
Abstract
We explored the molecular diversity of cytochrome P450 genes in the filamentous fungus Aspergillus oryzae using bioinformatic and experimental approaches. Based on bioinformatic annotation, we found 155 putative genes of cytochromes P450 in the whole genome sequence; however, 13 of 155 appeared to be pseudogenes due to sequence deletions and/or inframe stop codon(s). There are 87 families of A. oryzae cytochromes P450 (AoCYPs), indicating considerable phylogenetic diversity. To characterize A. oryzae AoCYPs, we attempted to isolate cDNAs using RT-PCR and determined their transcriptional capabilities. To date, we have confirmed gene expression of 133 AoCYPs and cloned 121 AoCYPs as full-length cDNAs encoding a mature open reading frame. Using experimentally deduced sequences and intron–exon organization, we analyzed AoCYPs phylogenetically. We also identified intronic consensus sequences in AoCYPs genes. The experimentally validated exonic and intronic sequences will be a powerful advantage in identification and characterization of novel P450s from various ascomycetous fungi.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytochromes P450 (P450s) constitute a large superfamily of heme-containing monooxygenases, which are distributed in a wide variety of organisms. The large-scale molecular variations among P450 species imply an evolutional trajectory in which a common ancestor extensively branched into various organisms (Gotoh 1993; Nelson et al. 1993; Lewis et al. 1998). The vast majority of P450s are thought to have specifically emerged and individually diversified during evolution of each organism. For instance, only CYP51, which is involved in the sterol biosynthesis pathway, is conserved across eukaryotic phyla (Yoshida 1993; Aoyama et al. 1996). Thus, the molecular and functional diversities of P450s enable specialization to meet the metabolic requirements of each organism, especially secondary metabolic pathways such as detoxification of xenobiotics and synthesis of secondary metabolites (Ortiz-de-Montellano 2005; Demain and Fang 2000). However, the degree of P450 divergence, such as the numbers of genes and gene families, differs significantly across biological kingdoms, phyla, and species. Such divergences presumably reflect an evolutionary driving force to develop survival strategies (Nelson 1999). In particular, the scale of divergence of P450s within the fungal kingdom is unprecedented (Park et al. 2008; Deng et al. 2007; Doddapaneni et al. 2005; Intikhab et al. 2007). In spite of their high sequence diversity, P450s share conserved overall protein architecture and contain several conserved sequences, such as the FxxGxxxCxG signature motif. Such motifs can be used to discover novel genes from genomic databases. Within the last few years, the sequence database of P450s has enlarged exponentially, and continues to increase (http://drnelson.utmem.edu/CytochromeP450.html; http://p450.riceblast.snu.ac.kr/index.php?a=view; Park et al. 2008). A compilation of P450 sequences will increase understanding of metabolic diversity and evolutionary history of living organisms. However, there are few detailed studies on transcriptional profiles and catalytic functions of P450s in metabolic processes, so their exact roles and functions are still poorly understood.
A wide variety of filamentous fungi is used to produce economically valuable consumer items. The filamentous fungus Aspergillus oryzae is one of the most widely used microorganisms, and has been used for more than 1,000 years in Japanese fermentation industries to produce indigenous products such as sake (rice wine), miso (soybean paste), and shoyu (soy sauce). Because of the long history of use in fermentation and food production, A. oryzae is listed as “generally recognized as safe” by the food and drug administration in the United States. Besides fermentation technologies, many studies on production of recombinant enzymes and primary and secondary metabolites have focused on A. oryzae (Tailor and Richardson 1979; Abe et al. 2006). Recently, the whole genomic sequence of A. oryzae (strain RIB40) was determined and made available to the public (Machida et al. 2005). A. oryzae has eight chromosomes with a total genome size of 37.6 Mb, which is 20–30% larger than the genomes of A. nidulans and A. fumigatus. In total, there are 12,074 predicted genes in A. oryzae, compared with 9,396 in A. nidulans and 9,009 in A. fumigatus (Machida et al. 2005; Galagan et al. 2005; http://www.bio.nite.go.jp/dogan/Top). The increased gene number in A. oryzae is mainly due to a gain of extra genes involved in secondary metabolic pathways, including P450s, suggesting that A. oryzae has unique metabolic processes that are absent in other Aspergillus species. Thus, genomic data will increase molecular understanding of previously uncharacterized metabolic processes in A. oryzae.
In the present study, we explored the molecular diversity of A. oryzae P450s (AoCYPs) using a bioinformatic annotation and experimental validation. To our knowledge, this is the first comprehensive transcriptional survey of ascomycetous P450s. The identified and isolated AoCYPs have potential benefits to improve bioinformatic algorithms, expand biochemical knowledge, and advance biotechnology.
Materials and methods
Microorganism and culture conditions
Aspergillus oryzae strain RIB40 (NBRC 100959) was cultured on YPD/agar plates (1% yeast-extract, 2% bacto-peptone, 0.04% adenine sulfate, 2% glucose, 1.5% bacto-agar) at 30°C for 3 days, and then inoculated into synthetic culture media as described previously (Kirk et al. 1978). The synthetic media contained 1% glucose, 1.2 mM (nitrogen-limited conditions) or 12 mM (nitrogen-enriched conditions) ammonium tartrate, 20 mM dimethylsuccinate (pH 4.5), and trace elements (100 mL/L culture). The trace elements (1 L) contained 20 g KH2PO4, 5.3 g MgSO47H2O, 1.3 g CaCl22H2O, 10 or 0 mg thiamine hydrochloride, 150 mg N(CH2COOH)2, 18 mg CoSO47H2O, 18 mg ZnSO47H2O, 1 mg CuSO45H2O, 1.8 mg AlK(SO4)12H2O, 1 mg H3BO3, 70 mg MnSO45H2O, 10 mg FeSO47H2O, 100 mg NaCl, and 1 mg Na2MoO42H2O. Fungal cells were grown with shaking (130 rpm) at 30°C for 2–21 days under aerobic conditions.
RNA extraction and first-strand cDNA synthesis
Total RNA was extracted individually from 5-, 10-, 18-, and 21-day-old mycelia using the acid guanidinium–phenol–chloroform method and further purified using an RNeasy Plant Mini Kit (QIAGEN). The concentration of RNA was calculated from the absorbance at 260 nm. Equal quantities of RNA isolated from mycelia of the four different ages were then mixed. The RNA cocktail was treated with DNase I (Takara), and first-strand cDNAs were synthesized with SUPERSCRIPT III™ reverse transcriptase (Invitrogen) in the presence of oligo(dT) primer (5′-TTTTTTTTTTTTTTTTTTV-3′; V = A, C, or G). The reaction mixtures (50 μL) contained 5 μg total RNA, 200 units SUPERSCRIPT™ III reverse transcriptase (Invitrogen), 40 units RNase-Out (Invitrogen), 4 mM DDT, 0.4 mM dNTPs, and 25 pmol oligo(dT) primer in 1× first strand buffer, and were incubated at 50°C (60 min) for the extension reaction. The reaction mixtures were stored at –20°C until PCR amplifications.
Bioinformatic annotation of P450 from A. oryzae
A possible coding sequence for AoCYPs was used to search the National Institute of Technology and Evaluation database based upon sequence similarity to known P450s (http://www.bio.nite.go.jp/dogan/Top). To evaluate annotation accuracy, we identified the P450 signature sequence (F-x-x-G-x-x-x-C-x-G) in the heme-binding domain, the E-x-x-R motif in the K-helix, a conserved Thr in the center of the I-helix, and the hydrophobic transmembrane domain (TMD) at the N-terminal region. TMD sequences were analyzed by the SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui/) and the TMHMM server v.2.0 (http://www.cbs.dtu.dk/services/TMHMM/). If candidates lacked sequences corresponding to these regions, their capability to encode P450 was judged by overall sequence similarity to known P450s.
cDNA amplification by RT–PCR
PCR amplification were carried out using gene-specific primers designed to anneal to 5′- and 3′-untranslated regions; basically, to the 2–30 bp upstream or downstream flanking sequence from the putative start and stop codons (Supplemental Table). Custom-synthesized oligonucleotide primers were obtained from SIGMA–ALDRICH. cDNA was amplified by Phusion DNA polymerase (New England Biolabs). The reaction mixture (50 μL) contained first-strand cDNA solution (1 μL), dNTP (200 μM), primers (2 μM each), DMSO (2%), and Phusion DNA polymerase (0.02 U/μL) in Phusion HF buffer. The reaction conditions were programmed as follows: denaturation at 96°C for 3 min; 40 cycles of 96°C for 30 s, 55°C for 20 s, and 72°C for 60 s; and final extension at 72°C for 2 min. The PCR products were separated by 1.5% agarose gel electrophoresis and visualized on a UV-transilluminator. Target cDNAs were purified using a QIAquick Gel Extraction Kit (QIAGEN), phosphorylated with T4 polynucleotide kinase (TaKaRa), cloned into the pUC18 SmaI site, and transformed into Escherichia coli strain JM109. Positive transformants were selected on LB-agar plates containing ampicillin (100 mg/L), IPTG (100 mg/L), and X-gal (100 mg/L). The positive clones were further grown in LB medium supplemented with ampicillin (100 mg/L). Plasmids harboring AoCYP cDNA were extracted using a QIAprep Spin Miniprep Kit (QIAGEN) and sequenced with an automated DNA Sequencer (CEQ 8000; Beckman) using a DTCS Quick Start Kit (Beckman).
Validation of alternative splicing events
Total RNA was recovered from fungal cells grown in synthetic medium with or without exogenous thiamine (0 or 1 mg/L). RT-PCR was then carried out using SUPERSCRIPT III™ reverse transcriptase and Phusion DNA polymerase. The cDNA fragment of CYP5076C1 was amplified by gene specific primers, primer-1 (5′-ATGGATATCAAGGAAAAGCCGA-3′), primer-2 (5′CTATATAGCACGTTTTTGAAAGTGTA-3′), and primer-3 (5′-AGGTCGGCAAGCTTGCG-3′). The reaction mixture (50 μL) contained first-strand cDNA solution (1 μL), dNTP (200 μM), primers (2 μM each), DMSO (2%), and Phusion DNA polymerase (0.02 U/μL) in Phusion HF buffer. The PCR products were separated by 1.5% agarose gel electrophoresis, stained with GelStar® Nucleic Acid Stain (TaKaRa), and visualized using Molecular Imager FX (BioRad). cDNA fragments were purified, cloned into the pUC18 plasmid, and sequenced.
Sequence alignment and phylogenetic analysis
Multiple alignment of AoCYPs was carried out using the ClustalW program with a gap penalty of 10, a gap extension penalty of 0.2, and GONNET as protein matrix series (Thompson et al. 1994). The phylogenetic tree was constructed by the Unweighted Pair Group Method with Arithmatic Mean (UPGMA) method with the Jones-Taylor-Thornton matrix using PHYLIP software (Felsenstein 1989), and visualized using the FigTree program.
Results and discussion
Genome-wide survey and molecular identification of AoCYPs
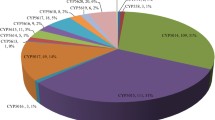
The filamentous fungus A. oryzae has eight chromosomes with an entire genome size of 37.6 Mb (Machida et al. 2005). The whole-genome sequence of A. oryzae strain RIB40 was released recently (http://www.bio.nite.go.jp/dogan/Top). According to the public database, there are several candidate genes assigned to P450s. However, some candidates have low sequence similarity to P450s but significantly higher similarity to other proteins, suggesting that annotational errors may be involved (data not shown). Several candidates have unexpected truncations of their N- and/or C-terminal sequence(s). Therefore, we further refined gene annotation accuracy based on the following sequence features: (1) conservation of F-x-x-G-x-x-x-C-x-G in the heme-binding domain, (2) conservation of E-x-x-R in the K-helix, (3) A/G-G-x-x-T at the center of the I-helix, and (4) a hydrophobic transmembrane domain (TMD) at the N-terminal region. After searching the database, 155 putative genes of P450 were identified from the whole-genome sequence (Fig. 1). However, sequence deletions and/or inframe stop codon(s) were found in 13 genes, suggesting that they are possibly pseudogenes that have originated from gene reorganizations and/or single mutations during fungal evolution (Fig. S1). Although some pseudogene-like AoCYPs were expressed, sequence deletions and/or inframe stop codon(s) were also verified from their transcripts by 3′- and 5′-RACE (Forhman 1993) and RT-PCR analyses (data not shown). Therefore, 142 AoCYPs from the A. oryzae genome were selected for further investigation.

Chromosomal localization of cytochrome P450 in Aspergillus oryzae. PG pseudogene
According to the P450 nomenclature committee, families share greater than 40% identity, and subfamilies share greater than 55% identity of amino acid sequences. The numbers following the root symbol CYP indicate the family and letters indicate the subfamily (Nelson et al. 1996). Based on sequence comparisons, 142 AoCYPs were assigned into 87 families. There were significantly more AoCYP genes than in other ascomycetous fungi, e.g., 122 in Magnaporthe grisea, 107 in Fusarium graminearum, 41 in Neurospora crassa, and 111 in A. nidulans (Deng et al. 2007; Kelly et al. 2008). A possible explanation for the marked increase in gene number may be that fungal P450s continuously diversified after separation of genuses, and perhaps after speciation as well, indicating that sequences diversified within a short evolutionary period (Deng et al. 2007). During the evolutionary history of Aspergillus spp., A. oryzae would have vigorously expanded its genome size to gain extra genes by horizontal gene transfer and duplication. For instance, both gene number and genomic size are 25–30% larger in A. oryzae than in A. nidulans and A. fumigatus. The extra genes are likely to be involved in secondary metabolism (Machida et al. 2005). Therefore, the substantial increase in P450 gene number might indicate adaptation to the specific metabolic requirements of A. oryzae (http://www.aspergillus.org.uk/index.html). In fact, several AoCYPs were located near genes involved in biosynthesis of secondary metabolites, such as polyketide synthase and non-ribosomal peptide synthase. For example, CYP655B1 and CYP5286A1 are flanked by polyketide synthase (protein ID; BAE56814.1) within 19 kbp distance on chromosome II, CYP5110A1 and CYP577A1 are flanked by polyketide synthase (protein ID; BAE58990.1) within 22 kbp distance on chromosome III, and CYP5099A1 is flanked by isopenicillin N synthase (protein ID; BAE56800.1) and non-ribosomal peptide synthase (protein ID; BAE56801.1) within 19 kbp distance on chromosome II. On the other hand, the genome of A. oryzae is strikingly similar to that of A. flavus; A. flavus has 12,197 genes in its 36.8 Mb genome (Payne et al. 2006). The presence of 159–167 A. flavus P450s has also been revealed by genomic annotation (Park et al. 2008; http://drnelson.utmem.edu/CytochromeP450.html, http://p450.riceblast.snu.ac.kr/index.php?a=view). Sequence comparisons indicated that 138 of the P450s in A. oryzae and A. flavus show orthologous relationships, sharing amino acid sequence identities of more than 95%. This raises the question as to whether the P450s of both species exhibit similar transcriptional and functional profiles. In contrast, 16 species were found in A. oryzae but not in A. flavus. Some of these 16 AoCYPs were located within 20 kbp distance, suggesting that a cluster of secondary metabolism genes has developed specifically in A. oryzae. In fact, CYP5119A1, CYP65AG1, CYP65AE1, and CYP5098A1 are distributed within an 18-kbp region on chromosome III, and are flanked by several metabolic genes such as the non-ribosomal peptide synthase module (Protein ID; BAE60013.1), which is also absent from A. flavus. Because CYP5119 is an orphan family that is only found in A. oryzae, the related gene cluster might play important roles in a secondary metabolic process that is unique to this fungus.
Insights into conserved sequences of AoCYPs
The protein architecture of P450s is generally well conserved, even though they show considerable sequence divergence (Graham and Peterson 1999). Structural conservation is presumably important for fundamental aspects of P450 activity, such as heme binding, acceptance of electrons, and activation of molecular oxygen. Therefore, the classical P450s contain conserved sequences that shape the core structure. The most characteristic P450 consensus sequence, F-x-x-G-x-x-x-C-x-G, is found in the heme-binding domain, where the conserved Cys serves as the fifth axial ligand to the heme. Consequently, we identified 142 AoCYPs-containing proximal Cys residues in the heme-binding domain. In addition, several possible functions of conserved amino acid residues have been proposed: (1) the first amino acid is Phe, whose phenyl group appears to protect the reactive cysteine ligand, (2) the fourth Gly appears to initiate the hairpin turn in the loop, and (3) the tenth Gly is small enough to come in close contact with the heme (Ortiz-de-Montellano 2005; Koymans et al. 1993). Amino acid substitutions in the heme-binding domain were identified from some AoCYPs, such as the first Phe to Trp/Tyr in 11 species and the tenth Gly to Ala in seven species; nevertheless, no significant detrimental effects on catalytic functions might be introduced due to their physicochemical characteristics similar to those in classical P450s. However, rare substitutions were found in several AoCYPs, such as the first Phe to Val (CYP5097A1) or Leu (CYP5116A1), and the fourth Gly to Ser (CYP660C1 and 660C2) or Trp (CYP5099A1), suggesting possible involvement of a unique and novel heme-binding feature in those AoCYPs. Moreover, a multiple alignment revealed that CYP5102A1 contained a substantially altered signature sequence, L-S-T-S–I-N-D-C–P-K. Although there is a paucity of literature on biochemical and functional characterization of fungal P450s, further research on AoCYPs could clarify the peculiar reaction mechanisms associated with unique sequences.
The core structure of the proximal side of P450s is additionally stabilized by a consensus sequence of ExxR in the K-helix and a coil known as the “meander” located between the K-helix and the heme-binding domain (Hasemann et al. 1995; Chen and Zhou 1992). The characteristic ExxR sequence is always conserved, and a possible “meander” sequence, normally PER, was found in each of the AoCYPs. In addition to the proximal side, many P450s have a conserved Thr residue in the distal I-helix that shapes the oxygen binding pocket, stabilizes the iron-oxo intermediate, and facilitates heterolytic cleavage of the O–O bond (Poulos et al. 1987; Koymans et al. 1993). A multiple alignment analysis revealed that 107 AoCYPs contain the Thr residue in the I-helix and nine AoCYPs substituted Thr to Ser at the appropriate position. It is possible that both OH-containing Thr and Ser exhibit the same function with regard to oxygen activation. However, neither Thr nor Ser was identified in the I-helix in 38 AoCYPs, suggesting possible involvements of unique reaction mechanisms. It has been known that some P450s such as plant allene oxide synthase (CYP74 family) and human prostaglandin I2 synthase (CYP8 family), which lack conserved Thr, catalyze isomerization reactions of hydroperoxide compounds (Howe et al. 2000; Ullrich 2003). Thus, abnormal sequences in AoCYPs would be an attractive target to better understand the unique structural and mechanistic characteristics of P450s.
Membrane topology of AoCYPs
Most eukaryotic P450s are likely to have an N-terminal TMD sequence, which is responsible for subcellular localization to the endoplasmic reticulum (Nelson and Strobel 1988). A typical TMD contains 20–30 hydrophobic amino acid residues that shape a helical structure as a membrane anchor. The TMD-associated subcellular localization to membranes should also be important for protein–protein interactions with the membrane-anchored cytochrome P450 oxidoreductase (CPR), which is the common redox partner of eukaryotic P450s. A possible TMD was distinguished from 133 AoCYPs using SOSUI and TMHMM servers (Hirokawa et al. 1998; http://bp.nuap.nagoya-u.ac.jp/sosui/; http://www.cbs.dtu.dk/services/TMHMM/), suggesting that most AoCYPs localize to the endoplasmic reticulum. A well-known soluble nitric oxide reductase (P450nor, CYP55A5) lacks the N-terminal TMD sequence (Nakahara et al. 1993). On the other hand, a distinctive N-terminal TMD was not found in several AoCYPs assigned to the P450 family CYP505 (CYP505A3, 505C3, and 505A14), CYP540 (CYP540A3, 540B9, and 540B10), CYP541 (CYP541B3), and CYP5053 (CYP5053C1). The CYP505 family contains self-sufficient P450s fused with a reductase domain, such as P450foxy, which lacks a distinctive TMD but is capable of loosely binding to the membrane (Kitazume et al. 2000; Nakayama et al. 1996). This suggests that AoCYPs assigned to the CYP505 family might also be expressed in membrane fractions. The CYP541 and CYP540 families are phylogenetically close to the CYP505 family even though they lack the reductase domain. These families probably emerged via disconnection of an ancestral fusion P450. If they are not membrane-bound proteins, the membrane-anchored CPR should still weakly transfer electrons CYP540 and CYP541 families. In A. oryzae, we also annotated TMD-containing and TMD-lacking CPRs and evaluated their gene expression (Figs. S2, S3). The possession of both TMD-containing and TMD-lacking CPR would be advantageous to interact with various AoCYPs as a redox partner (Lah et al. 2008).
Transcriptomic survey of cytochromes P450 from A. oryzae
The current sequence database of P450s exponentially enlarged as a result of several genome projects (http://drnelson.utmem.edu/CytochromeP450.html; http://p450.riceblast.snu.ac.kr/index.php?a=view; Park et al. 2008). In addition to bioinformatic studies, experimental approaches are also necessary to evaluate practical applications in a post-genomic era. In this study, our principal aim was to isolate and characterize full-length cDNAs to clarify transcriptional capabilities of AoCYPs. Expression profiles of genes involved in secondary metabolic systems of A. oryzae are very likely to be affected by cultivation conditions (Tamano et al. 2008; Machida et al. 2005). Therefore, we used YPD and synthetic liquid culture media for fungal growth (detailed in materials and methods). Figure 2 shows expression profiles of AoCYPs encoded on chromosome VI. When fungi were grown in YPD liquid culture medium, many genes were not amplified by RT-PCR, whereas some genes such as CYP51 and CYP58 families were confirmed to be expressed (Fig. 2a). No significant gene expression was also observed in a synthetic liquid culture medium under nitrogen-rich conditions (data not shown). P450s classified in CYP51 family are highly conserved across diverse organisms, and play important roles in steroid metabolism. Although CYP58 family in Fusarium spp. has been shown to be involved in biosynthesis of secondary metabolites, metabolic pathways associated with CYP58 family might also be important for fungal cells because a wide variety of fungal species possess homologous genes (Hohn et al. 1995; http://drnelson.utmem.edu/CytochromeP450.html; http://p450.riceblast.snu.ac.kr/index.php?a=view). Thus, we expected that AoCYPs involved in housekeeping pathways would be expressed in YPD and nitrogen-rich synthetic culture media. In contrast, a series of AoCYP genes were strongly expressed when A. oryzae was grown in a synthetic liquid culture medium under nitrogen-limited conditions (Fig. 2a), suggesting that transcriptional regulation of AoCYP responds to nitrogen limitation or starvation. Previously, P450-dependent metabolic pathways in white-rot basidiomycetes such as Phanerochaete chrysosporium and Coriolus versicolor have been shown to be activated under nitrogen-limited conditions (Ichinose et al. 1999; Matsuzaki and Wariishi 2004). Thus, there may be a unique mechanism that activates the fungal secondary metabolic system during nitrogen limitations. Although AoCYPs encoded on chromosome VI showed different time course of gene expression, a significant expression level of AoCYPs appeared after 5 days incubation and continued until 21 days incubation (Fig. 2b). Therefore, amplification and isolation of cDNAs by RT-PCR were carried out using a RNA cocktail which was prepared by mixing total RNA obtained from 5-, 10-, 18-, and 21-day-old mycelia grown in a synthetic liquid culture medium under nitrogen-limited conditions.

Transcriptomic survey of AoCYPs encoded on chromosome VI. a Effects of culture conditions on gene expression of AoCYPs. RT-PCR was carried out using total RNA extracted from A. oryzae grown for 5 days in YPD liquid culture medium (Lanes 1–10) or 10 days in a nitrogen-limited synthetic liquid culture medium (Lanes 11–20). cDNA fragments of CYP51F4 (Lanes 1 and 11), CYP58F1 (Lanes 2 and 12), CYP58G1 Lane (Lanes 3 and 13), CYP505C3 (Lanes 4 and 14), CYP531E1 (Lanes 5 and 15), CYP5080E1 (Lanes 6 and 16), CYP5087B1 (Lanes 7 and 17), CYP5106A1 (Lanes 8 and 18), CYP5107A1 (Lanes 9 and 19), and CYP5114A1 (Lanes 10 and 20) were separated by 1.5% agarose gel electrophoresis, stained with GelStar® Nucleic Acid Stain (TaKaRa), and visualized using Molecular Imager FX (BioRad). Lane M was loaded with a DNA marker. Arrows indicate amplified cDNA of AoCYPs. Primer sequences are listed in Supplemental Table. b Time course of gene expression in a nitrogen-limited synthetic liquid culture medium. RT-PCR was performed with total RNA individually extracted from A. oryzae grown for 5, 10, 18, or 21 days in a nitrogen-limited synthetic liquid culture medium. Analytical procedures were same as that for Fig. 2a
Using an RT-PCR technique, we determined transcriptional capabilities of 133 AoCYPs experimentally (Table 1). To our knowledge, this is the first report of experimental validation of AoCYPs expression, and our results provide evidence that a series of P450s can be expressed in ascomycetous fungi. So far, we isolated 121 full-length cDNAs encoding a mature open reading frame. Identification of these clones will be an advantage for generating recombinant systems, which can contribute to characterization and practical applications of AoCYPs. In addition, the experimentally deduced sequences will improve bioinformatic algorithms; in fact, we identified several mature AoCYPs with novel intron/exon boundaries, which were unexpected and miss-annotated in the database (http://www.bio.nite.go.jp/dogan/Top). The isolated and predicted cDNA sequences of 142 AoCYPs are listed in Figures S2 and S3. Interestingly, our results showed that several AoCYPs were alternatively spliced in response to different culture conditions. For example, CYP5076C1 was spliced to produce frame-shifted variants when A. oryzae was grown with exogenously added thiamine, while it was differently spliced to produce a mature variant when grown without thiamine (Fig. 3). Although further investigations are required to confirm whether exogenous thiamine directly affects splicing events, our results strongly suggest that unique splicing mechanisms, such as riboswitching, might be involved. In our experimental conditions, we isolated 12 immature AoCYPs whose open reading frames were shifted by illegal splicing events. Their functional expression might also be regulated by sophisticated maturation mechanisms at a post-transcriptional stage (Winkler et al. 2002; Cheah et al. 2002; Kubodera et al. 2003; Thore et al. 2006). Thus, a combination of quantitative and qualitative transcriptional profiling of AoCYPs is very important to understand physiological impacts on fungal metabolic activities.

Thiamine-dependent alternative splicing of AoCYP. a Alternative splicing of CYP5076C1 analyzed by RT-PCR. RT-PCR was carried out using total RNA extracted from A. oryzae grown for 18 days in a thiamin-containing synthetic liquid culture medium (Lanes 1 and 3) or thiamin-free synthetic liquid culture medium (Lanes 2 and 4). Lanes 1 and 2 were loaded with PCR products amplified by primer-1 and primer-2. Lanes 3 and 4 were loaded with PCR products amplified by primer-1 and primer-3. Fragments 1, 2, and 3 are illustrated in Fig. 3b. b Variant-1 was obtained from thiamin-free media. Variant-2 and variant-3 were obtained from thiamin-containing media. Columns indicate exons and introns. Lines indicate untranslated region. ORF indicates open reading frame
Several Aspergillus spp. are known to produce polyketide derivatives, i.e., aflatoxins, which have serious toxic, mutagenic, and carcinogenic activities. However, the non-aflatoxigenic status of A. oryzae has been firmly established, because the aflatoxin biosynthesis pathway is inactivated. CYP64A1, a gene homolog involved in the aflatoxin biosynthesis pathway in A. flavus (Prieto and Woloshuk 1997), was transcriptionally silent in the synthetic culture medium in which a number of AoCYPs were expressed. This result is consistent with the inability of the fungus to produce aflatoxin. However, A. oryzae expressed seven AoCYPs assigned to the CYP620 family, which is phylogenetically close to the CYP64 family, suggesting a possible involvement of another polyketide biosynthesis pathway. In terms of human health and economically important products, polyketides have both positive (e.g., antibiotics and cancer therapeutic drug) and negative (e.g. mycotoxin) effects. Therefore, further investigations on their catalytic functions would be of great interest to better understand the safety and capabilities of this fungus (Barbesgaard et al. 1992; Machida et al. 2008).
Phylogeny and gene structure of cytochromes P450 in A. oryzae
Evolutionary histories of eukaryotic genes involve various trajectories such as gains and losses of introns. Although mechanisms and contributions of intron gain/loss events remain elusive, fossil aspects of introns can be helpful to unravel the dynamics of gene evolution. Figure 4 shows the phylogenetic tree and intron–exon organization of AoCYPs accurately constructed with experimentally deduced sequences. Multiple alignments of the deduced sequences and intron–exon structures revealed phylogenetic diversity of AoCYPs. Intron–exon organizations of P450 genes are generally conserved in plants, animals, and basidiomycetous fungi (Doddapaneni et al. 2005; Paquette et al. 2000; Tijet et al. 2001). The extremely diverse gene structure of AoCYPs might indicate that AoCYPs have emerged from a number of parent genes in the fungal ancestor (Deng et al. 2007). The phylogenetic analysis also suggested an evolutionary trajectory in which gene duplication events were restricted in A. oryzae. This suggests that molecular mechanisms such as repeat-induced point mutations (RIP) may be involved (Galagan et al. 2003; Ikeda et al. 2002; Montiel et al. 2006). However, it appeared that several CYP families were generated or enlarged by evolutionary duplication, and some AoCYPs in such families were not expressed. Although RIP-like phenomena in A. oryzae are poorly understood, we can suggest a hypothetical scenario in which some AoCYPs are transcriptionally silenced by RIP-like mechanisms. For instance, the considerable similarities of sequences and gene structures between CYP64 and CYP620 families indicate evolutionary duplication events; however, CYP64A1 was not expressed under our experimental conditions, whereas CYP620 was abundantly expressed (Fig. 4). Recently, clan-level classification has been proposed for a higher order grouping of P450 families. The main concept of this analysis is that genes within a clan share a common ancestor gene and catalytic functions (Nelson 1998, 1999). Although common parameters for clan membership have not been clearly defined, CYP64 and CYP620 families would be classified into the same clan because these families were branched on a neighbor-joining tree with bootstrap values >70% (Deng et al. 2007). Since the isolated cDNAs of AoCYPs would be a powerful advantage to facilitate downstream applications such as functional characterization using recombinant enzymes, further research on AoCYPs should aim to clarify the relationships between phylogeny and functions.

Phylogenetic tree and gene structures of AoCYPs. Phylogenetic tree was constructed by UPGMA methods using experimentally deduced sequences of isolated AoCYPs and bioinformatically predicted sequences for non-isolated cDNAs. Gene structures of AoCYPs were illustrated by solid line for full-length gene, dashed line for frame-shifted gene, and dotted line indicates non-expressed gene. Green circle, blue diamond, and red square indicate phase-0 intron, phase-1 intron, and phase-2 intron, respectively. Self-sufficient CYP505A3, 505A14, and 505C3 show catalytic P450 domain but not reductase domain
To better understand the molecular aspects of AoCYPs, we analyzed 371 intronic sequences in 121 experimentally validated AoCYPs. The average number of introns was 3.3 in each AoCYP gene, which is higher than the overall average 1.9 among all A. oryzae genes (Wang et al. 2008). The average intron length was 63 bp, twofold shorter than the overall average length among all A. oryzae genes (Wang et al. 2008). These data suggest that AoCYPs genes were organized more vigorously than other genes in A. oryzae. The vast majority of introns conserved the dinucleotide GT at their 5′-end and AG at their 3′-end (Fig. 5a). In addition, the penultimate position from the 3′-end of introns was a pyrimidine nucleotide, and there was significant nucleotide consensus at the 5′-end. Furthermore, 365 introns encoded a characteristic lariat sequence in which the conserved T was usually located at the 18 position. Although the consensus sequences of introns are important for RNA maturation, some peculiar introns such as TA-CC and TG-CC in CYP540B10 and GC-AG in CYP620H9 were identified by comparing sequences of isolated cDNAs with those in genomic database. To the best of our knowledge, this is the first report describing TA-CC and TG-CC in introns from any organisms, whereas GC-AG introns are rarely found in several other organisms (Rep et al. 2006). As shown in Fig. 5b and c, intron length and lengths of lariat sequences showed clear distributions. This characteristic is potentially useful for identification of novel P450s in ascomycetous fungi. Besides intronic sequences, microexons consisting of 6 or 10 nucleotides were found in CYP5111A1, CYP682B2, and CYP5101A1. The lengths of introns flanking microexons (52–75) were close to average size, suggesting that these unique gene structures were probably generated by stepwise insertion of two introns.

Sequence conservation and molecular aspects of AoCYP introns. a Consensus sequences around 5′- and 3′-ends of intron. A, G, T, and C indicate adenosine, guanosine, thymidine, and cytidine, respectively. R and Y indicate purine nucleosides (A or G) or pyrimidine nucleosides (G or T), respectively. D indicates A, G, or T. Asterisk indicates conserved T in lariat sequence. Undiscriminated nucleotides for lariat sequences are listed as ND. b Distribution of introns length. c Distribution of distance for conserved T in lariat sequence
In conclusion, this study describes the molecular diversity of AoCYPs, investigated using experimental and bioinformatic approaches. The experimentally validated sequences and gene structures will enable molecular localization and characterization of novel P450s from ascomycetous fungi. Although further investigations are required to better understand transcriptional and post-transcriptional regulation of AoCYPs, it is clear that sophisticated molecular mechanisms enable superior metabolic performance of A. oryzae. To date, only a few fungal P450s have been functionally characterized. The isolated cDNAs will be useful in advanced studies on functional surveys of AoCYPs using recombinant systems. Potential practical applications of AoCYP will be explored in the near future.
References
Abe K, Gomi K, Hasegawa F, Machida M (2006) Impact of Aspergillus oryzae genomics on industrial production of metabolites. Mycopathologia 162:143–153
Aoyama Y, Noshiro M, Gotoh O, Imaoka S, Funae Y, Kurosawa N, Horiuchi T, Yoshida Y (1996) Sterol 14-demethylase P450 (P45014DM) is one of the most ancient and conserved P450 species. J Biochem 119:926–933
Barbesgaard P, Heldt-Hansen HP, Diderichsen B (1992) On the safety of Aspergillus oryzae: a review. Appl Microbiol Biotechnol 36:569–572
Cheah MT, Wachter A, Sudarsan N, Breaker R (2002) Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature 447:497–500
Chen S, Zhou D (1992) Functional domains of aromatase cytochrome P450 inferred from comparative analyses of amino acid sequences and substantiated by site-directed mutagenesis experiments. J Biol Chem 267:22587–22594
Demain AL, Fang A (2000) The natural functions of secondary metabolites. Adv Biochem Eng Biotechnol 69:1–39
Deng J, Carbone I, Dean RA (2007) The evolutionary history of cytochrome P450 genes in four filamentous Ascomycetes. BMC Evol Biol 7:30
Doddapaneni H, Chakraborty R, Yadav JS (2005) Genome-wide structural and evolutionary analysis of the P450 monooxygenase genes (P450ome) in the white rot fungus Phanerochaete chrysosporium: evidence for gene duplications and extensive gene clustering. BMC Genomics 6:92
Felsenstein J (1989) PHYLIP—phylogeny inference package (version 3.2). Cladistics 5:164–166
Forhman MA (1993) Rapid amplification of complementary DNA ends for generation of full-length complementary DNAs: thermal RACE. Methods Enzymol 218:340–356
Galagan JE, Calvo SE, Borkovich KA et al (2003) The genome sequence of the filamentous fungus Neurospora crassa. Nature 422:859–868
Galagan JE, Calvo SE, Cuomo C et al (2005) Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438:1105–1115
Gotoh O (1993) Evolution and differentiation of P450 genes. In: Omura T, Ishimura Y, Fujii-Kuriyama Y (eds) Cytochrome P-450, 2nd edn. Kodansha, Tokyo, pp 255–272
Graham SE, Peterson JA (1999) How similar are P450 s and what can their differences teach us. Arch Biophys Biochem 369:24–29
Hasemann CA, Ravichandran KG, Boddupalli SS, Peterson JA, Deisenhofer J (1995) Structure and function of cytochrome P450: a comparative analysis of three crystal structures. Structure 3:41–62
Hirokawa T, Boon-Chieng S, Mitaku S (1998) SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379
Hohn TM, Desjardins AE, McCormick SP (1995) The Tri4 gene of Fusarium sporotrichioides encodes a cytochrome P450 monooxygenase involved in trichothecene biosynthesis. Mol Gen Genet 248:95–102
Howe GA, Lee GI, Itoh A, Li L, DeRocher AE (2000) Cytochrome P450-dependent metabolism of oxylipins in tomato. Cloning and expression of allene oxide synthase and fatty acid hydroperoxide lyase. Plant Physiol 123:711–724
Ichinose H, Wariishi H, Tanaka H (1999) Biotransformation of recalcitrant 4-methyldibenzothiophene to water-extractable products using lignin-degrading basidiomycete Coriolus versicolor. Biotechnol Prog 15:706–714
Ikeda K, Nakayashiki H, Kataoka T, Tamba T, Hashimoto Y, Tosa Y, Mayama S (2002) Repeat-induced point mutation (RIP) in Magnaporthe grisea: implications for its sexual cycle in the natural field context. Mol Microbiol 45:1355–1364
Intikhab A, Hubbard SJ, Oliver SG, Rattray M (2007) A kingdom-specific protein domain HMM library for improved annotation of fungal genomes. BMC Genomics 8:97
Kelly DE, Kraevec N, Mullins J, Nelson DR (2008) The CYPome (Cytochrome P450 complement) of Aspergillus nidulans. Fung Genet Biol 46:S53–S61
Kirk TK, Schultz E, Connors WJ, Lorenz LF, Zeikus JG (1978) Influence of culture parameters on lignin metabolism by Phanerochaete chrysosporium. Arch Microbiol 117:277–285
Kitazume T, Takaya N, Nakayama N, Shoun H (2000) Fusarium oxysporum fatty-acid subterminal hydroxylase (CYP505) is a membrane-bound eukaryotic counterpart of Bacillus megaterium cytochrome P450BM3. J Biol Chem 275:39734–39740
Koymans L, Donne-Op Den Kelder GM, Koppele Te JM, Vermeulen NPE (1993) Cytochromes P450: their active site structure and mechanism of oxidation. Drug Metab Rev 25:325–387
Kubodera T, Watanabe M, Yoshiuchi K, Yamashita N, Nishimura A, Nakai S, Gomi K, Hanamoto H (2003) Thiamine-regulated gene expression of Aspergillus oryzae thiA requires splicing of the intron containing a riboswitch-like domain in the 5’-UTR. FEBS Lett 555:516–520
Lah L, Krasevec N, Trontelj P, Komel R (2008) High diversity and complex evolution of fungal cytochrome P450 reductase: cytochrome P450 systems. Fung Genet Biol 45:446–458
Lewis DFL, Watson E, Lake BG (1998) Evolution of the cytochrome P450 superfamily: sequence alignments and pharmacogenetics. Mutat Res 410:245–270
Machida M, Asai K, Sano M et al (2005) Genome sequencing and analysis of Aspergillus oryzae. Nature 438:1157–1161
Machida M, Yamada O, Gomi K (2008) Genomics of Aspergillus oryzae: learning from the history of koji mold and exploration of its future. DNA Res 15:173–183
Matsuzaki F, Wariishi H (2004) Functional diversity of cytochrome P450 s of the white-rot fungus Phanerochaete chrysosporium. Biochem Biophys Res Commun 324:387–393
Montiel MD, Lee HA, Archer DB (2006) Evidence of RIP (repeat-induced point mutation) in transposase sequence of Aspergillus oryzae. Fung Genet Biol 43:439–445
Nakahara K, Hatano K, Usuda K, Shoun H (1993) Cytochrome P-450 55A1 (P-450dNIR) acts as nitric oxide reductase employing NADH as the direct electron donor. J Biol Chem 268:8350–8355
Nakayama N, Takemae A, Shoun H (1996) Cytochrome P450foxy, a catalytically self-sufficient fatty acid hydroxylase of the Fusarium oxysporum. J Biochem 119:435–440
Nelson DR (1998) Metazoan cytochrome P450 evolution. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 121:15–22
Nelson DR (1999) Cytochrome P450 and the individuality of species. Arch Biochem Biophys 369:1–10
Nelson DR, Strobel HW (1988) On the membrane topology of vertebrate cytochrome P450 proteins. J Biol Chem 263:6038–6050
Nelson DR, Kamataki T, Waxman DJ et al (1993) The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial names of enzymes and nomenclature. DNA Cell Biol 12:1–51
Nelson DR, Koymans L, Kamataki T et al (1996) P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenet 6:1–42
Ortiz-de-Montellano PR (2005) Cytochrome P-450: structure, mechanism, and biochemistry, 3rd edn. Kluwer Academic/Plenum Publishers, New York
Paquette SM, Bak S, Feyereisen R (2000) Intron-exon organization and phylogeny in a large superfamily, the paralogous cytochrome P450 genes of Arabidopsis thaliana. DNA Cell Biol 19:307–317
Park J, Lee S, Choi J, Ahn K, Park B, Park J, Kang S, Lee YH (2008) Fungal cytochrome P450 database. BMC Genomics 9:402
Payne GA, Nierman WC, Wortman JR, Pritchard BL, Brown D, Dean RA, Bhatnagar D, Cleveland TE, Machida M (2006) Whole genome comparison of Aspergillus flavus and A. oryzae. Med Mycol 44:9–11
Poulos TL, Finzel BC, Haward AJ (1987) High-resolution crystal structure of P450cam. J Mol Biol 195:687–700
Prieto R, Woloshuk CP (1997) ord1, an oxidoreductase gene responsible for conversion of O-methylsterigmatocytin to aflatoxin in Aspergillus flavus. Appl Environ Microbiol 63:1661–1666
Rep M, Duyvesteijn RGE, Gale L, Usgaard T, Cornelissen BJC, Ma L-J, Ward TJ (2006) The presence of GC-AG introns in Neurospora crassa and other euascomycetes determined from analysis of complete genomes: implications for automated gene prediction. Genomics 87:338–347
Tailor MJ, Richardson T (1979) Applications of microbial enzymes in food systems and in biotechnology. Adv Appl Microbiol 25:7–35
Tamano K, Sano M, Yamane N, Terabayashi Y, Toda T, Sungawa M, Koike H, Hatamoto O, Umitsuki G, Takahashi T, Koyama Y, Asai R, Abe K, Machida M (2008) Transcriptional regulation of genes on the non-syntenic blocks of Aspergillus oryzae and its functional relationship to solid-state cultivation. Fung Genet Biol 45:139–151
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Thore S, Leibundgut M, Ban N (2006) Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science 312:1208–1211
Tijet N, Helvig C, Feyereisen R (2001) The cytochrome P450 gene superfamily in Drosophila melanogaster: annotation, intron-exon organization and phylogeny. Gene 262:189–198
Ullrich V (2003) Thoughts on thiolate tethering. Tribute and thanks to a teacher. Arch Biochem Biophys 409:45–51
Wang K, Ussery DW, Brunak S (2008) Analysis and prediction of gene splice sites in four Aspergillus genomes. Fung Genet Biol 46:S14–S18
Winkler W, Nahvi A, Breaker RR (2002) Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419:952–956
Yoshida Y (1993) Sterol biosynthesis. In: Omura T, Ishimura Y, Fujii-Kuriyama Y (eds) Cytochrome P-450, 2nd edn. Kodansha, Tokyo, pp 93–101
Acknowledgments
We are grateful to Hiroko Tsutsumi and Yoji Hata (Res. Inst., Gekkeikan Sake Co.) for providing A. oryzae strain RIB40 and for valuable technical advices. This research was supported in part by an NIRS Research Grant (Noda Institute for Scientific Research; to H. I.).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Axel Brakhage.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Nazmul Hussain Nazir, K.H.M., Ichinose, H. & Wariishi, H. Molecular characterization and isolation of cytochrome P450 genes from the filamentous fungus Aspergillus oryzae . Arch Microbiol 192, 395–408 (2010). https://doi.org/10.1007/s00203-010-0562-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-010-0562-z