
Abstract
Summary
IL-35 is a novel anti-inflammatory cytokine, but the exact role of IL-35 in the progression of RA remains unclear, especially associated with osteoporosis and bone erosion. The present research has not been reported. Our purpose is to study how IL-35 affects RA bone destruction.
Introduction
This study investigated the effect of interleukin-35 (IL-35) on OPG and RANKL expression in collagen-induced arthritis (CIA) in rats and in cultured fibroblast-like synoviocytes (FLS).
Methods
Thirty DBA/1J mice were randomly assigned to three groups (n = 10 per group): the control group, the CIA group, and the CIA + IL-35 group. Collagen-induced arthritis was induced by immunization with collagen. IL-35 was intraperitoneally injected daily for 10 days, starting from the 24th day after immunization. FLS cells were isolated and cultured from CIA. The expression of IL-17, RANKL, and OPG was determined by RT-PCR and Western blot. Each experiment was repeated three times.
Results
CIA mice exhibited arthritis symptoms on day 24, followed by a rapid progression of arthritis. The expression of IL-17 and RANKL was increased and the expression of OPG was decreased in CIA mice compared with control mice. IL-35 treatment inhibited the development of arthritis in CIA mice, accompanied by a decrease in the expression of IL-17 and RANKL and an increase in the expression of OPG. Furthermore, IL-35 dose-dependently inhibited the expression of RANKL and increased the expression of OPG in cultured FLS cells.
Conclusion
IL-35 inhibits RANKL expression and increases OPG expression in CIA mice. IL-35 may be used for treating rheumatoid arthritis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease in which chronic inflammation causes severe damage of the joints [1, 2].RA is associated with a significant mortality and morbidity rate, and if untreated, the 2–3-year morbidity rate of RA can be as high as 70 % [3]. Bone destruction contributes to the morbidity associated with RA [4]. Bone erosion occurs early following the onset of RA and progresses throughout the course of the disease [5, 6]. Osteoclasts are the primary bone resorptive cells that are responsible for bone resorption, and play an important role in bone destruction in RA [7].Targeting osteoclasts represents a promising strategy for treating RA [8].
The receptor activator of the nuclear factor κB ligand (RANKL) pathway is important for osteoclast differentiation [9]. RANKL binds to the RANK receptor on osteoclast precursors and subsequently drives osteoclastogenesis [10]. The RANKL receptor, osteoprotegerin (OPG), is expressed by osteoblasts and acts to inhibit the RANKL/RANK pathway, leading to inhibition of osteoclast differentiation and formation [11].Several studies have shown that RANKL is upregulated in animal models of RA and in patients with RA [12–14], suggesting that RANKL plays an important role in the pathogenesis of RA. Activation of the RANKL-RANK pathway can lead to increased osteoclast formation in the joints, thus promoting bone destruction in RA [15].
Interleukin-35 (IL-35) is a novel anti-inflammatory cytokine comprising of Epstein-Barr virus-induced protein (EBI)-3 and IL-12 p35. It belongs to the IL-12 family and is specifically secreted by regulatory T cells (Treg) [16]. IL-35 has been found to inhibit the progression of RA in a mouse model of collagen-induced arthritis (CIA) [17]. In addition, IL-35 has been shown to inhibit the proliferation of Th1 and Th17 cells. It has also been demonstrated to reduce the development of T cell-dependent colitis in mice [18]. Previous reports suggest that Th17 cells play an important role in osteoclast differentiation and bone destruction in RA [19]. IL-35 suppresses differentiation of Th17 cells [16, 17]. Therefore, IL-35 may play an important role in the pathogenesis of RA.
In this study, we investigated the effect of IL-35 on the expression of RANKL and OPG in a mouse model of collagen-induced arthritis (CIA) and in cultured FLS isolated from CIA mice. This study aimed to investigate the effect of IL-35 on the RANKL/OPG pathway in RA.
Materials and methods
Animals
The Institutional Animal Care and Use Committee of China Medical University approved all experimental protocols associated with this study. Thirty DBA/1J mice (male, 6–8 weeks old, weighing 22.8 ± 2.6 g) were obtained from HFK Bioscience Co. Ltd, Beijing, China. Mice were housed at room temperature (25 °C) with 60 % humidity and a 12 h light/dark cycle. Mice were fed standard rat chow and water. The animals were randomly assigned to three groups: the control group (n = 10), the CIA group (n = 10), and the CIA + IL-35 group (n = 10).
Reagents
Chicken Type II collagen and interleukin-35 (mouse):Fc (human IgG1) were purchased from Sigma Company, USA. The complete Freund’s adjuvant (CFA), incomplete Freund’s adjuvant (IFA), and goad anti-mouse IL-17 monoclonal antibodies were obtained from R&D Company, USA. Goat anti-mouse vimentin polyclonal antibodies were obtained from Proteintech, USA. Goat anti-mouse CD68 monoclonal antibodies, goat anti-mouse RANKL monoclonal antibodies, goat anti-mouse OPG monoclonal antibodies, rabbit anti-goat horseradish peroxidase-conjugated secondary antibodies, RT-PCR kits, TRizol reagents, total protein extraction kit, and BCA protein quantification kit were obtained from Abcam Company, UK.
Collagen-induced arthritis animal model
Mice were immunized 3 days after feeding commenced. Mice in the CIA and CIA + IL-35 groups were immunized subcutaneously at the base of the tail with 0.2 ml of chicken type II collagen (2 mg/ml) in acetic acid, and emulsified in CFA at a ratio of 1:1. Twenty-one days after primary immunization, mice were boosted subcutaneously at the base of the tail with 0.2 ml of chicken type II collagen (2 mg/ml) in IFA using a ratio of 1:1. The collagen-induced arthritis models were successfully generated on day 24 after primary immunization when the mice had an arthritis index (AI) > 4, and the volume of the footpad swelling below the angle on one paw had increased by >1.6-fold.
From day 24, the mice in the CIA + IL-35 group received a daily intraperitoneal injection of IL-35 (2 μg) in phosphate buffered saline (PBS) for 10 days. The mice in the control and CIA group received the same volume of PBS once a day for 10 days. The severity of arthritis in each paw was scored as follows: 0, normal; 1, mild swelling with or without erythema and with no joint deformities; 2, severe swelling without joint deformities; and 3, joint deformities and ankylosis. The cumulative score of all four paws was used as the AI. The maximum score was 12 points for each mouse. The severity of swelling was evaluated by measuring the thickness of the footpad.
Isolation and culture of fibroblast-like synoviocytes
Under aseptic conditions, the joint cavity of mice in the CIA group was opened up to expose the synovial tissues. After removal of the fat tissues and blood vessels, the synovial tissues were cut into small pieces (1 mm3). The tissue pieces (8–12 pieces) were then evenly placed on the bottom of a culture bottle with a distance of 5 mm between the pieces. The synovial pieces were cultured in H-DMEM supplemented with 15 % FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5 % CO2 for 24 h. The synovial pieces were removed, and culture media was changed every 2 days. The cells were passaged at 70–80 % confluence and cells from passages 3–5 were used for the following experiments. Fibroblast-like synoviocytes (FLS) were identified by immunocytochemistry for their vimentin-immunopositivity and CD68-immunonegativity.
The cells were seeded on a 24-well plate at a density of 5 × 104/ml, and were assigned to four groups (n = 4 per group): the control group without IL-35 treatment, the 25 ng/ml IL-35 group treated with 25 ng/ml IL-35, the 50 ng/ml IL-35 group treated with 50 ng/ml IL-35, and the 100 ng/ml IL-35 group treated with 100 ng/ml IL-35. The cells were cultured in H-DMEM supplemented with 15 % FBS at 37 °C in 5 % CO2 for 12 h. IL-35 was subsequently added into each well. Cells were further cultured for 48 h, and used for the following experiments.
Real-time RT-PCR
Total RNA was isolated from synovial tissues or FLS cells using TRizol reagents according to the manufacturer’s protocol. RNA was reverse transcribed into complementary DNA using a reverse transcription system. RT-PCR was performed in a final volume of 20 μl containing 2 μl of cDNA, 0.5 μl of each primer, and 10 μl of SYBR Green. For the synovial tissues of mice, the primers used were as follows: 5′-CTTCGTGCCTTGATGGA-3′ (forward) and 5′-TTGGGAAAGTGGGATGT-3′ (reverse) for OPG, 5′-ACCAAGATGGCTTCTATTACC-3′ (forward) and 5′-TCCCTCCTTTCATCAGGTTAT-3′ (reverse) for RANKL, and 5′-ACCGCAATGAAGACCCTGAT-3′ (forward) and 5′-ACACCCACCAGCATCTTCTC-3′ (reverse) for IL-17. The reaction conditions were as follows: 94 °C for 2 min; 30 cycles of 94 °C for 30 sec, 65 °C for 30 sec, 72 °C for 30 sec; and 72 °C for 2 min. The PCR products were analyzed by electrophoresis on an agarose gel containing ethidium bromide, and the gel was viewed with a digital imaging system. The relative expression of IL-17, RANKL, and OPG were normalized with a β-actin internal control.
For FLS cells, the primers used were as follows:5′-TGAGAGAACGAGAAAGACCTGC-3′ (forward) and 5′-CGGATTGAACCTGATTCCCTAT-3′ (reverse) for OPG, and 5′-CAACATTTGCTTTCGGCATCAT-3′ (forward) and 5′- TTTCGTGCTCCCTCCTTTCATC-3′ (reverse) for RANKL. The reaction conditions were as follows: 95 °C for 10 min; 40 cycles of 95 °C for 10 sec, 60 °C for 20 sec, and 72 °C for 30 sec; and 4 °C for 5 min. β-actin (size, 453 bp) was used as an internal control. The relative expression of RANKL and OPG normalized with the β-actin internal control was calculated using the 2-ddCT method.
Western blot
The synovial tissues of mice in each group or FLS cells were homogenized on ice in a lysis buffer. Proteins were resolved using SDS–PAGE, and transferred onto polyvinylidene fluoride membranes by electroblotting. Membranes were incubated with primary antibodies against RANKL (dilution 1:1,000), OPG (dilution 1:2,000), and IL-17(dilution 1:1,000) at 4 °C overnight. Membranes were incubated with secondary antibodies (horseradish peroxidase-conjugated rabbit anti-goat IgG, dilution, 1:2000) at room temperature for 2 h. A chemiluminescence detection system was used to detect protein bands. Band intensities were analyzed using Quantity one software.
Immunofluorescence
The FLS cells were placed on a 24-well plate at a density of 2 × 104/ml. After culture for 48 h, cells were fixed in 4 % paraformaldehyde and permeabilized with 0.1 % Triton X-100 for 15 min. The cells were then incubated in 5 % BSA for 20 min at room temperature to block non-specific protein binding sites. The cells were incubated with primary antibodies against RANKL (dilution 1:100) at 4 °C overnight. After the primary antibody was removed following washing with PBS, immunoreactivity was detected by incubation with Cy3-coupled secondary antibodies (dilution 1:200) at room temperature for 1 h. The cells were counterstained with DAPI and examined using a fluorescent microscope. The IOD and AA were measured using Image J software, and the mean IOD/AA ratio was used for comparison among groups.
Statistical analysis
Analyses were performed using SPSS 19.0. Data are presented as mean ± standard deviation. The difference of means among groups were compared by one-way analysis of variance (ANOVA) followed by post hoc LSD (least significant difference) tests. Probability values less than 0.05 were considered as statistically significant.
Results
IL-35 inhibits arthritis symptoms in CIA mice
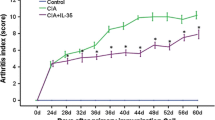
Compared with the control mice, the CIA mice exhibited arthritis symptoms on day 24. The AI score rapidly increased with time and reached a peak on day 48 (Fig. 1). The AI score was significantly reduced in CIA mice treated with IL-35 (P < 0.05), suggesting that IL-35 inhibited arthritis symptoms in CIA mice.

IL-35 reduced the arthritis index in mice with collagen-induced arthritis (CIA). IL-35 (2 μg/d, 0.1 mg/kg) was administered intraperitoneally on day 24 after primary immunization with collagen. n = 5.*P < 0.05 vs CIA mice
IL-35 inhibits the expression of IL-17 and RANKL and increases the expression of OPG in synovial tissues of CIA mice
We further examined the effect of IL-35 on the expression of IL-17, RANKL, and OPG using RT-PCR and Western blot. RT-PCR results showed that the expression of IL-17 and RANKL was significantly increased in CIA mice compared with control mice (Fig. 2a, b). IL-35 treatment significantly inhibited CIA-induced increases in the expression of IL-17 and RANKL (Fig. 2a, b). In contrast, the expression of OPG was significantly decreased in CIA mice compared with control mice (Fig. 2c). IL-35 treatment significantly increased the expression of OPG in CIA mice (Fig. 2c).Consistent with the RT-PCR results, Western blot analysis showed that IL-35 treatment significantly reduced the protein expression of IL-17 and RANKL (Fig. 3a, b) and increased the protein expression of OPG (Fig. 3c) in CIA mice.

The mRNA expression of IL-17, RANKL, and OPG in the synovial tissues of mice from the control, CIA, and CIA + IL-35 groups on day 48. a–c Representative agarose gel showing the expression of IL-17 (a), RANKL (b), and OPG (c) in mice from the control, CIA, and CIA + IL-35 groups. d–f The relative mRNA expression of IL-17 (d), RANKL (e), and OPG (f) normalized with the expression of β-actin. n = 5.*P < 0.05 vs control, # P < 0.05 vs CIA

Western blot analysis showing the protein expression of IL-17, RANKL, and OPG in the synovial tissues of mice from the control, CIA, and CIA + IL-35 groups on day 48. a–c Representative Western blot showing the expression of IL-17 (a), RANKL (b), and OPG (c) in mice from the control, CIA, and CIA + IL-35 groups. e–f Quantification of the expression of IL-17 (d), RANKL (e), and OPG (f). n = 5. *P < 0.05 vs control, # P < 0.05 vs CIA
IL-35 inhibits the expression of RANKL in cultured FLS cells
We then examined the effect of IL-35 on the expression of RANKL in cultured FLS cells. RT-PCR showed that IL-35 treatment for 48 h significantly decreased the messenger RNA (mRNA) expression of RANKL in cultured FLS cells (P < 0.05, Fig. 4a). IL-35 inhibited the expression of RANKL in a dose-dependent manner (Fig. 4a). Consistent with RT-PCR results, Western blot analysis showed that IL-35 inhibited the protein expression of RANKL in a dose-dependent manner (Fig. 4b, c). Similarly, IL-35 dose-dependently reduced RANKL-immunopositive staining in cultured FLS cells (Fig. 5).

IL-35 inhibited the expression of RANKL in cultured FLS cells. a RT-PCR results showing the relative expression of RANKL mRNA in FLS cells treated with 0, 25, 50, and 100 ng/ml IL-35. The relative mRNA expression of RANKL was normalized with the expression of β-actin. n = 4. *P < 0.05 vs 0 ng/ml IL-35, # P < 0.05 vs 25 ng/ml IL-35. b Representative Western blot showing the expression of RANKL in cultured FLS cells treated with 0, 25, 50, and 100 ng/ml IL-35. c Quantification of the protein expression of RANKL.n = 4. *P < 0.05 vs 0 ng/ml IL-35, # P < 0.05 vs 25 ng/ml IL-35

Representative immunofluorescence staining for RANKL in cultured FLS cells treated with 0, 25, 50 and 100 ng/ml IL-35. Magnification, ×400. Scale bar: 50 μm
IL-35 increases the expression of OPG in cultured FLS cells
We further investigated the effect of IL-35 on the expression of OPG in cultured FLS cells. RT-PCR showed that IL-35 treatment for 48 h significantly increased the expression of OPG in cultured FLS cells (P < 0.05, Fig. 6a). The effect of IL-35 on the expression of OPG occurred in a dose-dependent manner (Fig. 6a). Consistent with RT-PCR results, Western blot analysis showed that IL-35 increased the protein expression of OPG in a dose-dependent manner (Fig. 6b, c).

IL-35 increased the expression of OPG in cultured FLS cells. a RT-PCR results showing the relative expression of OPG mRNA in FLS cells treated with 0, 25, 50, and 100 ng/ml IL-35. The relative mRNA expression of RANKL was normalized with the expression of β-actin. n = 4. *P < 0.05 vs 0 ng/ml IL-35, # P < 0.05 vs 25 ng/ml IL-35. b Representative Western blot showing the expression of OPG in cultured FLS cells treated with 0, 25, 50, and 100 ng/ml IL-35. c Quantification of the protein expression of OPG.n = 4. *P < 0.05 vs 0 ng/ml IL-35, # P < 0.05 vs 25 ng/ml IL-35
Discussion
A hallmark of RA pathogenesis is bone destruction that is caused by an elevation of bone resorption by osteoclasts [4]. It is well known that osteoclasts are dependent on the presence of RANKL, a signal for osteoclast differentiation and formation [2]. It has been reported that RANKL is upregulated in the synovial tissues in patients with RA [12, 13], suggesting that RANKL contributes to the pathogenesis of RA. Targeting RANKL may be a novel therapeutic strategy for the treatment of RA. In the present study, we investigated the effect of IL-35 on the expression of RANKL and its receptor OPG in a mouse model of collagen-induced arthritis, a well-established animal model for human RA [20]. We found that IL-35 treatment inhibited the development of arthritis in CIA mice, and was accompanied by a decrease in the expression of RANKL and an increase in the expression of OPG. Furthermore, we found that IL-35 dose-dependently inhibited the expression of RANKL and increased the expression of OPG in cultured FLS cells. Our study suggests that IL-35 may inhibit osteoclast function by promoting the expression of OPG and inhibiting the expression of RANKL. IL-35 may be a potential therapeutic agent for the treatment of RA.
Niedhala et al. reported that IL-35 treatment inhibited arthritis symptoms in CIA mice [17]. Consistent with this report, we found that IL-35 inhibited the development of arthritis in CIA mice. Several studies have demonstrated that IL-35 inhibits proliferation and differentiation of effectors T cells, increases secretion of the anti-inflammatory cytokine IL-10, and promotes the proliferation and differentiation of regulatory T cells, including Th1, Th2, and Th17 [21–27]. Recent studies have shown that Th17 cells can regulate osteoclast differentiation and promote bone destruction and are therefore believed to be involved in the pathogenesis of RA [19, 28]. Th17 cells secrete many cytokines including IL-17, which can promote osteoclast differentiation [28]. IL-17 has been found in high concentrations in the synovium and synovial fluid of patients with RA and its expression levels are associated with joint damage progression in RA [29–31]. Furthermore, Niedbala et al. reported that IL-35 inhibited differentiation of Th17 cells and reduced secretion of IL-17 [17]. Our findings that IL-35 inhibited arthritis development and inhibited CIA-induced increases in the expression of IL-17 suggest that IL-35 may inhibit RA via reducing secretion of IL-17 from Th17 cells.
IL-17 has been found to promote the expression of RANKL, while inducing osteoclastogenesis [32, 33]. RANKL has been known to play an important role in osteoclast cell differentiation and bone resorption [9]. Both clinical and animal studies have shown that RANKL is upregulated in the synovial tissues [12–14]. Consistent with previous studies, we found that IL-17 and RANKL were significantly upregulated in synovial tissues in CIA mice, suggesting that IL-17 and RANKL may contribute to the pathogenesis of RA. Furthermore, we found that IL-35 treatment reduced the expression of IL-17 and RANKL in CIA mice, suggesting that IL-35 may inhibit arthritis progression by reducing the production of IL-17 and RANKL. The inhibitory effect of IL-35 on the expression of RANKL in vivo was further supported by the findings that IL-35 dose-dependently inhibited the expression of RANKL in cultured FLS cells. It has been reported that inhibition of RANKL prevents loss of bone and cartilage in mice [34]. Our findings suggest that IL-35 may be used to prevent bone loss via inhibition of RANKL, and thus may be a novel therapeutic agent for the treatment of RA.
OPG is a decoy receptor for RANKL with a high affinity and competes with RANK for RANKL binding. It thus functions as an inhibitor of RANK-RANKL interaction and inhibits osteoclast maturation and activation [11, 35]. In the present study, we found that IL-35 inhibited the development of arthritis in CIA mice, accompanied by an increase in the expression of OPG and a decrease in the expression of RANKL in synovial tissues of CIA mice, suggesting that IL-35 may inhibit arthritis by promoting the expression of OPG which inhibits the RANKL pathway. Furthermore, we found that IL-35 dose-dependently increased the expression of OPG in cultured FLS cells, suggesting that IL-35 induced OPG expression. Several studies have shown that OPG treatment slows joint destruction, decreases bone erosion, and prevents bone loss in animal models of arthritis [36–39]. Our findings suggest that IL-35 may prevent bone loss and destruction via promoting the expression of OPG.
Although the overall structure and function of the immune system is very similar in humans and mice, there are some significant differences between mouse and human immunology, including the expression and function of many cytokines and their receptors. It has been reported that the expression of IL-35 is different between mice and humans [25, 40]. IL-35 is constitutively expressed in non-stimulated mouse Tregs and stimulated human Tregs, but not in non-stimulated human Tregs [40–42]. The AU-rich element (ARE) binding proteins and microRNAs that regulate IL-35 subunit transcripts have been reported to contribute to the differential expression status associated with IL-35 [25], possibly leading to differences in IL-35 expression between humans and mice. Although the expression status of IL-35 is different between humans and mice, IL-35 has been reported to produce similar effects in suppressing effector T cells such as Th1, Th2, and Th17 cells in both humans and mice [16, 43]. In addition, it has been demonstrated that IL-35 can induce an increase in regulatory B cells in both humans and mice [44]. Therefore, it appears that IL-35 produces similar effects in both humans and mice.
In this study, we found that IL-35 inhibited the progression of arthritis in a mouse model of CIA, a well-established model of human RA. Similarly, several studies have reported that IL-35 inhibited inflammation in collagen-induced arthritis and experimental colitis in mice [16, 18], suggesting that IL-35 may exert anti-inflammatory effects in mice. However, to date, there are no clinical reports showing the anti-inflammatory effect of IL-35 in patients with RA. Our preliminary study has shown that the serum levels of IL-35 are significantly higher in patients with RA compared with healthy controls (unpublished data), suggesting that IL-35 may be involved in the pathogenesis of RA. Furthermore, Senolt et al. [45] reported that the synovial levels of IL-35 are significantly correlated with disease activity in patients with RA, further suggesting that IL-35 plays an important role in the development of RA. These results suggest that IL-35 may be a potentially novel therapeutic strategy for the treatment of RA. Further clinical studies are required to confirm our observations that IL-35 inhibited the development of arthritis by upregulating OPG and downregulating RANKL in a murine model.
Conclusions
In summary, we found that IL-35 inhibited the progression of arthritis. This was accompanied by a decrease in the expression of IL-17 and RANKL, and an increase in the expression of OPG in a mouse model of CIA. In cultured FLS cells, IL-35 dose-dependently inhibited the expression of RANKL and increased the expression of OPG. Our findings suggest that IL-35 may prevent the development of arthritis by upregulating OPG and downregulating RANKL.
Abbreviations
- RA:
-
Rheumatoid arthritis
- FLS:
-
Fibroblast-like synoviocytes
- IL-35:
-
Interleukin-35
- CIA:
-
Collagen-induced arthritis
- RANK:
-
Receptor activator of nuclear factor κB
- RANKL:
-
Receptor activator of nuclear factor κB ligand
- OPG:
-
Osteoprotegerin
- EBI:
-
Epstein-Barr virus-induced protein
- Treg:
-
Regulatory T cells
- Th17:
-
T helper type 17
- CFA:
-
Complete Freund’s adjuvant
- IFA:
-
Incomplete Freund’s adjuvant
- FBS:
-
Fetal bovine serum
- HE:
-
Hematoxylin and eosin
- H-DMEM:
-
High glucose Dulbecco’s Modified Eagle’s medium
- DAB:
-
3,3′-diaminobenzidine
- SDS–PAGE:
-
Sodium dodecylsulphate-polyarcylamide gel electrophoresis
- IOD:
-
Integratedoptical density
- AA:
-
Accumulated area
- ANOVA:
-
One-way analysis of variance
- SD:
-
Standard deviation
- LSD:
-
Least significant difference
References
McInnes IB, Schett G (2011) The pathogenesis of rheumatoid arthritis. N Engl J Med 365:2205–2219
Tanaka S (2013) Regulation of bone destruction in rheumatoid arthritis through RANKL-RANK pathways. World J Orthop 4:1–6
Jutley G, Raza K, Buckley CD (2015) New pathogenic insights into rheumatoid arthritis. Curr Opin Rheumatol 27:249–255
Goldring SR (2002) Bone and joint destruction in rheumatoid arthritis: what is really happening? J Rheumatol Suppl 65:44–48
Sharp JT, Wolfe F, Mitchell DM, Bloch DA (1991) The progression of erosion and joint space narrowing scores in rheumatoid arthritis during the first twenty-five years of disease. Arthritis Rheum 34:660–668
Kaarela K, Kautiainen H (1997) Continuous progression of radiological destruction in seropositive rheumatoid arthritis. J Rheumatol 24:1285–1287
Schett G (2007) Cells of the synovium in rheumatoid arthritis. Osteoclasts. Arthritis Res Ther 9:203
Choi Y, Arron JR, Townsend MJ (2009) Promising bone-related therapeutic targets for rheumatoid arthritis. Nat Rev Rheumatol 5:543–548
Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423:337–342
Xing L, Schwarz EM, Boyce BF (2005) Osteoclast precursors, RANKL/RANK, and immunology. Immunol Rev 208:19–29
Walsh MC, Choi Y (2014) Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol 5:511
Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR (2000) Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum 43:250–258
Shigeyama Y, Pap T, Kunzler P, Simmen BR, Gay RE, Gay S (2000) Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum 43:2523–2530
Stolina M, Adamu S, Ominsky M, Dwyer D, Asuncion F, Geng Z, Middleton S, Brown H, Pretorius J, Schett G, Bolon B, Feige U, Zack D, Kostenuik PJ (2005) RANKL is a marker and mediator of local and systemic bone loss in two rat models of inflammatory arthritis. J Bone Miner Res 20:1756–1765
Geusens P (2012) The role of RANK ligand/osteoprotegerin in rheumatoid arthritis. Ther Adv Musculoskelet Dis 4:225–233
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA (2007) The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450:566–569
Niedbala W, Wei XQ, Cai B, Hueber AJ, Leung BP, McInnes IB, Liew FY (2007) IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol 37:3021–3029
Wirtz S, Billmeier U, McHedlidze T, Blumberg RS, Neurath MF (2011) Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 141:1875–1886
Sato K (2008) Th17 cells and rheumatoid arthritis—from the standpoint of osteoclast differentiation. Allergol Int 57:109–114
Anthony DD, Haqqi TM (1999) Collagen-induced arthritis in mice: an animal model to study the pathogenesis of rheumatoid arthritis. Clin Exp Rheumatol 17:240–244
Kochetkova I, Golden S, Holderness K, Callis G, Pascual DW (2010) IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J Immunol 184:7144–7153
Bettini M, Castellaw AH, Lennon GP, Burton AR, Vignali DA (2012) Prevention of autoimmune diabetes by ectopic pancreatic beta-cell expression of interleukin-35. Diabetes 61:1519–1526
Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA (2011) Cutting edge: Human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol 186:6661–6666
Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, Finkelstein D, Forbes K, Workman CJ, Brown SA, Rehg JE, Jones ML, Ni HT, Artis D, Turk MJ, Vignali DA (2010) IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol 11:1093–1101
Li X, Mai J, Virtue A, Yin Y, Gong R, Sha X, Gutchigian S, Frisch A, Hodge I, Jiang X, Wang H, Yang XF (2012) IL-35 is a novel responsive anti-inflammatory cytokine—a new system of categorizing anti-inflammatory cytokines. PLoS One 7:e33628
Collison LW, Pillai MR, Chaturvedi V, Vignali DA (2009) Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J Immunol 182:6121–6128
Whitehead GS, Wilson RH, Nakano K, Burch LH, Nakano H, Cook DN (2012) IL-35 production by inducible costimulator (ICOS)-positive regulatory T cells reverses established IL-17-dependent allergic airways disease. J Allergy Clin Immunol 129(207–215):e201–e205
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H (2006) Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 203:2673–2682
Cho ML, Yoon CH, Hwang SY, Park MK, Min SY, Lee SH, Park SH, Kim HY (2004) Effector function of type II collagen-stimulated T cells from rheumatoid arthritis patients: cross-talk between T cells and synovial fibroblasts. Arthritis Rheum 50:776–784
Kohno M, Tsutsumi A, Matsui H, Sugihara M, Suzuki T, Mamura M, Goto D, Matsumoto I, Ito S, Suguro T, Sumida T (2008) Interleukin-17 gene expression in patients with rheumatoid arthritis. Mod Rheumatol 18:15–22
Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, Shnier R, Portek IJ (2006) Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum 54:1122–1131
Adamopoulos IE, Bowman EP (2008) Immune regulation of bone loss by Th17 cells. Arthritis Res Ther 10:225
Neumann E, Gay S, Muller-Ladner U (2005) The RANK/RANKL/osteoprotegerin system in rheumatoid arthritis: new insights from animal models. Arthritis Rheum 52:2960–2967
Wu Y, Liu J, Feng X, Yang P, Xu X, Hsu HC, Mountz JD (2005) Synovial fibroblasts promote osteoclast formation by RANKL in a novel model of spontaneous erosive arthritis. Arthritis Rheum 52:3257–3268
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–323
Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M, Lang S, Turk B, Pietschmann P, Woloszczuk W, Haralambous S, Kollias G, Steiner G, Smolen JS, Schett G (2002) Tumor necrosis factor alpha-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum 46:785–792
Schett G, Redlich K, Hayer S, Zwerina J, Bolon B, Dunstan C, Gortz B, Schulz A, Bergmeister H, Kollias G, Steiner G, Smolen JS (2003) Osteoprotegerin protects against generalized bone loss in tumor necrosis factor-transgenic mice. Arthritis Rheum 48:2042–2051
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A et al (1999) Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402:304–309
Bolon B, Campagnuolo G, Feige U (2002) Duration of bone protection by a single osteoprotegerin injection in rats with adjuvant-induced arthritis. Cell Mol Life Sci 59:1569–1576
Bardel E, Larousserie F, Charlot-Rabiega P, Coulomb-L’Hermine A, Devergne O (2008) Human CD4+ CD25+ Foxp3+ regulatory T cells do not constitutively express IL-35. J Immunol 181:6898–6905
Collison LW, Vignali DA (2008) Interleukin-35: odd one out or part of the family? Immunol Rev 226:248–262
Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA (2013) Retraction. Human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol 191:2018
Zeng JC, Zhang Z, Li TY, Liang YF, Wang HM, Bao JJ, Zhang JA, Wang WD, Xiang WY, Kong B, Wang ZY, Wu BH, Chen XD, He L, Zhang S, Wang CY, Xu JF (2013) Assessing the role of IL-35 in colorectal cancer progression and prognosis. Int J Clin Exp Pathol 6:1806–1816
Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, Wingfield PT, Kim SH, Egwuagu CE (2014) Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med 20:633–641
Senolt L, Sumova B, Jandova R, Hulejova H, Mann H, Pavelka K, Vencovsky J, Filkova M (2015) Interleukin 35 synovial fluid levels are associated with disease activity of rheumatoid arthritis. PLoS One 10:e0132674
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81172867 and 81471542).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The Institutional Animal Care and Use Committee of China Medical University approved all experimental protocols associated with this study.
Conflicts of interest
None.
Rights and permissions
About this article

Cite this article
Li, Y., Li, D., Li, Y. et al. Interleukin-35 upregulates OPG and inhibits RANKL in mice with collagen-induced arthritis and fibroblast-like synoviocytes. Osteoporos Int 27, 1537–1546 (2016). https://doi.org/10.1007/s00198-015-3410-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-015-3410-9