
Abstract
Great genetic diversity was revealed among 75 rhizobal isolates associated with Vicia faba grown in Chinese fields with AFLP, ARDRA, 16S rDNA sequencing, DNA–DNA hybridization, BOX-PCR and RFLP of PCR-amplified nodD and nodC. Most of the isolates were Rhizobium leguminosarum, and six isolates belonged to an unnamed Rhizobium species. In the homogeneity analysis, the isolates were grouped into three clusters corresponding to (1) autumn sowing (subtropical) region where the winter ecotype of V. faba was cultivated, (2) spring sowing (temperate) region where the spring ecotype was grown, and (3) Yunnan province where the intermediate ecotype was sown either in spring or in autumn. Nonrandom associations were found among the nod genotypes, genomic types and ecological regions, indicating an epidemic symbiotic gene transfer pattern among different genomic backgrounds within an ecological region and a relatively limited transfer pattern between different regions. Conclusively, the present results suggested an endemic population structure of V. faba rhizobia in Chinese fields and demonstrated a novel rhizobium associated with faba bean.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Faba bean (Vicia faba L.) is a leguminous crop cultivated in 57 countries around the world for as long as 6,000 years (Bond et al. 1985; Duke 1981; George 1999). Many botanical varieties have emerged in this species and they could be mainly categorized as winter and spring ecotypes (Ye 2003). V. faba forms nitrogen-fixing root nodules with Rhizobium leguminosarum bv. viciae (Jordan 1984). Previously, diversity of V. faba rhizobia has been investigated in various studies, and they were mainly for the rhizobial populations isolated from the same region (Laguerre et al. 1996, 2003) or for comparison with R. leguminosarum strains isolated from other legume species (Mutch and Young 2004; van Berkum et al. 1995). Comparison of rhizobial populations from different geographic regions was rarely studied (Mutch et al. 2003). There was no study of the rhizobia in association with faba bean ecotypes.
In the present study, we made a first comparative characterization of genomic and nod gene diversity of rhizobial populations associated with the spring, winter, and intermediate ecotypes (Ye 2003) of faba bean respectively in temperate region, subtropical region and plateau region in subtropics in China.
Vicia faba was introduced to China about 4,000–5,000 years ago according to archaeological remains (Bond 1985; Thompson and Kelly 1957; Ye 2003) and it has been cultivated in 25 Chinese provinces. The yield of dry faba bean in China covered about 46% of total world production in 2000–2002 (FAO). The faba bean producing area in China could be divided into two main ecological regions: autumn sowing region where the faba bean belongs to winter ecotype and is sown in autumn; and spring sowing region where the spring ecotype faba bean is sown in spring (Ye 2003). In addition, Yunnan province was considered as an intermediate region where the intermediate ecotype of faba bean was sown in either spring or autumn (Ye 2003).
Since the successful spread of legume crops depends critically on their microsymbionts (Howieson et al. 2000; Lohrke et al. 1996; Mutch et al. 2004; Weir et al. 2004), diverse rhizobial communities associated with V. faba might have established in Chinese soils. Considering that the rhizobia of V. faba in Chinese soils have not been systematically studied and that the rhizobia-legume mutualism is essential in the sustainable agriculture (Graham and Vance 2000, 2003; Resh et al. 2002), we were interested in investigating the rhizobia associated with V. faba grown in Chinese fields. The aim of this work was to clarify the genetic diversity of the rhizobia nodulating different ecotypes of V. faba grown in different regions of China.
Materials and methods
Isolation of rhizobia
Root nodules were collected from V. faba plants grown on 56 sites of 14 Chinese provinces (Supplementary Table A; Fig. 1). Four subtropical provinces Anhui (11 sites), Jiangxi (10 sites), Hubei (11 sites) and Guangxi (2 sites) are located in the autumn sowing region. The nine temperate provinces, with 1–5 sites each, are located in the spring sowing region. Located in a plateau, the subtropical province Yunnan (two sites) is the intermediate ecological region where the faba bean is sown either in autumn or in spring. The Tibetan nodules were obtained with plant trapping method by inoculating the soil sample to the seedlings (Vincent 1970).

Map of China showing the sampling sites. Sampling sites were marked with points. The numbers in parenthesis represent provinces of China: (1) Liaoning, (2) Hebei, (3) Inner Mongolia, (4) Shanxi, (5) Ningxia, (6) Gansu, (7) Qinghai, (8) Xinjiang and (9) Tibet are temperate provinces located in spring sowing region; (10) Yunan province is located in subtropical plateau region where intermediate ecotype of faba bean is sown either in autumn or spring; (11) Guangxi, (12) Jiangxi, (13) Hubei and (14) Anhui are subtropical provinces located in autumn sowing region
A standard procedure and YMA medium (Vincent 1970) were used to isolate rhizobia from root nodules (one nodule per plant). The isolates were purified by repeatedly streaking and their purity was checked by colony morphology and microscopic examination of cellular morphology. The nodulation ability was confirmed by inoculation of the isolates on V. faba seedlings (Vincent 1970). A total of 75 rhizobial isolates were obtained and all of them were fast-growing, acid producing bacteria (Jordan 1984). The isolates were incubated and stored as described previously (Liu et al. 2005).
AFLP genomic fingerprinting
The DNAs were extracted from bacteria, restricted by EcoRI and MseI and ligated with EcoRI adapter and MseI adapter according to the methods of Terefework et al. (2001). Primers EcoRI-gc (5′-GAC TGC GTA CCA ATT CGC-3′) and MseI-cg (5′-GAT GAG TCC TGA GTA ACG-3′) with two selective nucleotides at the 3′ ends were used for selective PCR amplification (Terefework et al. 2001). The amplified fragments were separated by electrophoresis in 5% denaturing polyacrylamide gel, and the resulting gels were stained with silver staining method (Terefework et al. 2001). The Pearson correlation coefficient and UPGMA method were used to generate dendrogram from the AFLP patterns with the GelCompar program.
Genomic fingerprinting of bacteria by BOX-PCR
DNAs extracted with the same method (Terefework et al. 2001) were used as templates for repetitive extragenic palindromic PCR with the BOXA1R primer (Koeuth et al. 1995) and the procedure of Nick et al. (1999). The amplified products (8 μl) were separated by electrophoresis in 2% (w/v) agarose gels containing ethidium bromide (0.5 μg ml−1) and photographed under UV light. The Dice similarity coefficient and UPGMA method were used to generate dendrogram from the BOX-PCR patterns with the GelCompar program.
Amplified 16S rDNA restriction analysis (ARDRA) and sequencing of 16S rDNA
Primers fD1 and rD1 and the PCR procedure described by Weisburg et al. (1991) were used to amplify almost complete 16S rDNA. The PCR products (5 μl) were digested separately by the restriction endonucleases AluI, MboI, MspI and HaeIII. The restriction patterns were separated by electrophoresis in 3% (w/v) of agarose gels supplied with ethidium bromide (0.5 μg ml−1) and photographed under UV light. The ARDRA patterns were analyzed as mentioned in BOX-PCR analysis.
Based on the results of BOX-PCR and 16S rDNA PCR-RFLP, representative strains were chosen for direct sequencing of the 16S rDNA PCR products (Vinuesa et al. 1998) with the same primers. Clustal W software (Jeanmougin et al. 1998; Thompson et al. 1994) was used to align the acquired sequences and the related sequences obtained from the GenBank database by blast searching. A neighbor-joining tree was reconstructed using the Jukes–Cantor distances and bootstrapped with 1,000 replications with program of MEGA3.1 (Kumar et al. 2004).
DNA–DNA hybridization
A phenol–chloroform extraction protocol (Marmur 1961) was used to isolate and to purify total DNAs from the V. faba rhizobia. The G + C content of V. faba rhizobia was determined by the thermal melting profile method (De Ley 1970) using Escherichia coli K12 as standard. DNA–DNA relatedness was determined by the initial renaturation rate method (De Ley et al. 1970).
Restriction fragment length polymorphism (RFLP) of PCR amplified nodulation genes
For most test bacteria, primers NBA12 and NBF12′ and the procedure described by Laguerre et al. (1996) were used to amplify the complete nodD gene. In several isolates, this primer pair was not functional and primers NBA12 and Y6 were used instead to amplify an internal nodD fragment (Mutch and Young 2004; Zézé et al. 2001). Primers NodCfor540 and NodCrev1160 and PCR procedure of Sarita et al. (2005) were used to amplify a 640 bp fragment of nodC gene. The PCR products (5 μl) were digested separately with the restriction endonucleases MspI, HinfI, AluI, MboI and DdeI (for nodD) or with AfaI, DdeI, HaeIII, HinfI, MspI and TaqI (for nodC). The electrophoresis and subsequent analysis of restriction fragments were the same as in ARDRA.
Homogeneity analysis
The relationships among ecological regions, genomic types and nod genotypes were estimated by homogeneity analysis with the HOMALS 1.0 program in SPSS 11.0 package (by Data Theory Scaling System Group, Faculty of Social and Behavioral Sciences, Leiden University, The Netherlands). In this analysis, the ecological region, nod genotype and genomic type were treated as three variables. Three levels were contained in variable of ecological region as mentioned in the section of rhizobia isolation: Yunnan, subtropical region, and temperate region. Thirteen levels corresponding to the 13 nod genotypes were included in the variable of nod genotype. The 12 genomic types were 12 levels in the variable of genomic type. The result of homogeneity analysis was presented in a two-dimension figure, in which different levels of the variables were grouped.
Results and discussion
Genomic diversity of V. faba rhizobia
AFLP, BOX-PCR, ARDRA, sequencing of 16S rDNA and DNA–DNA hybridization were used to estimate the genomic diversity of the rhizobial populations in the present study.
AFLP analysis
In studies of rhizobial diversity, similarities of 50 and 60% have been used to define the AFLP groups. However, these similarity levels did not produce meaningful taxonomic groups in many cases (Andronov et al. 2003). In the present study, AFLP analysis was applied to a subset of isolates because other 18 isolates were obtained later and added in subsequent analyses. Among the test isolates, no identical AFLP patterns were found, demonstrating that the method was powerful to differentiate closely related bacteria. The similarity level of 38% was used in this study to define the AFLP groups because the reference strains for different R. leguminosarum biovars were separated and the isolates formed clearly separated groups at this similarity. Four AFLP groups were drawn (Table 1 and Supplementary Fig. A) among the isolates. R. leguminosarum bv. viciae USDA 2370T (van Berkum et al. 1995) and bv. trifolii 162 × 68 were single strains distantly related to the isolates.
Using AFLP and other techniques, Andronov et al. (2003) showed that diversity in R. galegae bv. orientalis corresponds well with the host plant variation. However, the AFLP groups were not clearly related to the host plant ecotypes in our study, because AFLP groups 1, 3 and 4 were isolated from different ecotypes of faba bean, which was similar to the results of Wolde-meskel et al. (2004) with rhizobia of Acacia spp. and Sesbania sasban.
BOX-PCR
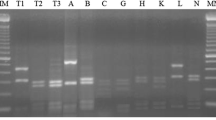
BOX-PCR was attractive for its high-resolution and good reproducibility, and had been widely used to reveal the genetic diversity of closely related bacterial strains (Cho et al. 2000; Healy et al. 2005; Rademaker et al. 2000; Vinuesa et al. 1998). In this study, 75 isolates were used and they were divided into 12 BOX clusters at similarity level of 80% (Table 1; Fig. 2). Similar to the AFLP analysis, USDA 2370T and 162 × 68 were ungrouped strains. BOX clusters 4, 6 and 12 contained isolates originated from two ecological regions. The remaining clusters contained isolates originated from either subtropical region or temperate region (Table 1). In this study, great genomic diversity was revealed in V. faba rhizobia with BOX PCR. In the case of BOX clusters 4 and 5, the isolates were completely the same as AFLP groups 1 and 2, respectively. Two and five BOX clusters were distinguished in AFLP groups 4 and 3, respectively.

Genomic diversity of Vicia faba rhizobia revealed by BOX-PCR. The dendrogram was constructed by the UPGMA method in the GelCompar program
ARDRA and sequencing analysis
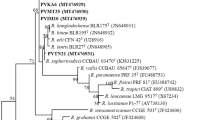
The 75 isolates and 19 reference strains of defined species in Rhizobium, Sinorhizobium, Mesorhizobium, Bradyrhizobium and Agrobacterium were used. Three rDNA types were defined in the isolates (Table 1) and they were grouped in the genus Rhizobium (Supplementary Fig. B). Except CCBAU 85003 and 6 isolates in AFLP group 2, the remaining 68 isolates were found in rDNA type 1 together with R. leguminosarum USDA 2370T and 162 × 68. CCBAU 85003 formed rDNA type 2 that was most similar to R. leguminosarum. The 6 isolates in AFLP group 2 were defined as rDNA type 3 that had RFLP patterns with similarity of 90% to those of R. leguminosarum and R. tropici type strains. The ARDRA relationships were supported by the phylogeny of 16S rDNA (Fig. 3). A total of 18 strains representing the AFLP groups and BOX clusters were used in sequence analysis. Most of the isolates had 99.7–100% of 16S rDNA sequence similarities with published R. leguminosarum biovars. Two isolates of AFLP group 2 had 99.5% of similarities with R. etli, 99.2–99.3% with R. leguminosarum biovars, and 98.7–95.5% with other Rhizobium species.

Phylogenetic tree of 16S rDNA of V. faba rhizobia isolated from Chinese fields (marked with asterisk). This tree was constructed from sequences of the 16S rRNA genes using the Neighbor-Joining method in MEGA3.1. Bootstrap probability values above 70% are indicated at the branch points. The bar represents a 0.5% nucleotide divergence
DNA–DNA hybridization
This analysis was performed only for the AFLP group 2 (BOX cluster 5) because these isolates were quite different from R. leguminosarum. The G + C mol% (Tm) of these isolates was about 62%, within the range of Rhizobium (Jordan 1984). In the DNA–DNA hybridization, DNA relatedness more than 70% was detected among the isolates within AFLP group 2 (BOX group 5), while the representative isolate CCBAU 33202 had 14 and 19% of DNA relatedness with R. leguminosarum USDA 2370T and R. etli CFN42T. These results indicated that the AFLP group 2 (BOX cluster 5) represented a genomic species different from both species.
Based upon the results above, the AFLP group 2 (BOX cluster 5) was a genomic species that might represent a novel species in Rhizobium. The other genomic groups were R. leguminosarum as indicated in RFLP and phylogenetic analysis of 16S rDNA (Supplementary Fig. B and Fig. 3). However, they were all easily distinguished from USDA 2370T and 162 × 68 with high-resolution method AFLP or BOX-PCR. This finding was coincident with van Berkum et al. (1995) that the faba bean rhizobia were genetically distinguishable from the R. leguminosarum strains isolated from other hosts. Our results in this study confirmed that most of the V. faba rhizobia were R. leguminosarum as documented earlier (Bond et al. 1985; Jordan 1984; Mutch et al. 2003; Mutch and Young 2004; Young et al. 2003), but another unnamed Rhizobium species also nodulated faba bean.
Diversity of nod genes of V. faba rhizobia
Nodulation gene characterization
In nod gene clusters, nodD is a regulation gene and it is related to the host specificity. The gene nodC is a common nodulation gene. In earlier studies, nodD genotype had been used to reveal the diversity of R. leguminosarum bv. viciae and specificity of symbiosis between this bacterium and its hosts (Laguerre et al. 1996, 2003; Louvrier et al. 1996; Mutch et al. 2003; Mutch and Young 2004; Zézé et al. 2001). The nodC gene was also recently used to study the nodulation gene diversity of soil rhizobial population (Sarita et al. 2005).
In this study, neither nodD nor nodC region was amplified from CCBAU53093-1. The nodC fragments were amplified from the remaining 74 isolates and the two reference strains for R. leguminosarum. With primers NBA12 and NBF12′, nodD regions (1,200–1,400 bp) were amplified from 71 isolates and from USDA 2370T. Fragments of nodD (about 900 bp) were amplified from isolates CCBAU 03058, CCBAU 65315, CCBAU 43235 and R. leguminosarum 162 × 68 with primers NBA12 and Y6. We combined the RFLP patterns generated from PCR-products with both primer sets because the amplification condition also reflected the genetic difference and typing rather than phylogenetic analysis was considered herein.
In RFLP analysis, the reference strains USDA 2370T and 162 × 68 had nodC and nodD gene types different from each other and from the isolates. A total of 13 nodD gene types were defined among the isolates (Fig. 4). The nodD types B and D dominated respectively in the subtropical region (33/37) and in the temperate region (15/30). The other nodD types were composed of 1–4 isolates. The nodD types A, B, C and N were found in two or three ecological regions. The remaining types were found in one of the three regions, temperate region, subtropical region, or Yunnan.

Simplified dendrogram showing the nodD diversity in V. faba rhizobia defined by RFLP of amplified nodD fragments. The dendrogram was generated with UPGMA method from the RFLP patterns in the GelCompar program. Asterisk RFLP patterns of about 900 bp nodD fragments amplified from isolates CCBAU 03058 (type N), CCBAU 65315 (type N), CCBAU 43235 (type E) and 162 × 68 (type O) with primers NBA12 and Y6 were included together with other patterns obtained with RFLP analysis of PCR products with primers NBA12 and NBF12′, because the amplification condition also reflected the genetic difference
A total of 5 nodC RFLP patterns were found in the isolates (Table 1 and supplementary Fig. C). The nodC gene types B, D and E were found in two or three ecological regions, while nodC types B, D, E was dominating respectively in Yunnan (5/7), temperate region (15/30) and subtropical region (36/37).
These results showed that the nodC gene was rather conserved and nodD was more diverse. A nodC gene type could combine with as much as five nodD types (in the case of nodC type B), but no reverse sample was detected. This situation could be explained as that the nodD and nodC had coevolved and nodD changed faster than nodC.
Previously, it was found that the V. faba rhizobia in European soils harbored the dominant nodD type g on the basis of its HaeIII restriction pattern (Laguerre et al. 2003; van Berkum et al. 1995). But Mutch et al. (2003) and Mutch and Young (2004) revealed that V. faba rhizobia in Jordan had nodD types quite different from those in European soils. In Chinese field, different dominating nod types were found in subtropical region, temperate region and Yunnan. Thus, we might suggest the endemicity of nod types of V. faba rhizobia.
Associations among nod gene types, genomic backgrounds and geographic origins of V. faba rhizobia
Homogeneity analysis
This analysis was performed to estimate the interactions among the genomic types, nod types and ecological regions. The genomic types were defined same as the BOX clusters because the combined grouping results of ARDRA, AFLP and BOX-PCR were identical to the BOX clusters (Table 1). The nod types were designed by combining the nodD and nodC gene types, and the letters representing nodD gene types were used to name the nod types since they were exactly the same (Table 1).
In the generated two-dimension figure (Fig. 5), three groups were found based upon the distances among the genomic types, nod types and ecological origins. Different levels in the same variable located in the same group meant that they might have similar characteristics in certain situations. For example, four nod types J, K, L and N had one common genomic background (genomic type 12), and isolates of genomic types 1, 2, 5 mainly harbored one common nod type B and all originated from subtropical region. The distance between different variables in the same cluster represented their relative correlation. The genomic types 3, 6, 7, 8, 9, 10, 11 and nod types A, C, D, F, G, I were related to temperate region. Genomic types 1, 2, 4, 5 and nod types B, E, M were related to subtropical region. Genomic type 12 and nod types L, N, J and K formed a group related to Yunnan. The nod types C, N and genomic type 6 were distantly related to the ecological regions in each of the two groups, because all of them were found in two different ecological regions.

Homogeneity analysis of ecological regions, genomic groups and nodulation genotypes. The Dimensions 1 and 2 were the linear combination of the three variables and may have no real meaning. Eigen value for Dimension 1 and 2 was 0.938 and 0.666, respectively, indicating both the dimensions were closely related to all the three variables. The discrimination measures were 0.942, 0,953 and 0.931, respectively for ecological region, genomic type, and nod type in Dimension 1; and were 0.759, 0.921 and 0.949, respectively for the three variables in Dimension 2. These discrimination measures indicated that the two dimensions could distinguish all the variables with high confidence
It had been revealed that symbiotic genotype of rhizobia isolated from faba bean appeared to be the determinant of the success in nodule occupancy of rhizobial genotypes independent of the associated genomic background (Laguerre et al. 2003; Louvrier et al. 1996; Mutch and Young 2004). However, our results in Fig. 5 indicated that the nod types were not randomly associated with the genomic types and the associations between nod type and genomic types displayed endemicity, despite few noises. For example, the nod type B was mainly harbored by subtropical isolates in five genomic types and by only a temperate isolate, while nod type D was only found in five genomic types in temperate region. According to the plasmid/chromosome evolutionary patterns proposed by Souza et al. (1997), an epidemic symbiotic gene transfer pattern among different genomic backgrounds within an ecological region and relatively limited transfer pattern between different regions could be deduced from these results. Moreover, as shown in Table 1, it was also revealed that one genomic type could harbor different nod types at different field sites, such as the cases of genomic types 4, 5, 6, 8, 9, 11 and 12. This phenomenon might be related to the survival strategy of V. faba rhizobia because different nod genotypes could help a genomic type to be competitive in nodulation with different cultivars of V. faba on different field sites.
In the homogeneity analysis, the ecological regions could be replaced by ecotypes of faba bean, as mentioned in introduction. Then the non-random correlation between the ecological regions and the genomic or nod types also represented the same correlation between faba bean ecotypes and the rhizobia genomic or nod genotypes. From Table 1, it was clear that different sampling sites in the same ecological region could harbor rhizobia with similar genomic background and nod type, such as BOX cluster 1 (with nod type B) included isolates originated from 11 sites in three provinces, BOX cluster 2 (with nod type B) from eight sites in four provinces, BOX cluster 3 (with nod type D) from three sites in two distant provinces. Moreover, the same site could harbor rhizobia in different genomic and nod types, as the cases of sites 9 and 11 in Shanxi and site 21 in Yunnan. Considering that the sampling sites covered a vast territory with many different soil types and other environmental characters, it could be estimated that the faba bean ecotype might be the determinant for the distribution of rhizobial genomic and nod types. However, further experiments were needed to clearly confirm this association or coevolution.
Conclusively, the present study revealed a novel microsymbiont (Rhizobium sp.) of V. faba in addition to R. leguminosarum. Furthermore, the population of V. faba rhizobia displayed endemic distributions in Chinese fields. The nonrandom association V. faba ecotypes (or ecological origins), rhizobial chromosomal backgrounds and nodulation genotypes should be considered in screening rhizobial inoculants in agricultural production.
References
Andronov EE, Terefework Z, Roumiantseva ML, Dzyubenko NI, Onichtchouk OP, Kurchak ON, Dresler-Nurmi A, Young JPW, Simarov BV, Lindrström K (2003) Symbiotic and genetic diversity of Rhizobium galegae isolates collected from the Galega orientalis gene center in the Caucasus. Appl Environ Microbiol 69:1067–1074
Bond DA, Lawes DA, Hawtin GC, Saxena MC, Stephens JH (1985) Faba bean (Vicia faba L.). In: Summerfield RJ, Roberts EH (eds) Grain legume crops Collins professional and technical books. William Collins Sons & Co. Ltd, London, pp 199–265
Cho JC, Tiedje JM (2000) Biogeography and degree of endemicity of fluorescent Pseudomonas strains in soil. Appl Environ Microbiol 66:5448–5456
De Ley J (1970) Reexamination of the association between melting point, buoyant density, and chemical base composition of DNA. J Bacteriol 101:738–754
De Ley J, Cattoir H, Reynaerts A (1970) The quantitative measurement of DNA hybridization from renaturation rates. Eur J Biochem 12:133–142
Duke JA (1981) Handbook of legumes of world economic importance. Plenum Press, New York, pp 275–279
George RAT (1999) Vegetable seed production, 2nd edn. CABI Publishing, Wallingford, pp 204–205
Graham PH, Vance CP (2000) Nitrogen fixation in perspective: an overview of research and extension needs. Field Crops Res 65:93–106
Graham PH, Vance CP (2003) Legumes: importance and constraints to greater use. Plant Physiol 131:872–877
Healy M, Huong J, Bittner T, Lising M, Frye S, Raza S, Schrock R, Manry J, Renwick A, Nieto R, Woods C, Versalovic J, Lupski JR (2005) Microbial DNA typing by automated repetitive-sequence-based PCR. J Clin Microbiol 43:199–207
Howieson JG, O’Hara GW, Carr SJ (2000) Changing roles for legumes in Mediterranean agriculture: developments from an Australian perspective. Field Crops Res 65:107–122
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ (1998) Multiple sequence alignment with Clustal x. Trends Biochem Sci 23:403–405
Jordan DC (1984) Genus I. Rhizobium Frank 1889, 338AL. In: Krieg NR, Holt G (eds) Bergey’s manual of systematic bacteriology, vol 1. Williams and Wilkins, Baltimore, pp 235–242
Koeuth T, Versalovic J, Lupski JR (1995) Differential subsequence conservation of interspersed repetitive Streptococcus pneumoniae BOX elements in diverse bacteria. Genome Res 5:408–418
Kumar S, Tamura K, Nei M (2004) MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Laguerre G, Mavingui P, Allard MR, Charnay MP, Louvrier P, Mazurier SI, Rigottier-Gois L, Amarger N (1996) Typing of rhizobia by PCR DNA fingerprinting and PCR-restriction fragment length polymorphism analysis of chromosomal and symbiotic gene regions: application to Rhizobium leguminosarum and its different biovars. Appl Environ Microbiol 62:2029–2036
Laguerre G, Louvrier P, Allard MR, Amarger N (2003) Compatibility of rhizobial genotypes within natural populations of Rhizobium leguminosarum biovar viciae for nodulation of host legumes. Appl Environ Microbiol 69:2276–2283
Liu J, Wang ET, Chen WX (2005) Diverse rhizobia associated with woody legumes Wisteria sinensis, Cercis racemosa and Amorpha fruticosa grown in the temperate zone of China. Syst Appl Microbiol 28:465–477
Lohrke SM, Orf JH, Sadowsky MJ (1996) Inheritance of host controlled restriction of nodulation by Bradyrhizobium japonicum strain USDA 110. Crop Sci 36:1271–1277
Louvrier P, Laguerre G, Amarger N (1996) Distribution of symbiotic genotypes in Rhizobium leguminosarum biovar viciae populations isolated directly from soils. Appl Environ Microbiol 62:4202–4205
Marmur J (1961) A procedure for the isolation of DNA from microorganisms. J Mol Biol 3:208–218
Mutch LA, Tamimi SM, Young JPW (2003) Genotypic characterisation of rhizobia nodulating Vicia faba from the soils of Jordan: a comparison with UK isolates. Soil Biol Biochem 35:709–714
Mutch LA, Young JPW (2004) Diversity and specificity of Rhizobium leguminosarum biovar viciae on wild and cultivated legumes. Mol Ecol 13:2435–2444
Nick G, Rasanen LA, de Lajudie P, Gillis M, Lindström K (1999) Genomic screening of rhizobia isolated from root nodules of tropical leguminous trees using DNA-DNA dot-blot hybridization and rep-PCR. Syst Appl Microbiol 22:287–299
Rademaker JLW, Hoste B, Louws FJ, Kersters K, Swings J, Vauterin L, Vauterin P, de Bruijn FJ (2000) Comparison of AFLP and rep-PCR genomic fingerpring with DNA-DNA homology studies: Xanthomonas as a model system. Int J Syst Evol Microbiol 50:665–677
Resh SC, Binkley D, Parrotta JA (2002) Greater soil carbon sequestration under nitrogen-fixing trees compared with Eucalyptus species. Ecosystem 5:217–231
Sarita S, Sharma PK, Priefer UB, Prell J (2005) Direct amplification of rhizobial nodC sequences from soil total DNA and comparison to nodC diversity of root nodule isolates. FEMS Microbiol Ecol 54:1–11
Souza V, Eguiarte LE (1997) Bacteria gone native vs. bacteria gone awry? Plasmidic transfer and bacterial evolution. Proc Natl Acad Sci USA 94:5501–5503
Terefework Z, Kaijalainen S, Lindström K (2001) AFLP fingerprinting as a tool to study the genetic diversity of Rhizobium galegae isolated from Galega orientalis and Galega officinalis. J Biotechnol 91:169–180
Thompson JD, Higgins DG, Gibson TJ (1994) Clustal-W—improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Thompson HC, Kelly WC (1957) Vegetable crops, 5th edn. Mograw-Hill Book Company, New York
van Berkum P, Beyene D, Vera FT, Keyser HH (1995) Variability among Rhizobium strains originating from nodules of Vicia faba. Appl Environ Microbiol 61:2649–2653
Vincent JM (1970) A manual for the practical study of root nodule bacteria. IBP handbook 15. Blackwell, Oxford
Vinuesa P, Rademaker JL, Bruijn FJ, Werner D (1998) Genotypic characterization of Bradyrhizobium strains nodulating endemic woody legumes of the Canary islands by PCR-restriction fragment length polymorphism analysis of genes encoding 16S rRNA (16S rDNA) and 16S-23S rDNA intergenic spacers, repetitive extragenic palindromic PCR genomic fingerprinting, and partial 16S rDNA sequencing. Appl Environ Microbiol 64:2096–2104
Weir BS, Turner SJ, Silvester WB, Park DC, Young JM (2004) Unexpectedly diverse Mesorhizobium strains and Rhizobium leguminosarum nodulate native legume genera of New Zealand, while introduced legume weeds are nodulated by Bradyrhizobium species. Appl Environ Microbiol 70:5980–5987
Weisburg WG, Barns SM, Pelletior DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Wolde-meskel E, Terefework Z, Lindström K, Frostegård Å (2004) Rhizobia nodulating African Acacia spp. and Sesbania sesban trees in southern Ethiopian soils are metabolically and genomically diverse Soil Biol Biochem 36:2013–2025
Ye Y (2003) “Zhong Guo Can Dou Xue” (Chinese faba bean) (in Chinese). China Agriculture Press, Beijing
Young JPW, Mutch LA, Ashford DA, Zézé A, Mutch KE (2003) The molecular evolution of host specificity in the rhizobium-legume symbiosis. In: Hails R, Godfray HCJ, Beringer J (eds) Genes in the environment. Blackwell, Oxford, pp 245–257
Zézé A, Mutch LA, Young JPW (2001) Direct amplification of nodD from community DNA reveals the genetic diversity of Rhizobium leguminosarum in soil. Environ Microbiol 3:363–370
Acknowledgments
We thank Dr. Min Qi Lu, Dr. Qiang Chen and other collaborators for their valuable contribution in collecting and isolating the rhizobia. This work was financial supported by the foundation of the State Key Basic Research and Development Plan of China (grant 2006CB100206), and by National Project for Basic S&T Platform Construction (grant 2005DKA21201-10).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Tian, C.F., Wang, E.T., Han, T.X. et al. Genetic diversity of rhizobia associated with Vicia faba in three ecological regions of China. Arch Microbiol 188, 273–282 (2007). https://doi.org/10.1007/s00203-007-0245-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-007-0245-6