
Abstract
A novel aerobic, non-motile, halotolerant, alkalitolerant, hydrocarbon degrading, and rod shaped bacterium, designated strain R160T, was isolated from soil in South Korea. Cells were Gram-staining-negative, catalase-positive, and oxidase-negative. This strain grew up to 7% of NaCl and in the pH range of 6–11 (optimum 7.0–10.0). The isolate degraded 51.7 ± 1.3% of hydrocarbon components (C-18, C-20, and C-22) and 45.8 ± 1.4% oil components (kerosene, diesel, and gasoline). Phylogenetic analysis based on 16 S rRNA gene sequences revealed that strain R160T formed a lineage within the genus Acinetobacter, and was closely related to ‘Acinetobacter oleivorans’ DR1T (97.47%, sequence similarity). Other closely related members have sequence similarity between 97.47 to 96.52%. The predominant respiratory lipoquinones of strain R160T were ubiquinone 9 (Q-9) and ubiquinone 8 (Q-8). The major polar lipids were phosphatidylethanolamine (PE), diphosphatidylglycerol (DPG), phosphatidylglycerol (PG), and phosphatidylcholine (PC). The major cellular fatty acids were 9-octadecenoic acid (C18:1 ω9c), hexadecanoic acid (C16:0), and summed feature (comprising C16:1 ω7c and/or C16:1 ω6c). The DNA G + C content of strain R160T was 44.9 mol%. On the basis of phenotypic, genotypic, chemotaxonomic, and phylogenetic characteristics, strain R160T represents a novel species of the genus Acinetobacter, for which the name Acinetobacter halotolerans sp. nov. is proposed. The type strain is R160T (= KEMB 9005-333T = KACC 18453T = JCM 31009T).
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The increased consumption of oil and its products cause a serious health and environmental problem. For removal of oil contaminants, several conventional methods, such as excavation, land-filling, recycling, incineration, stabilization, and vitrification, have been used. However, from the eco-friendly perspective, microbial oil pollutant removal method is most convenient, as it is a cost-effective and non-laborious technique (Fatajeva et al. 2014; Chaudhary 2016; Margesin and Schinner 2001). There are several reports on microbial degradation of oil from contaminated soil. Recently, a number of species of the genus Acinetobacter, such as ‘Acinetobacter oleivorans’, Acinetobacter calcoaceticus, Acinetobacter baumannii, and Acinetobacter venetianus, have been studied and characterized by their capacity to degrade and utilize oil and its products (Kang et al. 2011; Mishra et al. 2004; Wayne and Copper 1992; Yamahira et al. 2008; Vaneechoutte et al. 1999).
The genus Acinetobacter is a member of the family Moraxellaceae of the phylum Proteobacteria described in the 1950s (Brisou and Prévot 1954). This genus was first described by Brisou and Prévot in 1954 (Brisou and Prévot 1954). Members of the genus Acinetobacter have been isolated from a wide variety of habitats, including activated sludge, plant, cotton, clinical samples, raw milk, horse dong, zinc ore, sewage, fresh water, sea water, and soil (http://www.bacterio.net/acinetobacter.html). Furthermore, several strains of Acinetobacter can thrive well in extreme environmental condition, such as high altitudes lakes, dry surface, cold, and alkaline environment, high temperature, hyper-saline environment, organic solvent, and oil-contaminated soil (Yamahira et al. 2008; Fatajeva et al. 2014; Jawad et al. 1998; Vaneechoutte et al. 1999; Albarracin et al. 2012).
This study reports the hydrocarbon degradation capacity and growth properties in hyper-saline and high alkaline extreme environmental condition of strain R160T, which was isolated from reclaimed grassland.
Materials and methods
Sampling and cultivation
Bacteria were selected from soil sample obtained from reclaimed grassland, geographically located in Hwaseong, South Korea (37º16′10′′N and 126º45′42′′E), and a bacterial strain, named R160T, was isolated and subjected to polyphasic taxonomic characterization. Isolation, routine culture, and preservation of strain R160T were done as described previously by Dahal and Kim (2016a). ‘Acinetobacter oleivorans’ KCTC 23045T, Acinetobacter puyangensis JCM 18011T, Acinetobacter brisouii KACC 11602T, Acinetobacter soli KACC 14192T, and Acinetobacter baylyi KCTC 12413T were selected for comparative analysis, and were used as reference strains. All reference strains were grown on R2A agar at 28 °C for 3–4 days.
Cell morphology
The morphologies of cells grown on R2A agar for 2 days at 28 °C were visualized by light microscopy (BX50 microscope; Olympus, Japan), and scanning electron microscopy (S-4800 SEM; Hitachi, Japan). Colonial morphology was observed on R2A agar after incubation at 28 °C after 3 days using a Zoom Stereo Microscope (SZ61; Olympus, Japan). Gram-staining was performed according to the procedure described by Doetsch (1981).
Physiological and biochemical tests
Motility was tested in R2A medium containing 0.4% agar according to procedure described by Tittsler and Sandholzer (1936). Catalase activity was determined by production of bubbles after addition of 3% (v/v) hydrogen peroxide (H2O2). Oxidase activity was determined using 1% (w/v) tetramethyl-p-phenylenediamine dihydrochloride. The presence of flexirubin-type pigments was investigated using 20% (w/v) KOH solution (Reichenbach 1992). Growth on various bacteriological media was tested using R2A agar (MB Cell), nutrient agar (NA; Oxoid), tryptone soya agar (TSA; Oxoid), sorbitol MacConkey agar (Oxoid), Luria–Bertani agar (LBA; Oxoid), and marine agar (MA; Difco). Growth at various temperatures, ranging 4–45 °C (4, 10, 15, 20, 25, 28, 30, 32, 35, 37, 40, 41, 42, and 45 °C), was determined on TSA plates for 5 days. The pH range for the growth was determined in R2A broth at 28 °C. The pH of the medium was adjusted prior to sterilization to pH 4–12 (at intervals of 0.5 pH unit) using citrate/NaH2PO4 buffer (pH range, 4.0–5.5), phosphate buffer (pH range, 6.0–7.5), and Tris buffer (pH range, 8.0–10.0) (Breznak and Costilow 2007), and pH 10.5–12.0 was adjusted with 5 M NaOH. Verification of the pH after autoclaving revealed only minor changes. Growth with NaCl was examined in R2A broth containing 0–10% (w/v, at interval of 1%). Tween 80 hydrolysis was carried out according to Smibert and Krieg (1994). Anaerobic growth on TSA agar was assessed at 28 °C for 10 days using BBL (Becton Dickinson) anaerobic jar with GasPak™ EZ Gas Generating Container System (Becton Dickinson). Hydrolysis of starch and casein were determined as described previously (Tindall et al. 2007). Production of hydrogen sulfide was checked using sulfide indole motility (SIM) medium (Oxoid) according to procedure described by Macfaddin (1980). The Methyl Red and Voges-Proskauer (MR-VP) test was done with MR-VP broth (Vaughn et al. 1939). A DNase essay was performed with DNase agar (Oxoid). Haemolysis of sheep blood was determined on TSA containing 5% defibrinated sheep blood for 5 days. Susceptibilities of antibiotics were tested as described by Dahal and Kim (2016b). Endospore formation was investigated by staining with malachite green as described previously (Schaeffer and Fulton 1933). Other physiological and biochemical tests were performed using API 20NE and API ID 32 GN (bioMérieux) to evaluate basic chemical test and carbon source utilization. Enzyme activities were tested using an API ZYM kit (bioMérieux) according to the manufacturer’s instruction.
Phylogentic analysis
Genomic DNA of the strain R160T was extracted using an InstaGene Matrix kit (Bio-Rad, Hercules, CA, USA), and the 16S rRNA gene was amplified by PCR using universal bacterial primer 27F and 1492R (Frank et al. 2008). PCR product was purified with multiscreen-filter plate (Millipore Corp., Bedford, MA, USA), and was sequenced with an Applied Biosystems 3770XL DNA analyser using a BigDye Terminator cycle sequencing kit v.3.1 (Applied Biosystems, USA). A nearly complete sequence was complied with the SeqMan software (DNASTAR Inc., USA). Nearly full-length 16S rRNA gene (1468, bp) sequence was obtained, and the closest phylogenetic neighbours were identified using the EzTaxon-e database (Kim et al. 2012). Related 16S rRNA sequences were obtained from GenBank. Phylogenetic analysis was accomplished using MEGA6 (Tamura et al. 2013), after carrying a multiple alignment of the sequences using Clustal X 2.1 (Thompson et al. 1997) and analysis of the sequenced data with the software package BioEdit (Hall 1999). Evolutionary distances were calculated using the Kimura two-parameter model (Kimura 1980). Phylogenetic trees were constructed by the neighbour-joining method (Saitou and Nei 1987), maximum-parsimony (Fitch 1971), and maximum-likelihood (Felsenstein 1981). Bootstrap values were calculated based on 1000 replications (Felsenstein 1985).
For the confirmation of the relatedness of strain R160T to the genus Acinetobacter, and its separation from all recognized members of this genus, comparative sequence analysis of the rpoB gene was performed according to the method of La Scola et al. (2006) and Nemec et al. (2009), using two sets of primers to amplify two variable regions of the rpoB gene. Region 1 spans nucleotide positions 2916–3267 (primers Ac696F and Ac1093R), and region 2 spans nucleotide positions 3263–3773 (primers Ac1055F and Ac1598R). PCR amplifications were performed in 50 μL containing 14 μL Taq PCR Master Mix (Inclone biotech), 0.2 μM each primer, and 1.5 μL DNA template, with the following cycling conditions: 94 °C for 2 min; 32 cycles of 30 s at 94 °C, 30 s at 55 °C and 2 min at 72 °C; and 5 min at 72 °C (La Scola et al. 2006). Amplicons were purified for sequencing using Inclone Gel & PCR purification kit (Inclone biotech) following the manufacturers’ instruction. Sequencing analyses and phylogenetic trees were constructed as described earlier. The calculations were carried out for concatenated regions 1 and 2 using nucleotide positions 2917–3267 for region 1 and positions 3322–3723 for region 2. The positions numbers correspond to those of the rpoB encoding sequence of Acinetobacter baumannii (La Scola et al. 2006).
Comparative analysis of the partial sequence of the DNA gyrase subunit B (gyrB) gene was carried out to confirm the genotypic relationship between the strain R160T and the separation from the other members of the genus based on the rpoB sequences as described (Krizova et al. 2014). Nucleotide sequences of gyrB genes were determined from PCR fragments amplified using PCR primers M13R were appended at the 5′ end of the degenerated sequences of PCR primers UP-1E and APrU, respectively, as described by Yamamoto and Harayama (1995). Direct sequencing of the PCR fragments was performed using the universal sequencing primers M13R or M13(–21). PCR amplification of gyrB was performed in 50 μL containing 14 μL Taq PCR Master Mix (Inclone biotech), 0.2 μM each primer, and 1.5 μL target DNA. A total of 35 cycles of amplification were performed with template DNA denaturation at 94 °C for 1 min, primer annealing at 57 °C for 1 min, and primer extension at 72 °C for 2 min (Yamamoto et al. 1999). Purified amplicons were sequenced and constructions of phylogenetic trees were done as described previously. Names of primers are given in Table 1.
Chemotaxonomic characterization
For cellular fatty acid analysis, biomass of strain R160T and reference strains were harvested in late logarithmic phase (48 h) from TSA plate after incubation at 28 °C. Fatty acids were saponified, methylated, and extracted using the standard MIDI protocol (Sherlock Microbial Identification System, version 6.0B). The fatty acids were analyzed by GC (HP 6890 Series GC System; Hewlett Packard, USA) and identified using TSBA6 database of the Microbial Identification System (Sasser 1990). Polar lipid was extracted according to procedure described by Minnikin et al. (1984) and Card (1973). Isoprenoid quinone was extracted with chloroform:methanol (2:1), filtered through Whatman paper (No. 2), evaporated under vacuum at 50 °C, and extracted again with n-hexane. The crude n-hexane-quinone mixture was purified with a Sep-pak Vac silica cartride (Watters, Ireland). Purified n-hexane-quinone was evaporated, eluted with hexane:diethyl ether (98:2), re-extracted with acetone, and analyzed by HPLC (Hairaishi et al. 1996). The polar lipids were analyzed by two-dimensional TLC using chloroform/methanol/water (65:25:4; v/v/v) in the first direction, followed by chloroform/methanol/acetic acid/water (40:7.5:6:2, v/v/v/v) in the second. The appropriate detection reagents (Minnikin et al. 1984; Komagata and Suzuki 1987) were used to identify the spots.
Genotypic characterization
Genomic DNA of the strain R160T and other reference strains were extracted according to the method of Moore and Dowhan (1995). The DNA G + C content was performed and measured according to the procedure described by Mesbah et al. (1989). DNA–DNA hybridization was measured flourometrically according to the method developed by Ezaki et al. (1989), using photobiotin-labelled DNA probes and micro dilution plates.
Assessment of hydrocarbon degradation
Determination of hydrocarbon degradation capacity of strain R160T was done in a 50 mL capped bottle (each for hydrocarbons and oil) containing 10 mL of minimal salt medium (MSM; Pham et al. 2014, 2015) and 0.1 g of wet cells at pH 7.0 in which 900 ppm of hydrocarbons (300 ppm each octadecane, C-18; eicosane, C-20; and docosane, C-22) and 1500 ppm of oil (500 ppm each kerosene, diesel, and gasoline) were added. Control was adjusted without adding bacterial cells. All the bottles were incubated at 28 °C for 14 days in a shaking incubator (130 rpm). After incubation, 10 mL of dichloromethane (HPLC grade; Duksan, South Korea) was added to the bottles and again incubated for 24 h in a shaker at room temperature. After 24 h shaking, 2 mL of DCM extract was transferred into GC vials (Agilent). Residual concentrations of hydrocarbons were determined using gas chromatography (HP 5890 Series II; Agilent, USA) equipped with a flame ionization detector (FID), using an Ultra 2 capillary column (cross-linked 5% phenylmethylsilicone; length, 25 m; internal diameter, 0.32 mm; and film thickness, 0.17 m). Initially, oven temperature was set at 50 °C for 2 min and then increased by 8 °C/min to obtain final temperature of 320 °C. This temperature was maintained for 10 min. The carrier gas (N2) flow rate was set at 1.0 mL/min, and the temperatures of the injector and detector were 300 and 320 °C, respectively. The total sample volume injected was 2 μL. The hydrocarbon degradation percentage was calculated based on the difference between the initial concentration and final concentration of hydrocarbons, determined by a standard curve (Pham et al. 2014; Vermeulen 2007). All the experiments were carried out in triplicate.
Growth curve determination in MSM
Growth curves were determined in MSM broth at pH 7.0. Strain R160T was inoculated in media containing 900 ppm of C-18, C-20, and C-22 hydrocarbons in total (each 300 ppm) and 1500 ppm of oil (500 ppm each kerosene, diesel, and gasoline). The growth absorbance was measured at 600 nm in every 2 days after incubating at 28 °C (130 rpm) for 14 days.
Results and discussion
Morphological and physiological characteristics
Cells are 0.8–1.0 µm long and 0.5–0.7 µm wide (Supplementary Fig. S1). Cells grow optimally in the (0–4) % of NaCl but are able to tolerate up to 7% NaCl. Cells are catalase-positive and oxidase-negative. The KOH test results indicated that flexirubin-type pigment is absent. Casein and starch are hydrolysed. The DNase assay using the DNase agar test is negative. Hydrolysis of Tween 80, MR-VP tests, and haemolysis of sheep blood are negative. Strain R160T is sensitive to tetracycline, gentamycin, novobiocin, chloramphenicol, kanamycin, streptomycin, neomycin, rifampicin, and nalidixic acid, but resistant to cycloheximide, ampicillin, penicillin, trimethoprim, and sulfamethoxazole. The differential phenotypic features of strain R160T are presented with other closest strains (Table 2).
Phylogenetic analysis
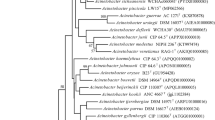
The nucleotide sequence of the 16S rRNA gene of strain R160T has been deposited in GenBank/EMBL/DDBJ under the accession number KT032155. Preliminary comparisons with 16S rRNA gene sequences deposited in the GenBank database indicated that strain R160T belongs to the genus Acinetobacter. The obtained 16S rRNA gene sequence analysis revealed that strain R160T was most closely related to ‘Acinetobacter oleivorans’ DR1T; Acinetobacter nosocomialis NIPH 2119T; and Acinetobacter seifertii NIPH 973T (97.47%, sequence similarity), A. puyangensis BQ4-1T (97.45%, sequence similarity), Acinetobacter pittii CIP70.29T (97.41%, sequence similarity), and A. baumannii ACTC 19606T (97.40%, sequence similarity). Neighbour–joining tree based on 16S rRNA gene sequences showed that strain R160T was clearly affiliated with the genus Acinetobacter and formed a phyletic line distinct from the clades of closely related species. The tree shows that the species with high similarities are not necessarily the closest neighbour (Fig. 1). The partial rpoB gene sequence (861 bp) of strain R160T showed the highest similarity with Acinetobacter haemolyticus NIPH 510T (88.07%) and lower than 88% similarities to other members of the genus Acinetobacter. The neighbour–joining phylogenetic tree based on rpoB gene (Supplementary Fig. S2) formed a separate lineage between Acinetobacter haemolyticus and A. puyangensis, and the neighbour–joining tree based on gyrB gene sequence (892 bp) (Supplementary Fig. S3) showed that strain R160T is different from recognized type strains of Acinetobacter.

Neighbour-joining tree based on nearly complete 16 S rRNA gene sequences showing the phylogenetic position of strain R160T with closely related species from the genus Acinetobacter. Filled circles indicate nodes recovered by all three treeing methods (neighbour–joining, maximum–likelihood, and maximum–parsimony). The numbers at the nodes indicate the percentage of 1000 bootstrap replicates; only values >50% are shown. Moraxella lacunata ATCC 17967T was used as an outgroup. The scale bar represents 0.01 substitutions per nucleotide position. GenBank accession number is given in the parentheses
Chemotaxonomic characteristics
Respiratory quinones of strain R160T are ubiquinone 9 (Q-9; 90%) and ubiquinone 8 (Q-8; 10%), which are consistence with the ubiquinone system of the genus Acinetobacter (Nishimura et al. 1988; Kim et al. 2008; Li et al. 2013). The total fatty acid compositions of strain R160T showed characteristic differences from other reference strains (Table 3). Major polar lipids of strain R160T are DPG, PE, PG, and PC, in addition to one unidentified phospholipid. No glycolipids were detected (Supplementary Fig. S4).
DNA G + C content and DNA–DNA hybridization
The DNA G + C content of strain R160T was 44.9 mol%. DNA–DNA hybridization of strain R160T with that of reference strains showed DNA similarities of 28.6 ± 2.4% with ‘Acinetobacter oleivorans’, 27.1 ± 1.3% with A. puyangensis, 24.2 ± 2.6% with Acinetobacter brisouii, 18.9 ± 2.2% with Acinetobacter soli, and 17.1 ± 3.1% with Acinetobacter baylyi. DNA–DNA relatedness between the species demonstrated that strain R160T differs sufficiently from other type strains of the genus Acinetobacter to warrant species status (Wayne et al. 1987).
Assessment of hydrocarbon degradation
In hydrocarbons degradation experiment, strain R160T degrades 51.7 ± 1.3% of hydrocarbon components (C-18, C-20, and C-22) and 45.8 ± 1.4% of the oil components (kerosene, diesel, and gasoline) after 14 days incubation under condition of pH 7.0 and cell density 0.1 g/10 ml media at 28 °C (Fig. 2). The growth absorbance measurement at 600 nm shows that the strain grows well in both hydrocarbon mixture and oil mixture using hydrocarbons as an energy and carbon source (Fig. 3). There are several reports of Acinetobacter spp. that describes the degradation and utilization of hydrocarbons for energy and carbon source (Kang et al. 2011; Yamahira et al. 2008; Chaudhary 2016; Ishige et al. 2000; Throne-Holst et al. 2006).

Hydrocarbon degradation efficiency of strain R160T in MSM broth containing 900 ppm of hydrocarbons (C-18, C-20, and C-22), and 1500 ppm of oil (kerosene, diesel, and gasoline) incubated for 7 and 14 days

Growth curve of strain R160T in MSM broth containing 900 ppm of hydrocarbons (C-18, C-20, and C-22), and 1500 ppm of oil (kerosene, diesel, and gasoline) incubated for 14 days. The growth absorbance was measured for every 2 days incubation
Strain R160T tolerates high salt concentration (7%) and alkali (pH 11.0). To the best of our knowledge, strain R160T is the first described halotolerant and alkalitolerant strain of the genus Acinetobacter that degrades the mixture of octadecane, eicosane, and docosane; and mixture of kerosene, diesel, and gasoline. This property of strain R160T could have a potential role in removing oil and other hydrocarbon pollutants from hyper-saline environments, such as marine, lakes, ponds, and rocky soils.
On the basis of physiological, chemotaxonomic, genotypic, and phylogenetic characteristics described here, strain R160T is clearly distinguished from the other species within the genus Acinetobacter and represents a novel species, for which the name Acinetobacter halotolerans sp. nov. is proposed.
Description of Acinetobacter halotolerans sp. nov
Acinetobacter halotolerans (ha.lo.to’le.rans. Gr. n. hals halos salt; L. part. adj. tolerans tolerating; N.L. part. adj. halotolerans, salt-tolerating, referring to the ability of the organism to tolerate high concentrations of salt).
Cells are coccobacilli, Gram stain-negative, non-motile and aerobic. They grow well on R2A, TSA, NA, LBA, MA, and sorbitol MacConkey agar. Colonies on R2A are circular, convex with entire margin, smooth, slightly opaque and milk-white in colour. The size of the colony is 1–2 mm in diameter after incubation at 3 days at 28 °C on R2A agar. Cells grow at a temperature of 10–41 °C (optimum, 20–32 °C) and pH 6.0–11.0 (optimum pH, 7.0–9.5). Hydrogen sulfide is not produced. Glucose fermentation is weak. Nitrate is not reduced. Gelatin is hydrolysed, but esculin and urea are not. For API ZYM kit, positive reactions were observed with esterase (C4), esterase lipase (C8), lipase (C14), leucine arylamidase, acid phosphatase, and napthol-AS-BI-phosphohydrolase, whereas alkaline phosphatase, cystine arylamidase, and α-galactosidase are weakly positive. The following substrates were utilized as a sole carbon source: sodium acetate, l-alanine, propionic acid, capric acid, malic acid, valerate, trisodium citrate, l-histidine, 4-hydroxy-butyrate, and l-proline. All the negative traits of commercial kits are given in Supplementary Table 1. The predominant respiratory lipoquinones of strain R160T were ubiquinone 9 (Q-9) and ubiquinone 8 (Q-8). The major polar lipids were phosphatidylethanolamine, diphosphatidylglycerol, phosphatidylglycerol, and phosphatidylcholine. The major cellular fatty acids were C18:1 ω9c, C16:0, and summed feature 3 (comprising C16:1 ω7c and/or C16:1 ω6c). The DNA G + C content of strain R160T was 44.9 mol%.
The type strain, R160T (= KEMB 9005-333T = KACC 18453T = JCM 31009T), was isolated from soil in Hwaseong, Gyeonggi-Do, South Korea.
References
Albarracin VH, Pathak GP, Douki T, Cadet J, Borsarelli CD, GÓ“rtner W, Farias ME (2012) Extremophilic Acinetobacter strains from high altitude lakes in Argentinean Puna: Remarkable UV-B and efficient DNA damage repair. Orig Life Evol Biosph 42:201–221
Breznak JA, Costilow RN (2007) Physicochemical factors in growth. In: Beveridge TJ, Breznak JA, Marzluf GA, Schmidt TM, Snyder LR (eds) Methods for General and Molecular Bacteriology, 3rd edn. American Society for Microbiology, Washington, D. C., pp 309–329
Brisou J, Prévot AR (1954) Etudes de systématique bactérienne. X. Révision des especes reunites dans le genre Achromobacter. Ann Inst Pasteur (Paris) 86:722–728 (French)
Card GL (1973) Metabolism of phosphatidylglycerol, phosphatidylethanolamide and cardiolipin of Bacillus stearothermaphilus. J Bacteriol 114:1125–1137
Chaudhary DK (2016) Bioremediation: an eco-friendly approach for polluted agriculture soil. Emer Life Sci Res 2:73–75
Dahal RH, Kim J (2016a) Pedobacter humicola sp. nov., a member of the genus Pedobacter isolated from soil. Int J Syst Evol Microbiol 66:2205–2211
Dahal RH, Kim J (2016b) Rhabdobacter roseus gen. nov., sp. nov., isolated from soil. Int J Syst Evol Microbiol 66:308–314
Doetsch RN (1981) Determinative methods of light microscopy. In: Gerhardt P (ed) Manual of methods for general bacteriology. American Society for Microbiology, Washington, DC, pp 21–33
Ezaki T, Hashimoto Y, Yabuuchi E (1989) Fluorometric DNA-DNA hybridization in microdilution wells as an alternative to member filter hybridization in which radioisotopes are used to determine genetic relatedness among bacterial strains. Int J Syst Evol Microbiol 39:224–229
Fatajeva E, Gailiütė I, Paliulis D, Grigiškis S (2014) The use of Acinetobacter sp. for oil hydrocarbon degradation in saline waters. Biologija 60:126–133
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evol Int J org Evol 39:783–791
Fitch WM (1971) Toward defining the course of evolution: minimum change for a specific tree topology. Syst Zool 20:406–416
Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ (2008) Critical evaluation of two primers commonly used for amplification of bacterial 16 S rRNA gene. Appl Environ Microbiol 74:2461–2470
Hairaishi A, Ueda Y, Ishihara J, Mori T (1996) Comparative lipoquinone analysis of influent sewage and activated sludge by high-performance liquid chromatography and photodiode array detection. J Gen Appl Microbiol 42:457–469
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Ishige T, Tani A, Saki Y, Kato N (2000) Long-chain aldehyde dehydrogenase that participates in n-alkane utilization and wax ester synthesis in Acinetobacter sp. strain M−1. Appl Environ Microbiol 66:3481–3486
Jawad A, Seifert H, Snelling M, Heritage J, Hawkey PM (1998) Survival of Acinetobacter baumannii on dry surfaces: comparison of outbreak and sporadic isolates. J Clin Microbiol 36:1938–1941
Kang SK, Jung J, Jeon CO, Park W (2011) Acinetobacter oleivorans sp. nov. is capable of adhering to and growing on diesel-oil. J Microbiol 49:29–34
Kim D, Baik KS, Kim MS, Park SC, Kim SS, Rhee MS, Kwak YS, Seong CN (2008) Acinetobacter soli sp. nov., isolated from forest soil. J Microbiol 46:396–401
Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, Park SC, Jeon YS, Lee JH, Yi H, Won S, Chun J (2012) Introducing EzTaxon-e: a prokaryotic 16 S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62:716–721
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Komagata K, Suzuki K (1987) Lipids and cell wall analysis in bacterial systematics. Methods Microbiol 19:161–203
Krizova L, Maixnerova M, Sedo O, Nemec A (2014) Acinetobacter bohemicus sp. nov. widespread in natural soil and water ecosystems in the Czech republic. Syst Appl Microbial 37:467–473
La Scola B, Gundi VA, Khamis A, Raoult D (2006) Sequencing of the rpoB gene and flanking spacers for molecular identification of Acinetobacter species. J Clin Microbiol 44:827–832
Li Y, Piao CG, Ma YC, He W, Wang HM, Chang JP, Guo LM, Wang XZ, Xie SJ, Guo MW (2013) Acinetobacter puyangensis sp. nov., isolated from healthy and diseased part of Populs xeuramericana canker bark. Int J Syst Evol Microbiol 63:2963–2969
Macfaddin JF (1980) Bacterial tests for identification of medical bacteria. 2nd edn. Williams and Wilkins, Baltimore, pp. 162–218
Margesin R, Schinner F (2001) Bioremediation (natural attenuation and bio-stimulation) of diesel-oil-contaminated soil in an alpine glacier skiing area. Appl Environ Microbiol 67:3127–3133
Mesbah M, Premachandran U, Whitman WB (1989) Precise measurement of the G + C content of deoxyribonucleic acid by high-performance liquid chromatography. Int Syst Bacteriol 39:159–167
Minnikin DE, O’Donnell AG, Goodfellow M, Alderson G, Athalye M, Schaal A, Parlett JH (1984) An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J Microbiol Methods 2:233–241
Mishra S, Sarma PM, Lal B (2004) Crude oil degradation efficiency of a recombinant Acinetobacter baumannii strain and its survival in crude oil-contaminated soil microcosm. FEMS Microbiol Lett 235:323–331
Moore DD, Dowhan D (1995) Preparation and analysis of DNA. In: Assubel FW, Brent R, Kingston RE, Moore DD, Seidman JD, Smith JA, Struhl K (eds) Current protocols in molecular biology. Wiley, New York, pp 2–11
Nemec A, Musilek M, Maixnerová M, De baere T, Van Der Reijden TJK, Vaneechoutte M, Dijkshroorn L (2009) Acinetobacter beijerinckii sp. nov. and Acinetobacter gyllenbergii sp. nov., haemolytic organisms isolated from humans. Int J Syst Evol Microbiol 59:118–124
Nishimura Y, Ino T, Iizuka H (1988) Acinetobacter radioresistens sp. nov. isolated from cotton and soil. Int J Syst Bacteriol 38:209–2011
Pham VHT, Jeong SW, Kim J (2014) Enhanced isolation and culture of highly efficient psychrophilic oil-degrading bacteria from oil-contaminated soils in South Korea. J Environ Biol 35:1145–1149
Pham VHT, Jeong SW, Kim J (2015) Psychrobacillus soli sp. nov., capable of degrading oil, isolated from oil-contaminated soil in Mongolia. Int J Syst Evol Microbiol 65:3046–3052
Reichenbach H (1992) The order Cytophagales. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer KH (eds) The prokaryotes, 2nd edn, vol 4. Springer, New York, pp 3631–3675
Saitou N, Nei M (1987) The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sasser M (1990) Identification of bacteria by gas chromatography of cellular fatty acids, MIDI Technical Note 101. MIDI Inc, Newark, DE
Schaeffer AB, Fulton M (1933) A simplified method o staining endospores. Science 77:194
Smibert RM, Krieg NR (1994) Phenotypic characterization. In: Gerhardt P, Murray RGE, Wood WA, Krieg NR (eds) Methods for general and molecular bacteriology. American Society for Microbiology, Washington, DC, pp. 607–654
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Throne-Holst M, Markussen S, Winnberg A, Ellingsen TE, Kotlar HK, Zotchev SB (2006) Utilization of n-alkanes by a newly isolated strain of Acinetobacter venetianus: the role of two AlkB-type alkane hydroxylases. Appl Microbiol Biotechnol 72:353–360
Tindall BJ, Sikorski J, Smibert RA, Krieg NR (2007) Phenotypic characterization and the principles of comparative systematics. In: Reddy CA, Beveridge TJ, Breznak JA, Marzluf GA, Schmidt TM, Snyder LR (eds) Methods for general and molecular bacteriology, 3rd edn. ASM Press, Washington, D. C., pp 330–393
Tittsler RP, Sandholzer LA (1936) The use of semi-solid agar for the detection of bacterial motility. J Bacteriol 31:575–580
Vaneechoutte M, Tjernberg I, Baldi F, Pepi M, Fani R, Sullivan ER, Toorn JV, Dijkshoorn L (1999) Oil-degrading Acinetobacter strain RAG-1 and strains described as Acinetobacter venetianus sp. nov. belong to the same genomic species. Res Microbiol 150:69–73
Vaughn RH, Mitchell NB, Levine M (1939) The Voges–Proskauer and methyl red reactions in the coli-aerogenes group. J Am Water Works Assoc 31:993–1001
Vermeulen J (2007) Ripening of PAH and TPH polluted sediments: determination and quantification of bioremediation parameters. PhD thesis. Wageningen University, Wageningen
Wayne AB, Copper DG (1992) Hydrocarbon degradation by Acinetibacter calcoaceticus RAG-1 using the self-cycling fermentation technique. Biotechnol Bioeng 40:797–805
Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, Moore LH, Moore WEC, Murray RGE, Stackebrandt E, Starr MP, Trüper HG (1987) Report of the ad hoc committee on reconciliation of approaches to bacterial systematic. Int J Syst Bacteriol 37:463–464
Yamahira K, Hirota K, Nakajima K, Morita N, Nodasaka Y, Yumoto I (2008) Acinetobacter sp. strain Ths, a novel psychrotolerant and alkalitolerant bacterium that utilizes hydrocarbon. Extremophiles 12:729–734
Yamamoto S, Harayama S (1995) PCR amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of Pseudomonas putida strains. Appl Environ Microbiol 61:1104–1109
Yamamoto S, Bouvet PJM, Harayama S (1999) Phylogenetic structures of the genus Acinetobacter based on gyrB sequence: comparison with the grouping by DNA–DNA hybridization. Int J Syst Bacteriol 49:87–95
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A1A09916982).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
The GenBank/EMBL/DDBJ accession number for the 16S rRNA, rpoB, and gyrB gene sequences of strain R160T are KT032155, KU958712, and KU958711 respectively.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dahal, R.H., Chaudhary, D.K. & Kim, J. Acinetobacter halotolerans sp. nov., a novel halotolerant, alkalitolerant, and hydrocarbon degrading bacterium, isolated from soil. Arch Microbiol 199, 701–710 (2017). https://doi.org/10.1007/s00203-017-1349-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-017-1349-2