relates to plant host evolution in maize and other Poaceae. Environ Microbiol 16:2804–2814. https://doi.org/10.1111/1462-2920.12442/full. http://onlinelibrary.wiley.com/
Bressan M et al (2009) Exogenous glucosinolate produced by Arabidopsis thaliana has an impact on microbes in the rhizosphere and plant roots. ISME J 3:1243. https://doi.org/10.1038/ismej.2009.68
Bulgarelli D et al (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95. https://doi.org/10.1038/nature11336
Bulgarelli D et al (2015) Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17:392–403. https://doi.org/10.1016/j.chom.2015.01.011
Costa R, Götz M, Mrotzek N, Lottmann J, Berg G, Smalla K (2006) Effects of site and plant species on rhizosphere community structure as revealed by molecular analysis of microbial guilds. FEMS Microbiol Ecol 56:236–249. https://doi.org/10.1111/j.1574-6941.2005.00026.x
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. https://doi.org/10.1038/nmeth.2604
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. https://doi.org/10.1093/bioinformatics/btr381
Edwards J et al (2015) Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci 112:E911–E920. https://doi.org/10.1073/pnas.1414592112
Halpin AL et al (2016) Intestinal microbiome disruption in patients in a long-term acute care hospital: a case for development of microbiome disruption indices to improve infection prevention. Am J Infect Control 44:830–836. https://doi.org/10.1016/j.ajic.2016.01.003
Hartmann M, Frey B, Mayer J, Mäder P, Widmer F (2014) Distinct soil microbial diversity under long-term organic and conventional farming. ISME J 9:1177. https://doi.org/10.1038/ismej.2014.210
Hua Z-S et al (2014) Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J 9:1280. https://doi.org/10.1038/ismej.2014.212
İnceoğlu Ö, Falcão Salles J, van Elsas JD (2012) Soil and cultivar type shape the bacterial community in the potato rhizosphere. Microb Ecol 63:460–470. https://doi.org/10.1007/s00248-011-9930-8
Jari Oksanen FGB, Friendly M, Kindt R, Legendre P, Pierre Legendre DM, Minchin PR, O’Hara RB, Simpson GL, Henry MHH, Szoecs E, Wagner H (2018) vegan: Community Ecology Package. R package version 2.5-2. 2018
Keesing F et al (2010) Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature 468:647–652. https://doi.org/10.1038/nature09575
Kõljalg U et al (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277. https://doi.org/10.1111/mec.12481
Ladygina N, Hedlund K (2010) Plant species influence microbial diversity and carbon allocation in the rhizosphere. Soil Biol Biochem 42:162–168. https://doi.org/10.1016/j.soilbio.2009.10.009
Leff JW, Lynch RC, Kane NC, Fierer N (2017) Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus. New Phytol 214:412–423. https://doi.org/10.1111/nph.14323
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. https://doi.org/10.1093/bioinformatics/btr507
Marques JM et al (2015) Bacterial endophytes of sweet potato tuberous roots affected by the plant genotype and growth stage. Appl Soil Ecol 96:273–281. https://doi.org/10.1016/j.apsoil.2015.08.020
Mayrose M, Kane NC, Mayrose I, Dlugosch KM, Rieseberg LH (2011) Increased growth in sunflower correlates with reduced defences and altered gene expression in response to biotic and abiotic stress. Mol Ecol 20:4683–4694. https://doi.org/10.1111/j.1365-294X.2011.05301.x
Mendes R, Raaijmakers JM (2015) Cross-kingdom similarities in microbiome functions. ISME J 9:1905. https://doi.org/10.1038/ismej.2015.7
Mendes R et al (2011) Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332:1097–1100. https://doi.org/10.1126/science.1203980
Mendes LW, Raaijmakers JM, de Hollander M, Mendes R, Tsai SM (2018) Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J 12:212–224. https://doi.org/10.1038/ismej.2017.158
Nallanchakravarthula S, Mahmood S, Alström S, Finlay RD (2014) Influence of soil type, cultivar and verticillium dahliae on the structure of the root and rhizosphere soil fungal microbiome of strawberry. Plos One 9:e111455. https://doi.org/10.1371/journal.pone.0111455
Nguyen NH et al (2016) FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol 20:241–248. https://doi.org/10.1016/j.funeco.2015.06.006
Ofek M, Voronov-Goldman M, Hadar Y, Minz D (2014) Host signature effect on plant root-associated microbiomes revealed through analyses of resident vs. active communities. Environ Microbiol 16:2157–2167. https://doi.org/10.1111/1462-2920.12228
Parks DH, Tyson GW, Hugenholtz P, Beiko RG (2014) STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30:3123–3124. https://doi.org/10.1093/bioinformatics/btu494
Perez-Jaramillo JE, Mendes R, Raaijmakers JM (2016) Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol Biol 90:635–644. https://doi.org/10.1007/s11103-015-0337-7
Pérez-Jaramillo JE et al (2017) Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J 11:2244–2257. https://doi.org/10.1038/ismej.2017.85
Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH (2013) Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol 11:789. https://doi.org/10.1038/nrmicro3109
Placido DF et al (2013) Introgression of novel traits from a wild wheat relative Improves drought adaptation in wheat. Plant Physiol 161:1806–1819. https://doi.org/10.1104/pp.113.214262
Prasifka JR, Spring O, Conrad J, Cook LW, Palmquist DE, Foley ME (2015) Sesquiterpene lactone composition of wild and cultivated sunflowers and biological activity against an insect pest. J Agric Food Chem 63:4042–4049. https://doi.org/10.1021/acs.jafc.5b00362
Pruesse E et al (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. https://doi.org/10.1093/nar/gkm864
Rodríguez-Blanco A, Sicardi M, Frioni L (2015) Plant genotype and nitrogen fertilization effects on abundance and diversity of diazotrophic bacteria associated with maize (Zea mays L.). Biol Fert Soils 51:391–402. https://doi.org/10.1007/s00374-014-0986-8
Santos-González JC, Nallanchakravarthula S, Alström S, Finlay RD (2011) Soil, but not cultivar, shapes the structure of arbuscular mycorrhizal fungal assemblages associated with strawberry. Microb Ecol 62:25–35. https://doi.org/10.1007/s00248-011-9834-7
Schlaeppi K, Dombrowski N, Oter RG, Ver Loren van Themaat E, Schulze-Lefert P (2014) Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc Natl Acad Sci 111:585–592. https://doi.org/10.1073/pnas.1321597111
Schlemper TR, van Veen JA, Kuramae EE (2018) Co-variation of bacterial and fungal communities in different sorghum cultivars and growth stages is soil dependent. Microb Ecol 76:205–214. https://doi.org/10.1007/s00248-017-1108-6
Schloss PD et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75:7537–7541. https://doi.org/10.1128/aem.01541-09
Schmalenbach I, Körber N, Pillen K (2008) Selecting a set of wild barley introgression lines and verification of QTL effects for resistance to powdery mildew and leaf rust. Theor Appl Genet 117:1093–1106. https://doi.org/10.1007/s00122-008-0847-7
Schreiter S et al (2014) Effect of the soil type on the microbiome in the rhizosphere of field-grown lettuce. Front Microbiol. https://doi.org/10.3389/fmicb.2014.00144
Shenton M, Iwamoto C, Kurata N, Ikeo K (2016) Effect of wild and cultivated rice genotypes on rhizosphere bacterial community composition. Rice 9:42–53. https://doi.org/10.1186/s12284-016-0111-8
Shi S et al (2018a) Impact of domestication on the evolution of rhizomicrobiome of rice in response to the presence of Magnaporthe oryzae. Plant Physiol Biochem 132:156–165. https://doi.org/10.1016/j.plaphy.2018.08.023
Shi S et al (2018b) The rhizomicrobiomes of wild and cultivated crops react differently to fungicides. Arch Microbiol. https://doi.org/10.1007/s00203-018-1586-z
Simko I et al (2013) Identification of QTLs conferring resistance to downy mildew in legacy cultivars of lettuce. Sci Rep 3:2875. https://doi.org/10.1038/srep02875
Szoboszlay M, Lambers J, Chappell J, Kupper JV, Moe LA, McNear DH (2015) Comparison of root system architecture and rhizosphere microbial communities of Balsas teosinte and domesticated corn cultivars. Soil Biol Biochem 80:34–44. https://doi.org/10.1016/j.soilbio.2014.09.001
Thijs S et al (2017) Comparative evaluation of four bacteria-specific primer pairs for 16S rRNA gene surveys. Front Microbiol. https://doi.org/10.3389/fmicb.2017.00494
Tian L, Zhou X, Ma L, Xu S, Nasir F, Tian C (2017) Root-associated bacterial diversities of Oryza rufipogon and Oryza sativa and their influencing environmental factors. Arch Microbiol 199:563–571. https://doi.org/10.1007/s00203-016-1325-2
Tian L et al (2018) Comparative analysis of the root transcriptomes of cultivated and wild rice varieties in response to Magnaporthe oryzae infection revealed both common and species-specific pathogen responses. Rice 11:26. https://doi.org/10.1186/s12284-018-0211-8
Turcotte MM, Lochab AK, Turley NE, Johnson MT (2015) Plant domestication slows pest evolution. Ecol Lett 18:907–915. https://doi.org/10.1111/ele.12467
Wagner MR, Lundberg DS, del Rio TG, Tringe SG, Dangl JL, Mitchell-Olds T (2016) Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun 7:12151. https://doi.org/10.1038/ncomms12151
Yan Y, Kuramae EE, Klinkhamer PGL, van Veen JA (2015) Revisiting the dilution procedure used to manipulate microbial biodiversity in terrestrial systems. Appl Environ Microb 81:4246–4252. https://doi.org/10.1128/aem.00958-15
Zachow C, Müller H, Tilcher R, Berg G (2014) Differences between the rhizosphere microbiome of Beta vulgaris ssp. maritima—ancestor of all beet crops—and modern sugar beets. Front Microbiol 5:41501–41513. https://doi.org/10.3389/fmicb.2014.00415
Zhang J-w, Long Y, Xue M-d, Xiao X-g, Pei X-w (2017) Identification of microRNAs in response to drought in common wild rice (Oryza rufipogon Griff.) shoots and roots. PLOS One 12:e0170330. https://doi.org/10.1371/journal.pone.0170330
Zhou X, Tian L, Zhang J, Ma L, Li X, Tian C (2017) Rhizospheric fungi and their link with the nitrogen-fixing Frankia harbored in host plant Hippophae rhamnoides L. J Basic Microb 57:1055–1064. https://doi.org/10.1002/jobm.201700312
Acknowledgements
We are very grateful to Dr. Zhiping Song for the supply of the seeds of wild rice and Dr. Xinhou Zhang for the supply of the seeds of wild soybean. In addition, we also thank Eiko E Kuramae and Johannes A. Van Veen for the revision of this manuscript. This work was financially supported by the Science Foundation of Chinese Academy of Sciences (XDB15030103), the National Key Research and Development Program of China (2016YFC0501202), the Key Deployment Project of the Chinese Academy of Sciences (KFZD-SW-112), the National Natural Science Foundation of China (41571255), and Key Laboratory Foundation of Mollisols Agroecology (2016ZKHT-05), the Key Technology Research and Development Program of CAS during the “13th Five-Year Plan” (Y6H2043001); The Excellent Researcher Award Program from Jilin province of China (20180520052JH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare no conflict of interest.
Additional information
Communicated by Erko Stackebrandt.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shi, S., Chang, J., Tian, L. et al. Comparative analysis of the rhizomicrobiome of the wild versus cultivated crop: insights from rice and soybean. Arch Microbiol 201, 879–888 (2019). https://doi.org/10.1007/s00203-019-01638-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-019-01638-8