
Abstract
Taurine (2-aminoethanesulfonate) is a widespread natural product whose nitrogen moiety was recently shown to be assimilated by bacteria, usually with excretion of an organosulfonate via undefined novel pathways; other data involve transcriptional regulator TauR in taurine metabolism. A screen of genome sequences for TauR with the BLAST algorithm allowed the hypothesis that the marine gammaproteobacterium Neptuniibacter caesariensis MED92 would inducibly assimilate taurine-nitrogen and excrete sulfoacetate. The pathway involved an ABC transporter (TauABC), taurine:pyruvate aminotransferase (Tpa), a novel sulfoacetaldehyde dehydrogenase (SafD) and exporter(s) of sulfoacetate (SafE) (DUF81). Ten candidate genes in two clusters involved three sets of paralogues (for TauR, Tpa and SafE). Inducible Tpa and SafD were detected in cell extracts. SafD was purified 600-fold to homogeneity in two steps. The monomer had a molecular mass of 50 kDa (SDS-PAGE); data from gel filtration chromatography indicated a tetrameric native protein. SafD was specific for sulfoacetaldehyde with a K m-value of 0.12 mM. The N-terminal amino acid sequence of SafD confirmed the identity of the safD gene. The eight pathway genes were transcribed inducibly, which indicated expression of the whole hypothetical pathway. We presume that this pathway is one source of sulfoacetate in nature, where this compound is dissimilated by many bacteria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Taurine (2-aminoethanesulfonate) is widespread in nature. It occurs in marine invertebrates and it is a major organic solute in mammals, which excrete it in urine (Allen and Garrett 1971; Huxtable 1992; Yancey et al. 2002). The compound is known to serve as a sole source of carbon or of sulfur for the growth of bacteria under aerobic conditions (e.g., Ikeda et al. 1963; Kondo et al. 1971; Uria-Nickelsen et al. 1993), and those processes have been largely elucidated at the biochemical and genetic levels (van der Ploeg et al. 1996; Eichhorn et al. 2000; Kahnert et al. 2000; Kertesz 2000; Cook and Denger 2002, 2006; Wiethaus et al. 2008): key reactions are the desulfonations catalyzed by sulfoacetaldehyde acetyltransferase (Xsc) and taurine dioxygenase (TauD) (van der Ploeg et al. 1996; Cook and Denger 2002), and the proven function of transcriptional regulator TauR (Wiethaus et al. 2008). Recently, taurine has been shown to serve as a sole source of nitrogen for bacterial growth, whereby most organisms remove the amino group and excrete an organosulfonate product into the medium (Denger et al. 2004b; Styp von Rekowski et al. 2005; Weinitschke et al. 2005; see also Chien et al. 1999; Denger et al. 2004a). In Rhodopseudomonas palustris CGA009 the inducible formation of ionic sulfoacetic acid (i.e., sulfoacetate) as a product of the assimilation of taurine-nitrogen was detected (Denger et al. 2004b).
Sulfoacetic acid was first recognized as a biological entity in 1944 in a set of widespread plant alkaloids (erysothiovine and erysothiopine) with curare-like effects (Folkers et al. 1944; Mondon 1970). Sulfoacetic acid was later shown to be a plant and an algal product from (derivatized) sulfoquinovose (the polar head group of the plant sulfolipid) and an algal product from l-cysteate (Shibuya et al. 1963; Lee and Benson 1972; Gupta and Sastry 1988). In mice, traces of sulfoacetate are formed from sulfolactate, which is in equilibrium with l-cysteate (Weinstein and Griffith 1988). Bacteria can also form sulfoacetate from (derivatized) sulfoquinovose (Martelli and Benson 1964). With the exception of the work with R. palustris (Denger et al. 2004b), almost no enzymic process (or the corresponding gene) has been established for any of these reactions or pathways.
Inducible assimilation of taurine-nitrogen by R. palustris CGA009 to yield sulfoacetate was presumed to involve TauR (RPA0105) (Denger et al. 2004b). The uptake system for taurine in R. palustris is unknown. Taurine dehydrogenase, sulfoacetaldehyde dehydrogenase (sulfoacetate formation, dehydrogenase; SafD) and the sulfoacetate exporter (sulfoacetate formation, exporter; SafE) were believed to be RPA0112a-0113, RPA0112 and RPA0111, respectively, (Denger et al. 2004b). However, the taurine dehydrogenase reaction to yield sulfoacetaldehyde could not be assayed in vitro: the enzyme requires a rhodopseudomonadal cytochrome c (Weinitschke et al. 2006). Further, the induction of sulfoacetaldehyde dehydrogenase (SafD) is such that the sulfoacetate and ammonium ions are released into the medium in a very short time, and the cells grow either with the excreted ammonium ion or by fixing dinitrogen (Denger et al. 2004b), which left very low levels of enzyme that were easily lost: the major protein in the separated SafD-fraction was catalase (Weinitschke, unpublished). In taurine degradation in other organisms there is an alternative to taurine dehydrogenase, namely a combination of taurine:pyruvate aminotransferase (Tpa) [EC 2.6.1.77] and alanine dehydrogenase (Ald) [EC 1.4.1.1] (e.g., Styp von Rekowski et al. 2005). Both enzymes in this metabolic context have been purified and characterized elsewhere (e.g., Laue and Cook 2000a, b; Denger et al. 2004a).
Given the experimental problems with R. palustris CGA009 (previous paragraph), we decided to search for an organism, which would allow us (1) to characterize the novel enzyme (sulfoacetaldehyde dehydrogenase; SafD) and (2) to formulate a complete pathway from external taurine to excreted sulfoacetate (Fig. 1a). Screens by means of the BLAST algorithm (Altschul et al. 1997) revealed that the genome of the aerobic, marine gammaproteobacterium Neptuniibacter caesariensis (formerly Oceanospirillum sp.) MED92 (Arahal et al. 2007) includes candidate genes to encode inducible SafD and SafE (see Results).

The hypothesized pathway of taurine uptake, deamination, oxidation and excretion (a) derived from our annotation of gene clusters on the genome of N. caesariensis MED92 (b). The gene clusters represent MED92_12196–MED92_13221 (tauC-tpa1) and MED92_03193–MED92_03208 (tpa2-safE2)
We now report that N. caesariensis MED92 does, indeed, excrete sulfoacetate when utilizing taurine as a source of nitrogen. The pathway is inducible and SafD has been purified and identified.
Materials and methods
Chemicals
Taurine (≥99.5%) was from Fluka and disodium sulfoacetate (>99%) from Acros Organics. Sulfoacetaldehyde, as the bisulfite addition complex, was synthesized and characterized previously (Denger et al. 2001). Phosphonoacetaldehyde was provided by H. B. F. Dixon (Cambridge University). Other chemicals were of the highest purity available and were purchased from Fluka, Merck, Roth, Serva and Sigma.
Organism, growth, harvesting of cells and preparation of cell-free extracts
N. caesariensis MED92T (CCUG 52065; previously Oceanospirillum sp.) (Arahal et al. 2007) was made available by J. Pinhassi (Department of Biology and Environmental Sciences, University of Kalmar, SE-39182 Kalmar, Sweden). Experimental requirements for (1) nitrogen-limiting conditions and (2) the ability to follow the fate of the sulfonate group caused two modifications to the published medium (MacLeod and Hori 1960; Baumann and Baumann 1981). First, the ammonium chloride was removed. Second, the concentration of magnesium sulfate was reduced to 1.0 mM: we found the organism to require the sulfate ion in excess of the sulfur required for growth. The basal medium thus contained: 200 mM NaCl, 50 mM MgCl2, 50 mM Tris–HCl, pH 7.5, 10 mM CaCl2, 10 mM KCl, 0.5 mM K2HPO4, 0.02 mM FeSO4 and trace elements SL6 (Pfennig 1974): after autoclaving, the solution was brought to 1 mM MgSO4. The carbon source was routinely 10 mM sodium succinate and the nitrogen source was routinely 2 mM taurine or 2 mM ammonium chloride. The combined nitrogen in the Tris buffer was not a source of nitrogen for growth.
Precultures (5 ml) of strain MED92 were grown at 30°C in 30-ml screw-cap tubes in a roller. Growth experiments were done on the 50-ml scale in 300-ml Erlenmeyer flasks shaken in a water bath in the dark. Samples were taken at intervals to measure optical density (at 580 nm; OD580), to assay protein, and to determine the concentrations of taurine, sulfoacetaldehyde, sulfoacetate, and the ammonium and sulfate ions. Similar cultures were used to generate small amounts of cells for enzyme assays or for molecular analyses.
Cells for the preparation of total RNA were harvested in the early exponential phase of growth (OD 580 = 0.15) and in late exponential phase (OD 580 = 0.5) by centrifugation at 5,000×g. RNA was extracted immediately.
Cultures (1 l) for protein purification were grown in 5-l Erlenmeyer flasks on a shaker. Cells were harvested at OD580 between 0.4 and 0.5 by centrifugation (15,000×g, 15 min, 4°C), washed with 20 mM Tris–HCl buffer, pH 9.0 (containing 5 mM MgCl2) and stored frozen. This buffer, augmented with 0.05 mg DNAse I ml−1, served as extraction buffer. Cell-free extracts were generated after disruption by three passages through a French pressure cell set at 140 MPa (Junker et al. 1994). The membrane/particulate fraction was pelleted by ultracentrifugation (340,000×g, 40 min, 4°C) and the supernatant fluid was called the soluble fraction.
Analytical methods
Growth was followed as OD580 (for N. caesariensis OD580 = 1 = 300 mg protein l−1) and quantified as protein in a Lowry-type reaction (Cook and Hütter 1981). Taurine and alanine were determined by HPLC after derivatization with 2,4-dinitrofluorobenzene (Laue et al. 1997). The ammonium ion was determined enzymically by its reaction with glutamate dehydrogenase (Bergmeyer and Beutler 1984). Sulfate was measured as the optical density of a suspension of insoluble BaSO4 (Sörbo 1987). Sulfoacetate was routinely determined by ion chromatography with suppression (Denger et al. 2004b) and its identity was confirmed by MALDI-TOF-MS used in the negative-ion mode (Denger et al. 2004b). Sulfoacetaldehyde was derivatized and assayed by HPLC (Cunningham et al. 1998). Protein in extracts was assayed by protein-dye binding (Bradford 1976). Denatured proteins were separated in 12% SDS-PAGE gels and stained with Coomassie-Brillant-Blue R250 (Laemmli 1970). The N-terminal sequence of a blotted protein was determined after Edman-degradation and HPLC separation under contract at TopLab (Martinsried, Germany).
Enzyme assays
Sulfoacetaldehyde dehydrogenase was routinely assayed spectrophotometrically (340 nm) as the reduction of NAD+ at room temperature (about 22°C) (Weinitschke et al. 2005): the reaction mixture (1 ml) contained 50 μmol Tris–HCl buffer pH 9.0, 5 μmol MgCl2, 4 μmol NAD+, 0.1–0.3 mg protein, and 400 nmol sulfoacetaldehyde, with which the reaction was started. Variants of this method were used to determine the stoichiometry of the reaction or to generate data which allowed kinetic constants to be derived by curve-fitting (Ruff et al. 2003). Tpa was assayed discontinuously; samples were taken at intervals, and taurine and alanine were determined (Weinitschke et al. 2005). The assays of SafD and Tpa involved the same buffer, and they could be combined to allow conversion of taurine to sulfoacetate. Alanine dehydrogenase (Ald) was assayed spectrophotometrically as reduction of NAD+ (Laue and Cook 2000b): the positive control was from Rhodococcus opacus ISO-5 (Denger et al. 2004a). Sulfoacetaldehyde acetyltransferase (Xsc) [EC 2.3.3.15] was assayed as release of sulfite from sulfoacetaldehyde with the enzyme from Cupriavidus necator H16 as positive control (Ruff et al. 2003; Weinitschke et al. 2007).
Purification of sulfoacetaldehyde dehydrogenase (SafD)
SafD was stable in Tris–HCl buffer, pH 9.0, which was also the condition of optimal enzyme activity. A two-step purification protocol was developed. The soluble fraction from cells harvested from 4 l of culture was divided into two portions, each of which was diluted 1:1 with 20 mM Tris buffer, pH 9.0 and loaded in separate runs on to an anion-exchange column (Mono Q 10/10, Pharmacia, Freiburg, Germany) equilibrated with the same buffer; the flow rate was 1 ml min−1, and, after ramping to 0.075 M sodium sulfate, proteins were eluted with a linear gradient to 0.325 M sodium sulfate, as described elsewhere (Ruff et al. 2003). SafD eluted at about 0.25 M sodium sulfate. Active fractions were pooled, concentrated using Vivaspin concentrators (10 kDa cut-off; Sartorius, Göttingen, Germany), rebuffered in 20 mM Tris buffer, pH 9.0, containing 1.7 M ammonium sulfate, and subjected to hydrophobic interaction chromatography on Phenyl Superose HR (5/5 column; Pharmacia) at a flow rate of 1.0 ml min−1. A linear decreasing gradient of ammonium sulfate (1.7–0.0 M) in 20 mM Tris buffer, pH 9.0, was applied over 40 min, and SafD eluted at 0 mM ammonium sulfate. Concentrated active fraction was loaded on to a gel filtration column (Superose 12 HR 10/30, Pharmacia) and eluted at a flow rate of 0.4 ml min−1 with 50 mM Tris buffer, pH 9.0 containing 0.15 M sodium sulfate.
Molecular methods
Oligonucleotides were synthesized by Microsynth (Balgach, Switzerland). Taq DNA polymerase and M-MuLV reverse transcriptase were from Fermentas (St Leon-Rot, Germany) and they were used as specified by the supplier. Chromosomal DNA was isolated from bacteria as described by Desomer (Desomer et al. 1991). Total RNA was isolated using the E.Z.N.A. bacterial RNA kit (Omega Bio-Tek, Doraville, USA). Contaminant DNA was removed with RNase-free DNase (Fermentas). The RNA was tested for residual DNA before reverse transcription (RT) by PCR using the primer set NcSafDf-NcSafDr (Table 1). RT and PCR were done as described elsewhere (Innis et al. 1990). The reverse PCR primers listed in Table 1 were used for RT reactions. Positive controls for cDNA integrity after RT were done using the 16S rRNA-specific primers 16S-27f and 16S-533r (Weisburg et al. 1991). As a positive control for PCR reactions the chromosomal DNA of N. caesariensis MED92 was used. PCR products were visualised on 1.5 or 2% agarose gels (Sambrook et al. 1989). A 50-bp DNA ladder (Fermentas) was used.
Software for sequence analyses and accession numbers
Analysis of the draft genome sequence (accession no. NZ_AAOW01000000) of N. caesariensis MED92 was done using the BLAST algorithm on the National Centre for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/). Sequence data were manipulated with different subroutines from the LASERGENE programme package (DNASTAR, Madison, USA). Primers for RT and PCR were designed using the programme Amplify (version 1.2). Transmembrane regions were predicted using the programme TMHMM at the Centre for Biological Sequence Analysis (CBS; http://www.cbs.dtu.dk/services/). The ‘Superfamily’ website (http://supfam.mrc-lmb.cam.ac.uk/SUPERFAMILY/) was used (Gough et al. 2001), as was the ‘Aldehyde dehydrogenase gene superfamily database’ (http://www.aldh.org/index.php) (Lindahl 1992).
Results
Deduction of the hypothetical pathway in N. caesariensis MED92
The regulator, TauR has been associated with (1) taurine degradative genes since their discovery (e.g., Ruff et al. 2003; Brüggemann et al. 2004; Denger et al. 2006; Gorzynska et al. 2006; Baldock et al. 2007), with (2) assimilation of taurine nitrogen in R. palustris CGA009 (Denger et al. 2004b) (where the same genes [RPB_1035-RPB_1039] are found in R. palustris strains HaA2 and TIE-1), and with (3) assimilation of taurine sulfur, where direct evidence of the function of TauR as a regulatory protein is available (Wiethaus et al. 2008). We detected two tauR–like genes (MED92_03198 (tauR2) and MED92_13211 (tauR1)) in the genome of N. caesariensis MED92, and we provisionally annotated the eight open reading frames (ORFs) in the flanking regions (Fig. 1b): the ald gene (MED92_06961) was also examined. The eight annotated genes should encode an ATP binding-cassette transporter for taurine uptake (TauABC) (cf. Eichhorn et al. 2000), two Tpa’s (see supplementary material to Laue et al. 2006), SafD (sulfoacetate formation, dehydrogenase; MED92_03203) (annotated by NCBI as NAD-dependent aldehyde dehydrogenase [superfamily]), and two potential sulfoacetate exporters, SafE1 and SafE2 (annotated by NCBI as members of the DUF81 family, membrane proteins with a domain of unknown function; see Weinitschke et al. 2007). There was no candidate gene to encode sulfoacetaldehyde acetyltransferase (Xsc) for the degradative cleavage of the carbon-sulfonate bond. Further, no candidate gene was detected to encode desulfonative taurine dioxygenase (TauD), which is involved in assimilation of taurine-sulfur (Eichhorn et al. 1997). We thus predicted that taurine would function as a sole source of nitrogen for growth of strain MED92, but not as a source of carbon (or sulfur), and that the taurine-carbon would be excreted by the cell as sulfoacetate (Fig. 1a).
Growth of N. caesariensis MED92 with taurine
N. caesariensis MED92 grew exponentially (μ = 0.42 h−1) in buffered marine-salts medium containing succinate as carbon source, and taurine as the sole source of combined nitrogen (Fig. 2a): similar growth was observed with the ammonium ion as the sole source of nitrogen (μ = 0.48 h−1). There was no growth in the absence of a source of combined nitrogen. Taurine did not serve as a sole source of carbon (not shown). Given the requirement of strain MED92 for 1 mM sulfate in the medium, taurine could not be tested as a source of sulfur for growth.

Growth of N. caesariensis MED92 with taurine as nitrogen source (a). Concentrations of substrate and product are shown as a function of growth (as protein concentration) (b): filled triangles taurine, filled squares sulfoacetate, open circles sulfate ion, open triangles ammonium ion, open squares sulfoacetaldehyde
The utilization of taurine-nitrogen by strain MED92 was concomitant with growth (Fig. 2b) and with the release of a product which was detected by ion chromatography and which co-eluted with authentic sulfoacetate. The identity of this product was confirmed to be sulfoacetate by MALDI-TOF-MS in the negative-ion mode: m/z = 139, identical to the data with authentic material (M = 140) and corresponding to [M – H]−. No sulfoacetaldehyde was detected in the medium (Fig. 2b) and the recovery of the sulfonate moiety of taurine as sulfoacetate was quantitative (Fig. 2b). Correspondingly, neither sulfate nor ammonium ion was released into the medium during growth (Fig. 2b). The overall molar growth yield was 73 ± 7 g protein (mol taurine)−1, identical to the yield with ammonium ion (75 ± 5 g protein (mol NH4 +)−1). There was, thus, mass balance for the nitrogen, carbon and sulfonate moieties of taurine. The specific rate of taurine utilization (and of sulfoacetate formation) was calculated from the specific growth rate and the molar growth yield to be 1.6 mkat (kg protein)−1.
Inducible enzymes in crude extracts of N. caesariensis MED92
Throughout the project, cells of strain MED92 grown with the ammonium ion as sole source of nitrogen (or extracts from these cells) were used in comparison with cells grown with taurine as sole source of nitrogen. Crude extracts of taurine-grown cells of strain MED92 were found to contain activity of Tpa as predicted above, whereas extracts of ammonium-grown cells did not (Table 2), so Tpa was considered to be inducible. Activity of Ald was not detected (Table 2), consistent with the absence of measurable ald-transcript in RT-PCR experiments (Table 2): activity of Ald was measurable in the positive control. No activity of Xsc was detected, consistent with the absence of an xsc-gene.
SafD was detected as an inducible, soluble enzyme (Table 2), which was dependent on the presence of both NAD+ and sulfoacetaldehyde for activity. The maximal specific activity of sulfoacetaldehyde dehydrogenase in extracts of induced cells was about 0.3 mkat (kg protein)−1 in the soluble fraction; there was no activity in the particulate fraction. These data confirmed our hypothesis that SafD is present in N. caesariensis MED92.
The routine assay of SafD involves the bisulfite addition complex of sulfoacetaldehyde, which can be completely oxidized to sulfoacetate (see below). Free sulfoacetaldehyde could be generated via Tpa, and this sulfoacetaldehyde was converted to sulfoacetate in crude extracts of strain MED92. Both free and complexed sulfoacetaldehyde are substrates for SafD.
Purification and some properties of SafD from N. caesariensis MED92
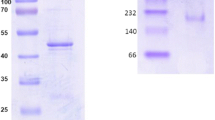
Proteins in the soluble fraction of taurine-grown cells were separated on an anion-exchange column. One peak of SafD activity, measured as sulfoacetaldehyde-dependent reduction of NAD+, was observed. The enzyme eluted with several proteins at about 250 mM sodium sulfate, and analysis by SDS-PAGE showed a 50-kDa protein to be present (Fig. 3), which corresponded to the prediction for SafD (see below). Further separation on a hydrophobic interaction column yielded an effectively homogeneous protein with an apparent molecular mass of about 50 kDa (Fig. 3). The overall recovery of the enzyme was 24%, and a purification factor of 610 was achieved (Table 3).

SDS-PAGE gel of denatured proteins from different stages of purification of SafD. Lanes: 1, 5 molecular mass marker, 2 soluble fraction of taurine-grown cells, 3 pooled fractions after anion-exchange column, 4 separated SafD after hydrophobic interaction chromatography
The N-terminal amino-acid sequence of SafD was determined to be SNTYSLV. This corresponded exactly with amino acids 2–8 in the sequence deduced from the candidate safD gene (MED92_03203). Cleavage of the fMet moiety indicates that the mature SafD has a deduced molecular mass of 49.1 kDa.
Native SafD eluted from a gel filtration column with the retention time corresponding to a molecular mass of about 190 kDa. This is considered to be preliminary evidence for a tetrameric SafD (calculated molecular mass = 196.5 kDa) as the native form of the enzyme.
When excess sulfoacetaldehyde (and NAD+) was added to an enzyme preparation, 0.12 mM sulfoacetate was detected as a product by ion chromatography, and 0.13 mM NADH was formed as determined (after dilution) by the absorption at 340 nm. In a different experiment, 1 mM sulfoacetaldehyde was converted to 1 mM sulfoacetate. Unit stoichiometry was thus observed. The K m-value for sulfoacetaldehyde at saturating concentrations of NAD+ was determined to be 0.12 ± 0.02 mM, so the cell is presumably maintaining the potentially reactive sulfoacetaldehyde at a low concentrations. The K m-value for NAD+ at saturating concentrations of sulfoacetaldehyde was 0.49 ± 0.05 mM, whereas the K m-value for NADP+ was 4.5 ± 1.2 mM. Substrate inhibition by NADP+ was observed at higher concentrations. We presume that the natural electron acceptor in the cell is NAD+, which is presumably present at about 1 mM, whereas NADP+ is usually present at about 0.1 mM (Cook and Schlegel 1978).
The substrate range of SafD was tested: no activity was observed with formaldehyde, acetaldehyde, betaine aldehyde, propionaldehyde, glyceraldehyde, phosphonoacetaldehyde, glyoxylate, glycolaldehyde or 2-oxobutyric acid. None of these compounds was an inhibitor of the enzyme reaction.
The pH optimum of enzyme activity was broad, around pH 9.0 in 50 mM Tris–HCl buffer: 70% of the optimal activity was found in Tris buffer at pH 8.0 and about 80% of optimum in 50 mM CAPS-NaOH buffer in the range pH 9.5–pH 10.5. Some 50% activity was observed in 50 mM potassium phosphate buffer at pH 7.2.
The pure enzyme was stored at 4°C with a daily loss of activity of about 5%. Freezing and thawing led to massive losses of activity.
Transcription of genes involved in sulfoacetate generation in N. caesariensis MED92
The intracellular conversion of taurine to sulfoacetate requires a transport system, presumably TauABC (Fig. 1a, b), to bring the highly ionic nitrogen source into contact with a Tpa(s): each of the candidate genes (tauABC) for the taurine ABC transporter was transcribed inducibly during growth (e.g., tauA in Fig. 4) (Table 2). The presence of Tpa(s), which are the nearest neighbours (in a dendrogram) of the Tpa purified from Bilophila wadsworthia (Laue et al. 2006, supplementary data), had been shown experimentally (Table 2): both genes were transcribed inducibly (Table 2), so both Tpa-paralogues were presumably active. Inducible SafD has been characterized (see above): correspondingly, inducible transcription of safD was detected (Table 2). The product of the SafD reaction is also highly ionic, so an exporter is necessary to remove this sulfoacetate and thus maintain ionic homeostasis in the cell. Transcription of safE1 early and late in the exponential phase was observed (Fig. 4), whereas, transcript from safE2 was detected only early in the growth phase (not shown).

Inducible transcription of tauA (333-bp fragment) and safE1 (229-bp fragment) from N. caesariensis MED92 during the late exponential phase of growth. Lanes: M marker, 50-bp DNA ladder, T cDNA from taurine-grown cells, A cDNA from ammonium-grown cells, + positive control (genomic DNA); − negative control (H2O as template)
Discussion
The physiological data (Fig. 2a, b) confirm the hypothesis (Fig. 1a, b) that N. caesariensis MED92 would utilize taurine as a sole source of nitrogen for growth without cleaving the carbon-sulfonate bond. Balanced growth was observed with quantitative utilization of taurine, and neither sulfate nor ammonium ion was excreted. Further, the sulfonated product was identified as sulfoacetate by mass spectrometry.
Enzyme data confirmed that taurine utilization was inducible (Table 2), as predicted from the presence of tauR gene(s). Indirect evidence of TauR as a regulator in carbon-limited and nitrogen-limited growth (cited above) has been upgraded recently, when a TauR in sulfur-limited growth with taurine was confirmed to be a regulator (Wiethaus et al. 2008). It is unclear whether one or other or both paralogues of TauR (Fig. 1b) is active. Paralogues of TauR are rare in the genomes of taurine-utilizing bacteria: there are about 40 relevant genome sequences, and only three other organisms, all alphaproteobacteria, share the phenomenon, Dinoroseobacter shibae DFL 12, Paracoccus denitrificans Pd1222 and Roseobacter denitrificans OCh 114.
The pathway (Fig. 1a) is apparently initiated by an ABC transporter for taurine (Table 2, Fig. 4), which is widespread (e.g., Eichhorn et al. 2000; Gorzynska et al. 2006). There is support for the presence of both Tpa1 and Tpa2 (Table 2). The key to the formation of sulfoacetate is SafD, which has been purified for the first time (Fig. 3, Table 3). The pathway is completed by release of sulfoacetate (Fig. 2). Both candidates for the exporter, SafE1 and SafE2 (DUF81 proteins), are transcribed inducibly (Table 2), and thus presumably translated. However, only safE1 is transcribed towards the end of growth, and SafE1 may be the main transporter of sulfoacetate. This is another example of DUF81 proteins being associated with the export (or import) of highly charged anions (e.g., Rückert et al. 2005; Weinitschke et al. 2007).
Previous experience with Tpa in a degradative pathway involved Ald, which released the ammonium ion originating from taurine and apparently allowed release of the ion to the medium via an ammonium-methylammonium transporter (Cook and Denger 2002; Gorzynska et al. 2006). Traces of the ammonium ion were detected when taurine served as sole added source of nitrogen for the growth of Klebsiella oxytoca TauN1, which also involves Tpa and Ald (Styp von Rekowski et al. 2005). We speculate that N. caesariensis MED92 does not express Ald (Table 2) to avoid the risk of losing ammonium to the ocean, and we presume that taurine-nitrogen is transferred to biosynthesis via transaminations.
This is the first proof of a pathway of formation of sulfoacetate in a marine bacterium: the similar pathway in terrestrial R. palustris CGA009 (Denger et al. 2004b) can be predicted in two other strains of the organism (HaA2 and TIE-1) given the genome sequences (accession numbers NC_007778 and NZ_ABJR01000003). The bacterial degradation of sulfoacetate, possibly by different pathways, is widespread (Martelli and Sousa 1970; King and Quinn 1997; Cook and Denger 2002) and is currently being elucidated in marine and terrestrial organisms (Weinitschke and Cook, unpublished).
The sulfoacetaldehyde dehydrogenase (SafD) identified here is encoded by MED92_03203 (Table 2, Fig. 1), as predicted. The enzyme acts on the aldehyde group of the electron donor with NAD+ as the electron acceptor. The enzyme thus belongs to the IUBMB Nomenclature Committee’s group EC 1.2.1. We propose the systematic name ‘2-sulfoacetaldehyde:NAD+ oxidoreductase’. The low level of sequence similarity to the presumed SafD in two strains of R. palustris (33%) indicates that the substrate specificity of some aldehyde dehydrogenases is poorly represented in the primary sequence. Nonetheless, the enzyme obviously belongs to the large aldehyde dehydrogenase superfamily with its homomultimers of subunits of about 50-kDa (http://www.aldh.org/index.php). The high specificity of SafD and its affinity for sulfoacetaldehyde (and its bisulfite addition complex) make the enzyme a candidate for the quantitative analysis of mixtures of both sulfoacetaldehyde and its bisulfite adduct, which is an advantage over the direct chemical method (Cunningham et al. 1998).
References
Allen JA, Garrett MR (1971) Taurine in marine invertebrates. Adv Mar Biol 9:205–253
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Arahal DR, Lekunberri I, González JM, Pascual J, Pujalte MJ, Pedros-Alio C, Pinhassi J (2007) Neptuniibacter caesariensis gen. nov., sp. nov., a novel marine genome-sequenced gannaproteobacterium. Int J Sys Evol Microbiol 57:1000–1006
Baldock MI, Denger K, Smits THM, Cook AM (2007) Roseovarius sp. strain 217: aerobic taurine dissimilation via acetate kinase and acetate-CoA ligase. FEMS Microbiol Lett 271:202–206
Baumann P, Baumann L (1981) The marine gram-negative eubacteria: genera Photobacterium, Beneckea, Alteromonas, Pseudomonas and Alcaligenes. In: Starr MP, Stolp H, Trüper HG, Balows A, Schlegel H-G (eds) The prokaryotes. Springer, Berlin, pp 1302–1331
Bergmeyer HU, Beutler H-O (1984) Ammonia. In: Bergmeyer HU (ed) Methods of enzymic analysis. Verlag Chemie, Weinheim, pp 454–461
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brüggemann C, Denger K, Cook AM, Ruff J (2004) Enzymes and genes of taurine and isethionate dissimilation in Paracoccus denitrificans. Microbiology (Reading, UK) 150:805–816
Chien C-C, Leadbetter ER, Godchaux WIII (1999) Rhodococcus spp. utilize taurine (2-aminoethanesulfonate) as sole source of carbon, energy, nitrogen and sulfur for aerobic respiratory growth. FEMS Microbiol Lett 176:333–337
Cook AM, Denger K (2002) Dissimilation of the C2 sulfonates. Arch Microbiol 179:1–6
Cook AM, Denger K (2006) Metabolism of taurine in microorganisms: a primer in molecular diversity? Adv Exp Med Biol 583:3–13
Cook AM, Hütter R (1981) s-Triazines as nitrogen sources for bacteria. J Agric Food Chem 29:1135–1143
Cook AM, Schlegel HG (1978) Metabolite concentrations in Alcaligenes eutrophus H16 and a mutant defective in poly-β-hydroxybutyrate synthesis. Arch Microbiol 119:231–235
Cunningham C, Tipton KF, Dixon HBF (1998) Conversion of taurine into N-chlorotaurine (taurine chloramine) and sulphoacetaldehyde in response to oxidative stress. Biochem J 330:939–945
Denger K, Ruff J, Rein U, Cook AM (2001) Sulfoacetaldehyde sulfo-lyase [EC 4.4.1.12] from Desulfonispora thiosulfatigenes: purification, properties and primary sequence. Biochem J 357:581–586
Denger K, Ruff J, Schleheck D, Cook AM (2004a) Rhodococcus opacus expresses the xsc gene to utilize taurine as a carbon source or as a nitrogen source but not as a sulfur source. Microbiology 150:1859–1867
Denger K, Weinitschke S, Hollemeyer K, Cook AM (2004b) Sulfoacetate generated by Rhodopseudomonas palustris from taurine. Arch Microbiol 182:254–258
Denger K, Smits THM, Cook AM (2006) Genome-enabled analysis of the utilization of taurine as sole source of carbon or nitrogen by Rhodobacter sphaeroides 2.4.1. Microbiology 152:3167–3174
Desomer J, Crespi M, Van Montagu M (1991) Illegitimate integration of non-replicative vectors in the genome of Rhodococcus fascians upon electrotransformation as an insertional mutagenesis system. Mol Microbiol 5:2115–2124
Eichhorn E, van der Ploeg JR, Kertesz MA, Leisinger T (1997) Characterization of α-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J Biol Chem 272:23031–23036
Eichhorn E, van der Ploeg JR, Leisinger T (2000) Deletion analysis of the Escherichia coli taurine and alkanesulfonate transport systems. J Bacteriol 182:2687–2795
Folkers K, Koniuszy F, Shavel J (1944) Erythrina alkaloids. XIV. Isolation and characterization of erysothiovine and erysothiopine, new alkaloids containing sulfur. J Amer Chem Soc 66:1083–1087
Gorzynska AK, Denger K, Cook AM, Smits THM (2006) Inducible transcription of genes involved in taurine uptake and dissimilation by Silicibacter pomeroyi DSS-3T. Arch Microbiol 185:402–406
Gough J, Karplus K, Hughey R, Chothia C (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol 313:903–919
Gupta SD, Sastry PS (1988) The biosynthesis of sulfoquinovosyldiacylglycerol: studies with groundnut (Arachis hypogaea) leaves. Arch Biochem Biophys 260:125–133
Huxtable RJ (1992) Physiological actions of taurine. Physiol Rev 72:101–163
Ikeda K, Yamada H, Tanaka S (1963) Bacterial degradation of taurine. Biochem J 54:312–316
Innis MA, Gelfand DH, Sninsky JJ, White TJ (1990) PCR protocols. A guide to methods and applications. Academic Press, Inc., San Diego
Junker F, Field JA, Bangerter F, Ramsteiner K, Kohler H-P, Joannou CL, Mason JR, Leisinger T, Cook AM (1994) Oxygenation and spontaneous deamination of 2-aminobenzenesulphonic acid in Alcaligenes sp. strain O–1 with subsequent meta ring cleavage and spontaneous desulphonation to 2-hydroxymuconic acid. Biochem J 300:429–436
Kahnert A, Vermeij P, Wietek C, James P, Leisinger T, Kertesz MA (2000) The ssu locus plays a key role in organosulfur metabolism in Pseudomonas putida S-313. J Bacteriol 182:2869–2878
Kertesz MA (2000) Riding the sulfur cycle - metabolism of sulfonates and sulfate esters in Gram-negative bacteria. FEMS Microbiol Rev 24:135–175
King JE, Quinn JP (1997) Metabolism of sulfoacetate by environmental Aureobacterium sp. and Comamonas acidovorans isolates. Microbiology 143:3907–3912
Kondo H, Anada H, Ohsawa K, Ishimoto M (1971) Formation of sulfoacetaldehyde from taurine in bacterial extracts. J Biochem 69:621–623
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London, UK) 227:680–685
Laue H, Cook AM (2000a) Biochemical and molecular characterization of taurine:pyruvate aminotransferase from the anaerobe Bilophila wadsworthia. Eur J Biochem 267:6841–6848
Laue H, Cook AM (2000b) Purification, properties and primary structure of alanine dehydrogenase involved in taurine metabolism in the anaerobe Bilophila wadsworthia. Arch Microbiol 174:162–167
Laue H, Denger K, Cook AM (1997) Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl Environ Microbiol 63:2016–2021
Laue H, Smits THM, Schumacher U, Claros M, Hartemink R, Cook AM (2006) Identification of Bilophila wadsworthia in enrichment cultures by specific PCR which targets the taurine:pyruvate aminotransferase gene. FEMS Microbiol Lett 261:74–79
Lee RF, Benson AA (1972) The metabolism of glyceryl [35S]sulfoquinovoside by the coral tree, Erythrina crista-galli, and alfalfa, Medicago sativa. Biochim Biophys Acta 261:35–37
Lindahl R (1992) Aldehyde dehydrogenases and their role in carcinogenesis. Crit Rev Biochem Mol Biol 27:283–335
MacLeod RA, Hori A (1960) Nutrition and metabolism of marine bacteria VIII: tricarboxylic acid cycle enzymes in a marine bacterium and their response to inorganic salts. J Bacteriol 80:464–471
Martelli HL, Benson AA (1964) Sulfocarbohydrate metabolism 1. Bacterial production and utilization of sulfoacetate. Biochim Biophys Acta 93:169–171
Martelli HL, Sousa SM (1970) Biochemistry of sulfonic compounds III. Formation of a two-carbon compound during the oxidation of sulfoacetate by a Pseudomonas strain. Biochim Biophys Acta 208:110–115
Mondon A (1970) Erythrina alkaloids. In: Pelletier SW (ed) Chemistry of the alkaloids. Van Nostrand Reihhold, New York, pp 173–198
Pfennig N (1974) Rhodopseudomonas globiformis, sp. n., a new species of the Rhodospirillaceae. Arch Microbiol 100:197–206
Rückert C, Koch DJ, Rey DA, Albersmeier A, Mormann S, Pühler A, Kalinowski J (2005) Functional genomics and expression analysis of the Corynebacterium glutamicum fpr2-cysIXHDNYZ gene cluster involved in assimilatory sulphate reduction. BMC Genomics 6:121
Ruff J, Denger K, Cook AM (2003) Sulphoacetaldehyde acetyltransferase yields acetyl phosphate: purification from Alcaligenes defragrans and gene clusters in taurine degradation. Biochem J 369:275–285
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbour
Shibuya I, Yagi T, Benson AA (1963) Sulfonic acids in algae. In: Japanese-society-of-plant-physiologists (ed) Studies on microalgae and photosynthetic bacteria. The University of Tokyo Press, Tokyo, pp 627–636
Sörbo B (1987) Sulfate: turbidimetric and nephelometric methods. Methods Enzymol 143:3–6
Styp von Rekowski K, Denger K, Cook AM (2005) Isethionate as a product from taurine during nitrogen-limited growth of Klebsiella oxytoca TAU-N1. Arch Microbiol 183:325–330
Uria-Nickelsen MR, Leadbetter ER, Godchaux WIII (1993) Sulfonate utilization by enteric bacteria. J Gen Microbiol 139:203–208
van der Ploeg JR, Weiss M, Saller E, Nashimoto H, Saito N, Kertesz MA, Leisinger T (1996) Identification of sulfate starvation-regulated genes in Escherichia coli: a gene cluster involved in the utilization of taurine as a sulfur source. J Bacteriol 178:5438–5446
Weinitschke S, Denger K, Cook AM, Smits THM (2007) The DUF81 protein TauE in Cupriavidus necator H16, a sulfite exporter in the metabolism of C2-sulfonates. Microbiology 193:3055–3060
Weinitschke S, Denger K, Smits THM, Hollemeyer K, Cook AM (2006) The sulfonated osmolyte N-methyltaurine is dissimilated by Alcaligenes faecalis and by Paracoccus versutus with release of methylamine. Microbiology 152:1179–1186
Weinitschke S, Styp von Rekowski K, Denger K, Cook AM (2005) Sulfoacetaldehyde is excreted quantitatively by Acinetobacter calcoaceticus SW1 during growth with taurine as sole source of nitrogen. Microbiology 151:1285–1290
Weinstein CL, Griffith OW (1988) Cysteinesulfonate and β-sulfopyruvate metabolism. Partitioning between decarboxylation, transamination, and reduction pathways. J Biol Chem 263:3735–3743
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Wiethaus J, Schubert B, Pfander Y, Narberhaus F, Masepohl B (2008) The GntR-like regulator TauR activates expression of taurine utilization genes in Rhodobacter capsulatus. J Bacteriol 190:487–493
Yancey PH, Blake WR, Conley J (2002) Unusual organic osmolytes in deep-sea animals: adaptations to hydrostatic pressure and other perturbants. Comp Biochem Physiol A Mol Integr Physiol 133:667–676
Acknowledgments
We are grateful to Stefanie Eschenbächer for data generated during a practical course for advanced students, to Jarone Pinhassi (Kalmar, Sweden) for making N. caesariensis MED92 available to us with advice on its growth, and to Hal Dixon (Cambridge, England) for phosphonoacetaldehyde. We are also grateful to many sequencing organizations, especially to the Moore Foundation for sequencing the genome of N. caesariensis and making the data generally available. This work was supported by the University of Konstanz, IAESTE, the LBS Stiftung ‘Umwelt und Wohnen’, the Centre for Applied Genomics (Prague) (Z.K.) and the DFG (S.W.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Friedrich Widdel.
Rights and permissions
About this article
Cite this article
Krejčík, Z., Denger, K., Weinitschke, S. et al. Sulfoacetate released during the assimilation of taurine-nitrogen by Neptuniibacter caesariensis: purification of sulfoacetaldehyde dehydrogenase. Arch Microbiol 190, 159–168 (2008). https://doi.org/10.1007/s00203-008-0386-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-008-0386-2