
Abstract
Bacteria in Lactobacillus casei group, including Lactobacillus casei (L. casei), Lactobacillus paracasei (L. paracasei), and Lactobacillus rhamnosus (L. rhamnosus) are important lactic acid bacteria in the production of fermented dairy products and are faced with the controversial nomenclatural status due to their close phylogenetic similarity. To probe the evolution and phylogeny of L. casei group, 100 isolates of lactic acid bacteria originated from naturally fermented dairy products in Tibet of China were subjected to multilocus sequence typing (MLST). The MLST scheme, based on analysis of the housekeeping genes fusA, ileS, lepA, leuS, pyrG, recA and recG, revealed that all the isolates belonged to a group containing the L. paracasei reference strains and were clearly different from the strains of L. casei and L. rhamnosus. Although nucleotide diversity (π) was low for the seven genes (ranging from 0.00341 for fusA to 0.01307 for recG), high genetic diversity represented by 83 sequence types (STs) with a discriminatory index of 0.98 was detected. A network-like structure based on split decomposition analysis, and the high values of the relative effect of recombination and mutation in the diversification of the lineages (r/m = 4.76) and the relative frequency of occurrence of recombination and mutation (ρ/θ = 2.62) indicated that intra-species recombination occurred frequently and homologous recombination played a key role in generating genotypic diversity amongst L. paracasei strains in Tibet. The discovery of 51 new STs and the results of STRUCTURE analysis suggested that the L. casei group in Tibet had an individual and particular population structure in comparison to European isolates. Overall, this research might be the first report about genetic diversity and population structure of Lactobacillus populations isolated from naturally fermented dairy products in Tibet based on MLST scheme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lactobacillus species are facultative heterofermentative bacteria naturally found in a variety of environmental habitats (Hammes and Vogel 1995), and are widely used for the production of fermented dairy food (Corsetti et al. 2008; Steele et al. 2007; Tan et al. 2012). As probiotics, some Lactobacillus casei strains have been implemented in alleviation of diarrhea (Nagata et al. 2011; Wong et al. 2014), modulation of immune response (Lim et al. 2009; Aragón et al. 2014) and reduction of infection risk (Guillemard et al. 2010; Gleeson et al. 2011). Based on the sequence analysis of 16S rRNA gene, L. casei, Lactobacillus paracasei and Lactobacillus rhamnosus are generally regarded as the L. casei group (Salvetti et al. 2012). Although the comparative analysis of 16S rRNA gene sequences is a conventional molecular method for bacterial identification and phylogenetic analysis (Petti et al. 2005), it is not suitable to distinguish between the species of the L. casei group. For example, the strains of BD-II, BL23, LC2W, and Zhang, previously defined as L. casei by 16S rRNA gene sequence analysis, have been reclassified as members of L. paracasei based on phylogeny of concatenated amino acid sequences of different proteins (Toh et al. 2013).
To improve the discrimination of closely related lactobacilli species, many molecular typing methods have been developed, such as pulsed-field gel electrophoresis (PFGE) of plasmid and genomic macrorestriction (Herrerofresno et al. 2012), randomly amplified polymorphic DNA (RAPD) (Capra et al. 2011), amplified fragment length polymorphisms (AFLP) and ribotyping (Coudeyras et al. 2008). However, these methods are difficult to facilitate data-sharing between laboratories and to investigate microbial population biology. To overcome the inherent limitations of the typing methods above, multilocus sequence typing (MLST) based on a set of 6–10 housekeeping genes has been developed to characterize the genetic diversity and phylogenetic structure of lactic acid bacteria (Aanensen and Spratt 2005; Bao et al. 2016; Cai et al. 2007; Diancourt et al. 2007; Ramachandran et al. 2013; Tanigawa and Watanabe 2011; Sun et al. 2015).
High diversity and specificity have been discovered in different niches in L. casei group. Previously, 36 sequence types (STs) and frequent intra-species recombination were found based on MLST method among 40 L. casei strains isolated from cheese, human GI tract and fermented plant products in different geographical origins (Cai et al. 2007). Similarly, 31 STs were detected among 52 strains of L. casei group from different sources (such as human, dairy food and fermented plant products), which indicated homologous recombination was not frequent enough to break the population structure of L. casei (Diancourt et al. 2007). Recently, 224 L. casei isolated from naturally fermented foods from different regions in China and Mongolia were divided into 171 STs. The results confirmed that recombination occurred more frequently than mutation during the evolution of L. casei, and clearly showed most of the strains isolated from acidic gruel, pickles, and traditional fermented dairy products formed different subgroups, respectively (Bao et al. 2016). These previous studies generated controversial conclusions about the frequency of homologous recombination and indicated that the L. casei population was specific to different ecological niches (Kimura 1979).
The Tibet region of China lies in the southwest of Qinghai-Tibet plateau, where the traditional fermented dairy products from yaks and goats are popular local food (Wu 2001). Due to the peculiar and individual state of climate (temperature, moisture, pressure, oxygen concentration, etc.) and geographic conditions, these dairy products exhibit unique flavour, probiotic properties and diverse microbial resource (Luo et al. 2011). However, no study on the population structure and diversity of single Lactobacillus group has been reported in this particular region. In this study, 100 lactic acid bacteria were isolated from traditional fermented dairy products in Tibet and the MLST scheme was used to understand the genetic characteristics of them.
Materials and methods
Sampling of fermented dairy products and bacterial isolation
For isolation of bacteria, traditional fermented dairy products, including cow yogurt, cow milk residue, dairy foods, ghee, goat yogurt, goat-yak yogurt, kurut, starter, urum, and yak milk residue were sampled from rural Tibetan households, at 13 towns and cities of Tibet in August of 2014 (Supplementary Table S1). The solid samples were sealed in sterilized plastic bags and liquid samples were introduced into sterilized test tubes with crew cap, unless the products have been packed by the producer. All the samples stored on ice and transported to our laboratory by plane. In the laboratory, those were stored at 4 °C until isolation of the bacterial strains.
Each of the samples was used to prepare a decimal dilution up to 10−6 and 0.1 mL of the last three dilutions were individually spread on MRS agar (Difco Laboratories, Detroit, MI, USA) plate and incubated at 28 °C for 48 h under an anaerobic environment created using Anaeropack system. After incubation L. casei colonies characterized with round, smooth and creamy surfaces (Vinderola and Reinheimer 2000) were picked up and purified by streaking repeatedly on the same medium. All the pure isolates were initially confirmed as members of the L. casei group using carbohydrate fermentation tests (API 50CHL), using as inoculants of cultures grown overnight at 37 °C in MRS Broth (Difco Laboratories, Detroit, Mich).
DNA extraction
A total of 100 isolates belonging to L. casei group were obtained from traditional fermented dairy products in Tibet (Supplementary Table S1). To further identify the isolates by phylogenetic analyses, the genomic DNA was extracted from each isolate using a modified Chelex method (Phillips et al. 2012). Briefly, the bacteria collected from 5 mL of overnight culture at 37 °C in MRS Broth by centrifugation were resuspended in 200 μL of digestion buffer (50 mM Tris–HCl, 1 mM EDTA, 0.5% Nonidet P-40) containing 25% w/v Chelex®100 and incubated in a water bath at 56 °C for 30 min followed by boiling for 10 min. The lysates were stored at − 20 °C as DNA extracts until further analysis.
Amplification and phylogeny of 16S rRNA genes
The 16S rRNA gene fragments of all strains were amplified with primers 27F and 1492R (Kim et al. 2007) using the DNA extracts mentioned above as templates. The obtained PCR products were analyzed by electrophoresis in 1% (w/v) agarose gels (Schneider et al. 1993). Both DNA strands of the extracted gene fragments were directly sequenced in Genewiz (Beijing, China) by following the procedures as described by Cui et al. (2014). The acquired sequences were aligned using ClustalW (Chenna et al. 2003) with 16S rRNA gene sequences of the type or reference strains in L. casei group obtained from the GenBank database and Danone Research, Centre de Recherche Daniel Carasso (Diancourt et al. 2007). The phylogenetic analysis of these sequences was processed by MEGA 6.0 software (Tamura et al. 2013) to construct phylogenetic trees with the neighbor-joining method and the Kimura 2-parameter model (Kimura 1979) with 1000 bootstrap replications.
Multilocus sequence typing (MLST)
The MLST scheme proposed by Diancourt et al. was used to characterize the L. casei strains. The following seven housekeeping genes were: fusA (elongation factor EF-2), ileS (isoleucyl-tRNA synthetase), lepA (GTP-binding protein LepA), leuS (leucyl-tRNA synthetase), pyrG (CTP synthetase), recA (recombinase A) and recG (ATP-dependent DNA helicase). The PCR conditions used for the amplification reactions were as follows: 94 °C for 5 min, followed by 30 cycles at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 5 min. The PCR products were examined by agarose gel electrophoresis and purified with the QIAquick PCR purification kit (Qiagen, Hilden, Germany). Both strands of the purified PCR products were sequenced by Genewiz (Beijing, China). Similar to the 16S rRNA gene analysis, sequences of the seven genes obtained in this study, together with those extracted from database (Diancourt et al. 2007), were processed by MEGA 6.0 software (Tamura et al. 2013) to construct phylogenetic trees with the neighbor-joining method and the Kimura 2-parameter model (Kimura 1979) with 1000 bootstrap replications for all the single gene sequences, as well as for the combined sequences.
Data analysis
For MLST analysis, novel allelic sequences were assigned consecutive numbers above those in the existing allele sequences. On the basis of their allelic profile, isolates were assigned to an ST that had already been described or to a new ST if the allelic profile was novel. Clonal complexes (CCs) were defined as groups of related STs differing by no more than one of the seven loci, which were considered as descendants from a common ancestor (Feil et al. 2004). The Simpson’s index of diversity was calculated to evaluate the discriminatory power of the MLST scheme (Hunter 1990). The equation is D = 1 − 1/[N(N − 1)]\(\sum\nolimits_{{j=1}}^{S} {n_j(n_j - 1)}\), where N is the number of bacterial strains, S is the number of sequence types, and nj is the number of strains belonging to the type j, j = 1 to S. Nucleotide diversity (π) and nonsynonymous substitutions to synonymous substitutions (dN/dS) were calculated using DnaSP 5.0 with Jukes–Cantor correction (Rozas et al. 2003). The Tajima’s D value and GC value were estimated with DnaSP 5.0 (Jukes and Cantor 1969; Rozas et al. 2003). To explore the relationship among the detected STs on the basis of allelic profiles, clonal complexes were analysed using the eBURST v3.0 program (http://eburst.mlst.net) (Feil et al. 2004). Neighbor-joining tree analysis was constructed by MEGA 6.0 with 1000 bootstrap replications (Tamura et al. 2013). Split decompositions and phi test for recombination were performed with SplitsTree4 (Huson and Bryant 2006). The r/m and ρ/θ were accounted by ClonalFrame with three independent runs of 100,000 Markov Chain Monte Carlo (MCMC) iterations plus 10,000 burn-in iterations (Didelot and Falush 2006). Structure v2.3 with linkage model (Falush et al. 2007) was performed to identify the number of ancestral subpopulations (K) with three independent runs consisted of 100,000 (MCMC) iterations.
Nucleotide sequence accession numbers
The GenBank accession numbers for the 16S rRNA, fusA, ileS, lepA, leuS, pyrG, recA and recG gene sequences determined in this study are KU955744–KU955843, KU511387–KU511486, KU511387–KU511486, KU511487–KU511586, KU511587–KU511686, KU511687–KU511786, KU511787–KU511886, and KU511887–KU511986, respectively.
Results
16S rRNA analysis of the isolates
The 16S rRNA sequence similarities varied from 98.7 to 99.9% among the isolates. In the 16S rRNA phylogenetic tree (Supplementary Fig. S2), all the isolates formed a cluster together with reference of L. rhamnosus JCM 1136T, L. zeae ATCC 15820T, L. casei ATCC 393T, L. paracasei JCM 8130T, that were distinct from L. sakei DSM 20017T and L. plantarum JCM 1149T. These results confirmed that all the isolates were members of the L. casei group.
MLST loci and allelic diversity of the isolates and reference strains
The seven genes were successfully amplified for all 100 isolated strains. Supplementary Fig. S1 presented polymorphic nucleotide sites for each locus including the original and the newly discovered, and only the variable sites were shown. A total of 132 polymorphisms sites in the 132 strains were detected based upon the 7 sequenced gene fragments. Across the seven genes, the frequency of polymorphic sites ranged from 4.51% (pyrG) to 21.8% (lepA) and the number of alleles varied from 7 (pyrG) to 24 (lepA). The length of the sequences for the MLST scheme ranged from 315 bp for recA to 663 bp for fusA (Table 1). The average nucleotide diversity (π) was 0.00786, ranging from 0.00341 (fusA) to 0.01307 (recG). The ratio of nonsynonymous to synonymous substitutions (dN/dS) was operated to estimate the selection pressure on each locus. The results remained less than 1 for each locus and ranged between 0.073 (lepA) and 0.426 (recA). Accordingly, the values from Tajima’s D test were between − 2.0075 (recA) to − 1.4005(leuS). The GC value of all alleles ranged from 46.8% (pyrG) to 50.4% (lepA).
Allelic profile of the isolates and reference strains
In this study, a total of 54 STs were assigned, based on the allele combination for the seven MLST gene fragments, in the 100 isolates from naturally fermented dairy products in Tibet (Supplementary Table S1). Four strains belonging to three STs (ST9, ST16 and ST19) found earlier in the findings of Diancourt et al. (2007), and 51 STs (ST33–83) were not previously identified. ST33, ST36, ST39, ST40, ST65 and ST71 represented new combinations of existing alleles, while ST34, ST35, ST37, ST38, ST41–64, ST66–69, ST70 and ST72–83 contained a variable number of new alleles not previously documented (Supplementary Table S1).
The Simpson’s index of diversity with 98% confidence intervals manifested that the L. casei group could be typed by the MLST scheme. As shown in Fig. 1, the resolution power and the number of STs also decreased with the number reduction of locus. When one locus was removed in the MLST scheme, the discriminatory power was although not less than the 0.95 threshold (Belkum et al. 2007), the number of STs was decreased based on concatenated sequence of remaining six loci (Fig. 1). Among 35 traditional fermented milk samples studied, each included more than one STs. Approximately 86% STs of isolates comprised only one isolate, 6% STs were represented by at least three sample sources. ST33 was remarkably prevalent type in this study and contained 13 strains, followed by ST69 (8 strains), and ST45 (7 strains).

Discriminatory power calculated with Simpson’s index of diversity, and the number of sequence types (STs) based on the various combinations of housekeeping genes of MLST scheme. When the seven MLST genes were concatenated, the Simpson’s index of diversity achieved to the highest confidence intervals
An eBURST population snapshot was performed to investigate the clonal relatedness of 132 strains on the basis of allelic profiles (Fig. 2). The sequences of 83 STs were divided into 16 CCs and 23 singletons, the latter were all from fermented dairy products in Tibet, the only two exceptions correspond to the strains D699 (ST30) and SB3888 (ST31) isolated from traditional food and human in Europe, respectively. The 16 CCs comprised 72% STs, which were differed by at least four out of the seven loci from all other profiles. Eight CCs (CC4, CC6 and CC11–CC16) were all from Tibet, and stains of five CCs (CC3 and CC7–CC10) were all from Europe. The predominant clonal complex of L. casei group was CC1, consisting of 13 (16%) STs with 40 (30%) strains, 95% of which were isolate strains in addition to the two strains D661 (ST16) and D645 (ST17) from European dairy products. ST45 was assigned as the common ancestry of CC1 on the basis of the highest number of single-locus variants (SLVs), which comprised six SLVs and two double-locus variants (DLVs). Moreover, ST33 was identified as predicted subgroup founder associated with five SLVs and three DLVs. The CC2 was the second largest and comprised seven STs representing 8 L. paracasei strains, half of which were isolated from Tibet. Moreover, L. paracasei ATCC 334 (ST32) was assigned to CC5 which were all isolated from dairy products with three isolates in Tibet and one in Europe.

Population structure of 132 L. casei–paracasei strains based on allelic profiles of seven housekeeping genes by eBURST analysis. Each circle corresponds to a sequence type (ST), and the sizes of the circle are associated with the number of isolates. Blue circles are indicated of group founder, and yellow circles represent subgroup founder of the clone complex. (Color figure online)
Phylogenetic and population structure analysis of strains in L. casei group
The individual genes fusA, ileS, lepA, leuS, pyrG, recA, and recG were successfully amplified for the isolates and strains of L. casei group, but they were not amplified in species L. fermentum and L. sakei. The phylogenetic trees were constructed from seven housekeeping genes using the neighbor-joining method, respectively. In all these seven phylogenetic trees, the 132 studied strains were clearly clustered together with L. paracasei strains ATCC 334 and JCM 8130T with 99 or 100% similarity. This group was obviously distinct from L. rhamnosus JSM 1136T and L. casei ATCC 393T (Supplementary Fig. S3).
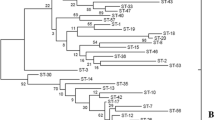
In the neighbor-joining tree constructed based on the concatenated sequences of the seven protein-coding genes (Fig. 3a), strains representing the 83 STs were divided into four clusters, which were consistent with four major ancestral populations revealed by the STRUCTURE. Cluster 1 (blue) contained three CCs (CC2, CC5 and CC6) and six singletons, 78.5% of these isolates were from Tibet and included reference strain L. paracasei ATCC 334. Cluster 2 (yellow) contained two subclusters. Cluster 2a contained 2CCs (CC3 and CC8) and three singletons, and all of these isolates were derived from Europe. Cluster 2b consisted of 4CCs (CC1, CC12, CC14 and CC16) and five singletons were all isolated from dairy products derived from Tibet except ST16 and ST17. Cluster 3 (green) was composed of 5CCs (CC7, CC9, CC10, CC15 and CC16) and eight singletons. Cluster 3a and Cluster 3c contained seven STs with isolates from Tibet except D699 (ST30). Cluster 3b included twelve STs, all of which were from Europe except TD059 (ST57). Cluster 4 (orange) contained 3CCs (CC4, CC11 and CC13) and one singleton, all of which were from traditional fermented dairy products in Tibet.

NJ tree and Ancestry of 83 STs of Lactobacillus casei group. a Neighbor-joining tree based on the concatenated partial sequences of 83 STs. The bootstrap values are shown for all branches. The bootstrap values are shown through the numbers at nodes based on analysis of 1000 replicates. ST are colored in terms of their affiliation to one of the four ancestral lineages. b The sources of ancestry of each unique ST from five ancestral populations by the linkage model of STRUCTURE. Each ST is represented by a single line with the ST designation at the top consisting of colour stacked bars that indicate the proportion of ancestry from each of four populations (blue, yellow, green, and orange). STs in the red box represented admixture of ancestral sources. (Color figure online)
In the analysis of average proportion of genetic material of each ST according to STRUCTURE with the linkage model, a maximum posterior probability of K = 4 was found within the genetic population of 83 STs (132 strains), revealing that the primary source of Cluster 1 to Cluster 4 were blue, yellow, green and orange colours ancestral population, respectively (Fig. 3b). The percentage of assignment to a cluster lower than 85% was considered as threshold for admixture. On the whole, 22.9% of all STs (Marked by red box in Fig. 3b) contained a mixture of ancestral sources, representing a high degree of heterogeneity. Little admixture of ancestral sources was observed among the 100 isolates from Tibet, which suggested that high degree of genetic homogeneity of these STs in each lineage.
Gene recombination in the tested populations
Split-decomposition analysis was used to examine the influence of recombination on the evolution of each locus. As shown in the Supplementary Fig. S4, the split graphs for all seven loci showed diverse parallelogram structures, except for the gene of pyrG and recA, indicating that intragenic recombination occurred during the evolution of the other five loci, especially lepA loci. In addition, the combined split graph of alleles for all seven MLST loci displayed a network-like structure (Fig. 4). On the whole, the phylogeny obtained using the neighbor-joining method was very similar to that of split network.

Split-decomposition analysis of 83 STs for the concatenated sequences of seven loci of L. casei–paracasei strains. Parallelogram structures indicate the recombination events
Furthermore, the P value for the concatenated sequences of strains was considered to be significant (P = 1.812E−7) by the phi test. In addition, the relative contribution of recombination and mutation for population structure were assessed by ClonalFrame. The value of ρ/θ was 2.62 and the r/m rate for nucleotides was 4.76, indicating that nucleotides are approximately fourfold more likely to change by recombination than by mutation during the evolution.
Discussion
As previously reported (Salvetti et al. 2012), the three species in L. casei group, L. casei, L. paracasei, and L. rhamnosus, as well as the reference species L. zeae, were undistinguishable by 16S rRNA gene phylogeny in the present study. In contrast, MLST was recognized as the most powerful technique for studying typing and population structure of bacteria. This approach had been widely applied to the studies of diversity and evolution of various lactobacillus, such as Leuconostoc lactis (Tong et al. 2014), Leuconostoc mesenteroides (Zhang et al. 2014), Lactococcus lactis (Xu et al. 2014) and L. plantarum (Xu et al. 2015). Up to now, three different MLST schemes had been employed for studying the evolution of L. casei group (Bao et al. 2016; Cai et al. 2007; Diancourt et al. 2007). To facilitate comparison and show more clearly the distinction of genetic diversity of Tibetan strains, we used the same system of ST designation, and data from the study of Diancourt et al. were also included to provide a broader perspective (Diancourt et al. 2007). In this study, all the isolates were identified as members of the L. casei group (Supplementary Fig. S2). Of which, the strains belonging to L. casei, L. paracasei, and L. rhamnosus were clearly differentiated from each other based on the single gene tree. Furthermore, all the isolates were clustered with L. paracasei reference strains, which illustrated that the isolates were a single group belonging to L. paracasei (Supplementary Fig. S3).
Although only one L. paracasei was detected in this study, the dividing of 100 isolates from fermented dairy products in Tibet into 54 different STs based on 7 genetic loci and 44 STs representing by only a single isolate revealed a high intraspecific diversity. The high genetic diversity also was proved by the four ancestral sources of L. paracasei populations in the analysis of STRUCTURE (Fig. 3b). The similar results of genetic diversity were found in the MLST analysis of L. helveticus (Sun et al. 2015), L. plantarum (Zhang et al. 2015) and L. fermentum (Tong et al. 2015). Significantly, Tibet isolates of cluster 4 only contained one novel orange ancestral population (Fig. 3b), which was distinguished from the other three clusters. It indicated that L. casei group in Tibet had an individual type of population structure in comparison to European isolates. The discovery of many new polymorphic sites and STs also reinforced that point. However, it remained to be determined due to the limitation of the number of isolates.
Combined with the previous studies, our results demonstrated that MLST is a suitable method for studying genetic diversity in populations, with high-resolution power (0.98) displayed by the discriminatory index. The number of STs was decreased when either gene loci removed. This phenomenon indicated that the current MLST scheme was a reliable method for studying genetic diversity and population structure of L. casei group from Tibet in this study. Based on the MLST scheme, the phylogenetic tree contained four major branches which were supported by low bootstrap values (Fig. 3a). According to the ancestral source of genetic diversity, the poor bootstrap values of the topology could be explained by the high degree of admixture. Therefore, the results showed that neighbor-joining tree was not suitable for analyzing the phylogenetic relationship of L. casei group in our study. The conclusion was consistent with previous reports for L. casei (Bao et al. 2016). In other words, this approach was more sensitive to the effects of genetic recombination. Instead, the approach such as eBURST could avoid the problem as much as possible (Diancourt et al. 2007; Bao et al. 2016).
The range of nucleotide diversity (π) from 0.00334 for fusA to 0.01337 for recG estimated for the 100 noncommercialized dairy isolates of L. paracasei (except the reference and type strains) from Tibet revealed relative high nucleotide diversity than those obtained from L. casei in 2007 (Diancourt et al. 2007), but little bit lower than those of L. helveticus (0.00373 for murE to 0.06233 for pepX) (Sun et al. 2015), L. fermentum (0.00393 for murC to 0.01421 for dnaA) (Tong et al. 2015) and L. plantarum (0.00401 for clpX to 0.03220 groEL) (Xu et al. 2015). These differences in distinct studies might be related to the variations in geographic and sample origins of the isolates because our isolates in this study were only isolated from naturally fermented dairy products in Tibet. However, the factors affecting the diversity of bacteria were multiple and complicated, including homologous recombination, gene decay and acquisition, horizontal gene transfers (HGTs) (Cai et al. 2009), and geographic isolation environment (Tong et al. 2014).
The homologous recombination has a significant impact on the accuracy of phylogenetic analysis and evolutionary stability of population structure (Delétoile et al. 2010; Feil and Spratt 2001). However, the rates of homologous recombination varied in different bacterial species (Fraser et al. 2007). In our study, the split graph with parallelograms (Fig. 4) and phi value (P = 2.452E−6) provided the evidence that homologous recombination occurred in the L. paracasei populations. In addition, the parallelogram-shaped structures were found in individual split graphs of fusA, ileS, lepA, leuS and recG (Supplementary Fig. S4). Thus it can be inferred that the homologous recombination was mainly affected by the five loci. The split-decomposition method for detecting recombination events was also implemented to other lactic acid bacteria, such as Bifidobacterium species (Delétoile et al. 2010), L. sakei (Chaillou et al. 2013) and Carnobacterium maltaromaticum (Rahman et al. 2014).
In our study, 22.9% of STs shared a high degree of admixture, which indicated that recombination has contributed to genetic heterogeneity of L. paracasei populations. Furthermore, the high value of r/m and ρ/θ in our study indicated that recombination had a major contribution to shape the population structure of L. paracasei populations than point mutation. Moreover, split-decomposition analysis also provided evidences for intra-species recombination among L. paracasei populations according to the network-like topology (Fig. 4). These results were consistent with the previous reports for L. casei group (Bao et al. 2016; Cai et al. 2007; Diancourt et al. 2007). And, this phenomenon could be interpreted by the research of Broadbent et al. that large shifting of genes was in relation to the evolution of L. casei isolates in nutrient-rich dairy niche (Broadbent et al. 2012). In addition, the incongruence of branching of Cluster3a and Cluster3c between the two topology structures could be attributed to HGT. Makarova and Koonin (2007) confirmed that genome diversity of L. casei group was mainly driven by horizontal gene transfer from other bacterial species particularly lactobacilli. The similar HGT events were also detected in the MLST analysis of Listeria monocytogenes because of the distinct branching between the trees with and without correction for recombination (Ragon et al. 2008). The low dN/dS values in the MLST analysis indicated that the neutral mutations in the selective pressure dominated the large accumulation of nucleotide changes in the evolution of L. paracasei populations. The conclusion was also confirmed by the Tajima’s D values across seven loci which did not present a significant deviation from neutral expectation (Tajima 1989). Besides, the housekeeping genes from L. casei group were driven by neutral selection, which was in accordance with the previous report (Bao et al. 2016; Cai et al. 2007; Diancourt et al. 2007).
The classification and nomenclature of L. casei group have always wavered between L. casei and L. paracasei due to the high genetic similarity of these species (Tindall 2008). The protein-coding genes with a sufficient number of sequence polymorphisms have successfully discriminated the species within L. casei group (Felis et al. 2001; Huang and Lee 2011), which are concordant with the phylogenetic results in our study. Thus, the individual genes of the MLST scheme can be proposed as alternative markers for the identification of species in the L. casei group. However, the sequence analysis of individual housekeeping genes was hardly performed for intraspecies identification or evolutionary biology compared with MLST. As a suitable alternative molecular marker, the recA gene could fully identify the species in L. casei group (Felis et al. 2001).
Conclusions
This research might be the first report about genetic diversity and population structure of Lactobacillus populations isolated from naturally fermented dairy products in Tibet based on MLST scheme. All the 100 isolates were identified as L. paracasei and 54 STs were among the isolates. High degree of homologous recombination was detected within the studied populations. Our results provided a better understanding of the evolution and phylogeny of L. casei group of Tibet and contributed for the meaningful information for future studies. Nevertheless, the isolated strains in our study were the only one from the fermented dairy products. It would be necessary to monitor the phylogenetic relationships of L. casei group from diverse niches by combined use of MLST and whole-genome resequencing techniques.
References
Aanensen DM, Spratt BG (2005) The multilocus sequence typing network: mlst.net. Nucleic Acids Res 33:W728–W733
Aragón F, Carino S, Perdigón G, De Moreno De Leblanc A (2014) The administration of milk fermented by the probiotic Lactobacillus casei CRL 431 exerts an immunomodulatory effect against a breast tumour in a mouse model. Immunobiology 219:457–464
Bao Q, Song Y, Xu H, Yu J, Zhang W, Menghe B, Zhang H, Sun Z (2016) Multilocus sequence typing of Lactobacillus casei isolates from naturally fermented foods in China and Mongolia. J Dairy Sci 99:5202–5213
Belkum AV, Tassios PT, Dijkshoorn L, Haeggman S, Cookson B, Fry NK, Fussing V, Green J, Feil E, Gerner-Smidt P (2007) Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Eur J Clin Microbiol 13(Suppl 3):1–46
Broadbent JR, Neenoeckwall EC, Stahl B, Tandee K, Cai H, Morovic W, Horvath P, Heidenreich J, Perna NT, Barrangou R (2012) Analysis of the Lactobacillus casei supragenome and its influence in species evolution and lifestyle adaptation. BMC Genom 13:533
Cai H, Rodríguez BT, Zhang W, Broadbent JR, Steele JL (2007) Genotypic and phenotypic characterization of Lactobacillus casei strains isolated from different ecological niches suggests frequent recombination and niche specificity. Microbiology 153:2655–2665
Cai H, Thompson R, Budinich MF, Broadbent JR, Steele JL (2009) Genome sequence and comparative genome analysis of Lactobacillus casei: insights into their niche-associated evolution. Genome Biol Evol 1:239–257
Capra ML, Mercanti DJ, Rossetti LC, Reinheimer JA, Quiberoni A (2011) Isolation and phenotypic characterization of Lactobacillus casei and Lactobacillus paracasei bacteriophage-resistant mutants. J Appl Microbiol 111:371–381
Chaillou S, Lucquin I, Najjari A, Zagorec M, Champomiervergès MC (2013) Population genetics of Lactobacillus sakei reveals three lineages with distinct evolutionary histories. PLoS One 8:e73253
Chenna R, Sugawar H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497
Corsetti A, Caldini G, Mastrangelo M, Trotta F, Valmorri S, Cenci G (2008) Raw milk traditional Italian ewe cheeses as a source of Lactobacillus casei strains with acid-bile resistance and antigenotoxic properties. Int J Food Microbiol 125:330–335
Coudeyras S, Marchandin H, Fajon C, Forestier C (2008) Taxonomic and strain-specific identification of the probiotic strain Lactobacillus rhamnosus 35 within the Lactobacillus casei group. Appl Environ Microbiol 74:2679–2689
Cui J, Wei T, Liu LN, Zhang X, Qi X, Zhang ZF, Wang ZQ (2014) Molecular characterization of a Spirometra mansoni antigenic polypeptide gene encoding a 28.7 kDa protein. Parasitol Res 113:3511–3516
Delétoile A, Passet V, Aires J, Chambaud I, Butel MJ, Smokvina T, Brisse S (2010) Species delineation and clonal diversity in four Bifidobacterium species as revealed by multilocus sequencing. Res Microbiol 161:82–90
Diancourt L, Passet V, Chervaux C, Garault P, Smokvina T, Brisse S (2007) Multilocus sequence typing of Lactobacillus casei reveals a clonal population structure with low levels of homologous recombination. Appl Environ Microbiol 73:6601–6611
Didelot X, Falush D (2006) Inference of bacterial microevolution using multilocus sequence data. Genetics 175:1251
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7:574–578
Feil EJ, Spratt BG (2001) Recombination and the population structures of bacterial pathogens. Annu Rev Microbiol 55:561
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG (2004) eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 186:1518
Felis GE, Dellaglio F, Mizzi L, Torriani S (2001) Comparative sequence analysis of a recA gene fragment brings new evidence for a change in the taxonomy of the Lactobacillus casei group. Int J Syst Evol Microbiol 51:2113–2117
Fraser C, Hanage WP, Spratt BG (2007) Recombination and the nature of bacterial speciation. Science 315:476–480
Gleeson M, Bishop NC, Oliveira M, Tauler P (2011) Daily probiotic’s (Lactobacillus casei Shirota) reduction of infection incidence in athletes. Int J Sport Nutr Exerc 21:55–64
Guillemard E, Tondu F, Lacoin F, Schrezenmeir J (2010) Consumption of a fermented dairy product containing the probiotic Lactobacillus casei DN-114001 reduces the duration of respiratory infections in the elderly in a randomised controlled trial. Br J Nutr 103:58–68
Hammes WP, Vogel RF (1995) The genus Lactobacillus. Springer US 2:19–54
Herrerofresno A, Martínez N, Sánchezllana E, Díaz M, Fernández M, Martin MC, Ladero V, Alvarez MA (2012) Lactobacillus casei strains isolated from cheese reduce biogenic amine accumulation in an experimental model. Int J Food Microbiol 157:297–304
Huang CH, Lee FL (2011) The dnaK gene as a molecular marker for the classification and discrimination of the Lactobacillus casei group. Antonie Van Leeuwenhoek 99:319–327
Hunter PR (1990) Reproducibility and indices of discriminatory power of microbial typing methods. J Clin Microbiol 28:1903–1905
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Jukes TH, Cantor CR (1969) Evolution of protein molecules. Mamm Protein Metab 3:21–132
Kim PI, Jung MY, Chang YH, Kim S, Kim SJ, Park YH (2007) Probiotic properties of Lactobacillus and Bifidobacterium strains isolated from porcine gastrointestinal tract. Appl Microbiol Biotechnol 74:1103–1111
Kimura M (1979) The neutral theory of molecular evolution. Sci Am 241:98
Lim LH, Li HY, Huang CH, Lee BW, Lee YK, Chua KY (2009) The effects of heat-killed wild-type Lactobacillus casei Shirota on allergic immune responses in an allergy mouse model. Int Arch Allergy Immunol 148:297–304
Luo F, Feng S, Sun Q, Xiang W, Zhao J, Zhang J, Yang Z (2011) Screening for bacteriocin-producing lactic acid bacteria from kurut, a traditional naturally-fermented yak milk from Qinghai-Tibet plateau. Food Control 22:50–53
Makarova KS, Koonin EV (2007) Evolutionary genomics of lactic acid bacteria. J Bacteriol 189:1199–1208
Nagata S, Asahara T, Ohta T, Yamada T, Kondo S, Bian L, Wang C, Yamashiro Y, Nomoto K (2011) Effect of the continuous intake of probiotic-fermented milk containing Lactobacillus casei strain Shirota on fever in a mass outbreak of norovirus gastroenteritis and the faecal microflora in a health service facility for the aged. Br J Nutr 106:549–556
Petti CA, Polage CR, Schreckenberger P (2005) The role of 16S rRNA gene sequencing in identification of microorganisms misidentified by conventional methods. J Clin Microbiol 43:6123–6125
Phillips K, Mccallum N, Welch L (2012) A comparison of methods for forensic DNA extraction: Chelex-100 and the QIAGEN DNA Investigator Kit (manual and automated). Forensic Sci Int Gen 6:282–285
Ragon M, Wirth T, Hollandt F, Lavenir R, Lecuit M, Monnier AL, Brisse S (2008) A new perspective on Listeria monocytogenes evolution. Plos Pathog 4:e1000146
Rahman A, Cailliezgrimal C, Bontemps C, Payot S, Chaillou S, Revoljunelles AM, Borges F (2014) High genetic diversity among strains of the unindustrialized lactic acid bacterium Carnobacterium maltaromaticum in dairy products as revealed by multilocus sequence typing. Appl Environ Microbiol 80:3920–3929
Ramachandran P, Lacher DW, Pfeiler EA, Elkins CA (2013) Development of a tiered multilocus sequence typing scheme for members of the Lactobacillus acidophilus complex. Appl Environ Microbol 79:7220–7228
Rozas J, Sánchezdelbarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Salvetti E, Torriani S, Felis GE (2012) The genus Lactobacillus : A taxonomic update. Probiotics Antimicrob 4:217–226
Schneider B, Ahrens U, Kirkpatrick BC, Seemüller E (1993) Classification of plant-pathogenic mycoplasma-like organisms using restriction-site analysis of PCR-amplified 16S rDNA. J Gen Appl Microbiol 139:519–527
Steele J, Broadbent J, Shah NP, Ustunol Z (2007) Invited review: advances in starter cultures and cultured foods. J Dairy Sci 90:4005–4012
Sun Z, Liu W, Song Y, Xu Y, Yu J, Bilige M, Zhang H, Chen Y (2015) Population structure of Lactobacillus helveticus isolates from naturally fermented dairy products based on multilocus sequence typing. J Dairy Sci 98:2962–2972
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tan WS, Budinich MF, Ward R, Broadbent JR, Steele JL (2012) Optimal growth of Lactobacillus casei in a Cheddar cheese ripening model system requires exogenous fatty acids. J Dairy Sci 95:1680–1689
Tanigawa K, Watanabe K (2011) Multilocus sequence typing reveals a novel subspeciation of Lactobacillus delbrueckii. Microbiology 157:727–738
Tindall BJ (2008) The type strain of Lactobacillus casei is ATCC 393, ATCC 334 cannot serve as the type because it represents a different taxon, the name Lactobacillus paracasei and its subspecies names are not rejected and the revival of the name ‘Lactobacillus zeae’ contravenes Rules 51b (1) and (2) of the International Code of Nomenclature of Bacteria. Opinion 82. Int J Syst Evol Microbiol 58:1764–1765
Toh H, Oshima K, Nakano A, Takahata M, Murakami M, Takaki T, Nishiyama H, Igimi S, Hattori M, Morita H (2013) Genomic adaptation of the Lactobacillus casei group. PLoS One 8:e75073
Tong D, Liu W, Sun Z, Qiang L, Xu H, Song Y, Zhang H (2014) A novel multi-locus sequence typing (MLST) protocol for Leuconostoc lactis isolates from traditional dairy products in China and Mongolia. BMC Microbiol 14:1–9
Tong D, Liu W, Song Y, Xu H, Menghe B, Zhang H, Song Y (2015) The evolution and population structure of Lactobacillus fermentum from different naturally fermented products as determined by multilocus sequence typing (MLST). BMC Microbiol 15:107
Vinderola CG, Reinheimer JA (2000) Enumeration of Lactobacillus casei in the presence of L. acidophilus, bifidobacteria and lactic starter bacteria in fermented dairy products. Int Dairy J 10:271–275
Wong S, Jamous A, O’Driscoll J, Sekhar R, Weldon M, Yau CY, Hirani SP, Grimble G, Forbes A (2014) A Lactobacillus casei Shirota probiotic drink reduces antibiotic-associated diarrhoea in patients with spinal cord injuries: a randomised controlled trial. Br J Nutr 111:672–678
Wu T (2001) The Qinghai-Tibetan plateau: how high do Tibetans live? High Alt Med Biol 2:489
Xu H, Sun Z, Liu W, Yu J, Song Y, Lv Q, Zhang J, Shao Y (2014) Multilocus sequence typing of Lactococcus lactis from naturally fermented milk foods in ethnic minority areas of China. J Dairy Sci 97:2633–2645
Xu H, Liu W, Zhang W, Yu J, Song Y, Menhe B, Zhang H, Sun Z (2015) Use of multilocus sequence typing to infer genetic diversity and population structure of Lactobacillus plantarum isolates from different sources. BMC Microbiol 15:241
Zhang W, Liu W, Song Y, Xu H, Menghe B, Zhang H, Sun Z (2014) Multilocus sequence typing of a dairy-associated Leuconostoc mesenteroides population reveals clonal structure with intragenic homologous recombination. J Dairy Sci 98:2284–2293
Zhang W, Liu W, Song Y, Xu H, Menghe B, Zhang H, Sun Z (2015) Use of multilocus sequence typing to infer genetic diversity and population structure of Lactobacillus plantarum isolates from different sources. J Dairy Sci 15:241
Acknowledgements
This study was supported by “Academic Backbone” Project of Northeast Agricultural University (15XG26), National Natural Science Foundation of China (No. 31401512), Science Foundation for Distinguished Young Scholars of Heilongjiang Province (JC201415).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. S1
Polymorphic nucleotide sites found among the stains. Only the variable sites are shown. The red font represents the newly discovered locus and the newly identified allele number (DOC 618 KB)
Supplementary Fig. S2
Phylogenetic tree of 16S rRNA sequence of the Lactobacillus casei group (DOC 80 KB)
Supplementary Fig. S3
Phylogenetic trees of individual gene (fusA, ileS, lepA, leuS, pyrG, recA, and recG) sequences of the Lactobacillus casei group. The red font represents the newly identified allele number (DOC 370 KB)
Supplementary Fig. S4
Split decomposition analysis based on individual gene (fusA, ileS, lepA, leuS, pyrG, recA, and recG) for the Lactobacillus casei group (DOC 234 KB)
Supplementary Table S1
Characteristics of strains of Lactobacillus casei group. ST1 to ST32 were reported by Diancourt et al, and ST33 to ST83 were identified in this study (DOC 383 KB)
Rights and permissions
About this article

Cite this article
Feng, J., Jiang, Y., Li, M. et al. Diversity and evolution of Lactobacillus casei group isolated from fermented dairy products in Tibet. Arch Microbiol 200, 1111–1121 (2018). https://doi.org/10.1007/s00203-018-1528-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-018-1528-9